

Acute lymphoblastic leukemia (ALL) is a disease of either B-cell (80–85%) or T-cell (20–25%) derivation. Several molecular aberrations (i.e. BCR-ABL1, MLL/AFF1, SIL/TAL1 and E2A/PBX1) confer an overall poor outcome.1,2 However, a proportion of patients do not carry known genetic abnormalities and have a heterogeneous clinical course.
P53 plays a crucial role in cell cycle regulation and apoptosis after DNA damage, and its role in tumorigenesis is well-recognized in solid and hematologic malignancies, particularly acute myeloid leukemia (AML) and chronic lymphocytic leukemia (CLL), in which its deregulation represents an important predictor of poor outcome.3–10
In ALL, TP53 mutations have been poorly investigated, mainly in children, for whom the incidence is low at diagnosis, increases at relapse, and is associated with poor outcome.8–10 In adult ALL, the few studies performed include mostly relapsed cases and small cohorts of patients.11,12
We evaluated 98 newly diagnosed adult ALL cases to address the incidence and prognostic impact of TP53 mutations; in 10 cases, paired material collected at relapse was also available.
Sixty-two cases had B-cell ALL (B-ALL) and 36 were TALL. Within B-ALL cases, 25 were BCR-ABL1, 9 MLL/AFF1+ and 4 E2A/PBX1+; within T-ALL cases, 7 were SIL/TAL1+, 1 NUP214/RAP+ and 1 SET/NUP214+. The remaining 24 B-ALL and 27 T-ALL cases did not carry the above mentioned fusion genes (defined “negative for recurrent fusion genes”).
TP53 mutation analysis was performed using the AmpliChip p53 Research Prototype Test (Roche Molecular Systems Inc., Pleasanton, CA, USA).13 Results were validated by Sanger Sequencing.14
Statistical tests were two-sided and P≤0.05 was considered significant. P values are not reported since significance was not reached because of the small number of TP53 mutated patients. Further details are given in the Online Supplementary Appendix.
At diagnosis, TP53 mutations were detected in 8 of 98 patients (8.2%) (Figure 1A), similar to figures reported in AML at diagnosis5 and in CLL at the time of progression and first line treatment.6
Figure 1.

Distribution of TP53 mutations and clinical features of TP53 mutated patients. (A) Distribution of TP53 mutations in the whole cohort (n=98). (B) Distribution of TP53 mutations in B-ALL (n=62) and in T-ALL (n=36). (C) Clinical and biological features of TP53 mutant and wild-type patients. (D) Correlation between TP53 mutations and response to induction treatment.
The incidence of TP53 mutated ALL appears slightly higher in adults than in children: in fact, while in childhood B-ALL the reported prevalence is 2%8 in our cohort of 62 adult patients, 4 had a TP53 mutation (6.4%) (Figure 1B). In pediatric T-ALL,9TP53 mutations were detected in 5% of cases, while in our cohort of 36 adult cases, 4 carried TP53 mutations (11.1%) (Figure 1B).
Notably, all TP53 mutated T-ALL cases were negative for recurrent fusion genes. Of the 4 B-ALL cases with TP53 mutations, 3 did not harbor any recurrent fusion gene and one had a BCR-ABL1 rearrangement. Thus, a TP53 mutation was found only in one of 38 (2.6%) molecularly positive cases. This contrasts with Hof and colleagues10 who reported a significant association between TP53 mutations and MLL/AFF1 in a cohort of relapsed children. The differences may be explained by the fact that the analysis was performed at disease recurrence.
TP53 mutations appear, therefore, the most frequent mutation in adult ALL negative for recurrent fusion genes.
All mutations affected the DNA-binding domain and known hotspots (Table 1). All but one were confirmed by Sanger sequencing; the reasons for this single discordance are not clear, but it is likely that the clone harboring the TP53 mutation was too small to be detected using this technique.
Table 1.
Details of TP53 mutations identified at diagnosis.
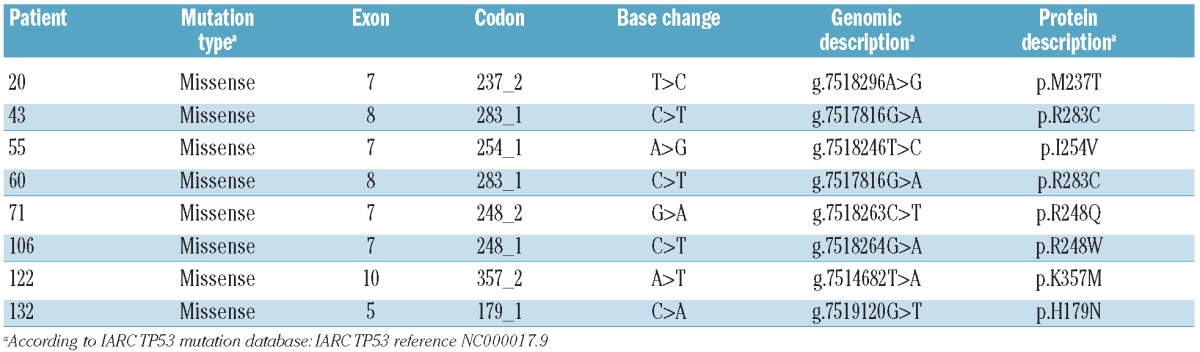
The majority of TP53 mutated patients were males, tended to be younger and had lower median white blood cell and higher platelet counts (Figure 1C).
Of the 8 TP53 mutated cases at diagnosis, 4 (50%) failed to achieve complete remission (CR). Since most of the mutations (n=7) occurred in cases negative for recurrent fusion genes, a detailed response to induction chemotherapy was evaluated in this subset, including 51 patients. Forty-three patients were evaluable: 30 achieved a CR, 6 were refractory, and 7 died during induction therapy.
When response to induction was correlated with TP53 status, 3 of 6 (50%) refractory cases harbored a TP53 mutation, suggesting a negative impact on CR achievement (P=0.059); one further TP53 mutated patient died during induction therapy (Figure 1D).
Of the 3 TP53 mutated patients who obtained a CR, 2 relapsed after 8 and 9 months, and one patient is in continuous CR 10 months later. Interestingly, this patient was enrolled in a pediatric-like regimen. Although the numbers are too small to draw definitive conclusions, it is tempting to speculate that more aggressive regimens may overcome TP53 disruption. A larger series of cases is needed to conclusively document the impact of TP53 mutations in adult ALL.
Paired genomic material at diagnosis and 1st relapse was available for 10 TP53 wild-type at diagnosis (7 B-ALL and 3 T-ALL). Two patients acquired the mutation at relapse: one patient had a BCR-ABL1+ B-ALL and the other a TALL: in one, ultra-deep sequencing of the diagnostic sample confirmed the absence of the mutation. Overall, 10 TP53 mutations were identified in the current study: 8 at diagnosis and 2 at relapse. These results confirm previous reports10–12 that TP53 mutations increase at disease reappearance. The number of mutations at relapse is lower than that reported for childhood ALL.12 This was probably because of sample selection criteria since in the pediatric series a backtracking of mutated cases was carried out, whereas in our study paired analysis only included TP53 wild-type cases at diagnosis. Furthermore, we analyzed a small cohort of paired diagnosis-relapse cases.
In conclusion, to the best of our knowledge, this is the largest adult ALL series studied so far and the results indicate that TP53 screening could be useful in a patient stratification algorithm for adult ALL. Furthermore, they indicate that TP53 mutations are more frequent in T-ALL than in B-ALL and can be detected in 14% of cases negative for recurrent fusion genes, regardless of lineage derivation. Given the negative influence on response to induction chemotherapy, we suggest that TP53 status should be investigated at diagnosis particularly in patients negative for recurrent fusion genes, where the genetic-based prognostic stratification is still limited.15 This is particularly relevant considering that, nowadays, the therapeutic algorithms for adult ALL are progressively being tailored on the basis of specific molecular lesions. Prospective analyses on larger cohorts are warranted.
Acknowledgments
We are also grateful to Roche Molecular Systems for providing part of the reagents and the AmpliChip p53 arrays. Finally, we are grateful to Dr. Irene Della Starza for part of the molecular analyses and Dr. Alfonso Piciocchi for providing help for part of the statistical analysis.
Footnotes
The online version of this article has a Supplementary Appendix.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
Funding: supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC), Special Program Molecular Clinical Oncology, 5 x 1000 Milan, Italy, the Ministero dell’Università e Ricerca (MIUR), Fondo per gli Investimenti della Ricerca di Base (FIRB), Rome, Italy, Fondazione Internazionale di Ricerca in Medicina Sperimentale (FIRMS), Torino, Italy, the Fondazione Buzzati-Traverso, Pavia, Italy, and the Compagnia di S. Paolo, Turin, Italy.
References
- 1.Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371(9617):1030–43 [DOI] [PubMed] [Google Scholar]
- 2.Bassan R, Hoelzer D. Modern therapy of acute lymphoblastic leukemia. J Clin Oncol. 2011;29(5):532–43 [DOI] [PubMed] [Google Scholar]
- 3.Peller S, Rotter V. TP53 in hematological cancer: low incidence of mutations with significant clinical relevance. Hum Mutat. 2003; 21(3):277–84 [DOI] [PubMed] [Google Scholar]
- 4.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358 (6381):15–6 [DOI] [PubMed] [Google Scholar]
- 5.Fenaux P, Preudhomme C, Quiquandon I, Jonveaux P, Laï JL, Vanrumbeke M, et al. Mutations of the P53 gene in acute myeloid leukaemia. Br J Haematol. 1992;80(2):178–83 [DOI] [PubMed] [Google Scholar]
- 6.Zenz T, Eichhorst B, Busch R, Denzel T, Häbe S, Winkler D, et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol. 2010;28(29):4473–9 [DOI] [PubMed] [Google Scholar]
- 7.Wattel E, Preudhomme C, Hecquet B, Vanrumbeke M, Quesnel B, Dervite I, et al. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood. 1994:84(9):3148–57 [PubMed] [Google Scholar]
- 8.Wada M, Bartram CR, Nakamura H, Hachiya M, Chen DL, Borenstein J, et al. Analysis of p53 mutations in a large series of lymphoid hematologic malignancies of childhood. Blood. 1993;82(10): 3163–9 [PubMed] [Google Scholar]
- 9.Kawamura M, Ohnishi H, Guo SX, Sheng XM, Minegishi M, Hanada R, et al. Alterations of the p53, p21, p16, p15 and RAS genes in childhood T-cell acute lymphoblastic leukemia. Leuk Res. 1999; 23(2):115–26 [DOI] [PubMed] [Google Scholar]
- 10.Hof J, Krentz S, van Schewick, Körner G, Shalapour S, Rhein P, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29(23):3185–93 [DOI] [PubMed] [Google Scholar]
- 11.Tang JL, Tien HF, Lin MT, Chen PJ, Chen YC. Frequent p53 mutation in relapsed acute lymphoblastic leukemia with cytogenetic instability: a longitudinal analysis. Anticancer Res. 1998;18(2B):1273–8 [PubMed] [Google Scholar]
- 12.Zhu YM, Foroni L, McQuaker IG, Papaioannou M, Haynes A, Russell HH. Mechanisms of relapse in acute leukaemia: involvement of p53 mutated subclones in disease progression in acute lymphoblastic leukaemia. Br J Cancer. 1999;79(7–8):1151–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiaretti S, Tavolaro S, Marinelli M, Messina M, Del Giudice I, Mauro FR, et al. Evaluation of TP53 mutations with the AmpliChip p53 Research Test in chronic lymphocytic leukemia: correlation with clinical outcome and gene expression profiling. Genes Chromosomes Cancer. 2011;50(4):263–74 [DOI] [PubMed] [Google Scholar]
- 14.Del Giudice I, Mauro FR, De Propris MS, Santangelo S, Marinelli M, Peragine N, et al. White blood cell count at diagnosis and immunoglobulin variable region gene mutations are independent predictors of treatment-free survival in young patients with stage A chronic lymphocytic leukemia. Haematologica. 2011;96(4):626–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J, Mullighan CG, Harvey RC, Wu G, Chen X, Edmonson M, et al. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood. 2011;118(11):3080–7 [DOI] [PMC free article] [PubMed] [Google Scholar]