

Colony-stimulating factor 3 (CSF3), also known as granulocyte-colony stimulating factor (G-CSF), is the main growth factor driving neutrophil production under physiological conditions and during episodes of microbial infections. A tight but dynamic balance between signal activation and attenuation of CSF3R is essential to achieve a well-adjusted neutrophil output both under steady-state and infectious conditions.1,2 CSF3 is used in the clinic to treat patients with severe congenital neutropenia (SCN), a bone marrow failure syndrome characterized by peripheral absolute neutrophil counts below 0.5 million per liter.3 Inherited or congenital mutations in ELANE and HAX1 are the most frequent cause of SCN.4 Acquisition of nonsense CSF3R mutations that truncate the carboxyl-terminus from the CSF3R protein is a common phenomenon in SCN patients.5 Studies in a variety of in vitro and in vivo models have established that these truncated CSF3R proteins confer elevated CSF3-induced proliferative responses to myeloid progenitors.5
CSF3R nonsense mutations have been reported in approximately 30% of SCN patients in the neutropenia phase, which increases to approximately 80% after progression to acute myeloid leukemia (AML).6–8 In contrast, such CSF3R nonsense mutations have not been reported in de novo AML.9–11 We recently identified a new extracellular CSF3R mutation (T595I) in the leukemic blasts of a SCN patient after progression to AML.12 This mutation, located on the CSF3R allele that already carried the nonsense mutation, causes fully autonomous, i.e. colony-stimulating factor independent, proliferation of myeloid progenitors in semi-solid colony assays.12 Because most SCN patients receive life-long CSF3 therapy, we asked whether the acquisition of CSF3R-T595I mutations is unique for SCN/AML or whether this mutation can also be found in AML patients who did not receive CSF3 treatment. To address this, we investigated the prevalence of CSF3R-T595I mutations in 1446 consecutive de novo AML patients, none of whom had a documented history of CSF3 treatment. The study was performed with the approval of the Institutional Review Board of the Erasmus MC (registration n. MEC-2008-387). Amplicons were generated from cDNA using 5’-GCTCAGAACCAGTC-CTTCTC-3’ and 5’-CTGCTGTGAGCTGGGTCTG-3’ primers and analyzed with denaturing high-performance liquid chromatography (dHPLC) using a WAVE device (Transgenomics, Omaha, NE, USA). Amplicons showing aberrant dHPLC patterns compared to wild-type control were analyzed by Sanger sequencing using the 5’-GCTCA-GAACCAGTCCTTCTC-3’ primer. We identified 5 AML patients who carried a CSF3R-T595I mutation identical to the SCN/AML case. Clinical, molecular and cytogenetic characteristics of these patients are listed in Table 1. In addition, we identified 2 patients with a CSF3R mutation substituting a threonine at amino acid position 617 for an isoleucine (T617I) or an asparagine (T617N) in the CSF3R transmembrane domain. Like T595I, T617N leads to ligand independent activation of CSF3R and has previously been reported as a germ line single nucleotide variation in hereditary chronic neutrophilia and in 2 out of 555 de novo AML patients.10,13 As germline material was not available we cannot confirm whether the mutations listed in Table 1 are acquired mutations. However, the fact that the T595I was acquired in the previously investigated SCN/AML patient12 highly suggests that the identified mutations listed in Table 1 were acquired as well. In none of the AML patients carrying CSF3R-T595I, T617N or T617I, CSF3R truncating mutations were present (Table 1). Substitution of a threonine for an isoleucine at amino acid position 595 results in a structural change as well as an alteration in charge, i.e. from polar to hydrophobic. To investigate which of these features caused ligand independent activation, we deliberately constructed mutant CSF3R-T595V, in which substitution of threonine for valine introduces a hydrophobic residue that is structurally similar to threonine. Introduction of CSF3R-T595V into mouse bone marrow cells resulted in growth factor independent progenitor cell proliferation similar to CSF3R-T595I, strongly suggesting that hydrophobicity rather than an altered primary structure was responsible for the autonomous signaling properties of CSF3R-T595I (Figure 1, Online Supplementary Design and Methods and Online Supplementary Table S1). It is likely that CSF3R-T595I dimers or oligomers adopt a conformation that autonomously activates intracellular signaling, similar to the T617N mutation.13 In line with this, HeLa cell transfectants expressing CSF3R-T595I spontaneously accumulated STAT3 and STAT5 in the nucleus, a process that normally requires CSF3-induced activation of JAKs (data not shown).5
Table 1.
Clinical and cytogenetic characteristics of AML patients with CSF3R mutations.
Figure 1.
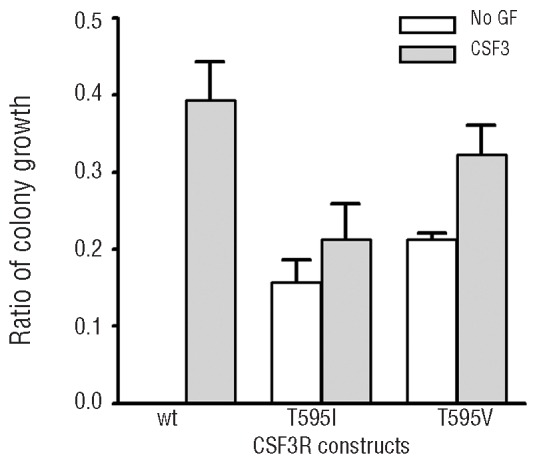
Functional analysis of CSF3R-T595V mutant in myeloid progenitor cell assay. In vitro colony growth of Csf3r deficient murine hematopoietic progenitor cells expressing the wild-type human CSF3R receptor (wt), the T595I and the T595V mutant, substituting a threonine at amino acid position 595 for an isoleucine or a valine, respectively. CSF3R expressing constructs in pBABE-puro were generated as previously described.12 The T595V mutation was introduced in pBABE-puro using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA, USA). Murine colony assays were performed as previously described.12 Colonies were grown in the presence of puromycin, either without growth factor (no GF) or with CSF3. The transduction efficiency was corrected for by dividing the number of CSF3-induced colonies by the number of CSF2 (granulocyte macrophage-colony stimulating factor, GM-CSF) induced colonies under puromycin selection as the CSF3R constructs confer puromycin resistance, but do not affect CSF2 induced colony growth. See Online Supplementary Design and Methods and Table S1.
Although the CSF3R self-activating mutations are rare, important questions are how the CSF3R mutations contribute to the pathogenesis of AML, and whether sustained administration of pharmacological dosages of CSF3 supports the evolution and expansion of clones harboring these mutations in SCN. Answering these questions will lead to a further understanding of the molecular mechanisms underlying leukemogenesis in these patients. The acquisition of the CSF3R-T595I mutation in the documented SCN/AML patient was a late event, detected in the AML that emerged 17 years after the initiation of CSF3 therapy, but absent in a bone marrow sample taken when the patient was treated with CSF3 for approximately eight years.12 Furthermore, we were unable to detect CSF3R-T595I mutations in 21 SCN patients in the neutropenia phase and in 5 patients who underwent CSF3 therapy and progressed to acute leukemia (data not shown). These results, combined with the findings in de novo AML patients argue against a relationship between the emergence of CSF3R-T595I mutations and CSF3 treatment. However, acquisition of this and other (T617I, T617N) mutations leading to autonomous proliferation of myeloid progenitors likely contributes to disease behavior and possibly has prognostic impact for AML.14
CSF3R nonsense mutations remain the most prevalent acquired abnormalities identified to date in both ELANE-SCN and HAX1-SCN. Because clones harboring these mutations emerge before leukemia becomes overt, it is conceivable that abnormal signaling from the truncated CSF3R proteins aggravates the accumulating damage in SCN stem and progenitor cells caused by ELANE or HAX1 mutations. Elevated production of reactive oxygen species, known to be genotoxic and potentially carcinogenic, is one of the potential explanations as to why truncated CSF3R contribute to leukemic progression of SCN.15 Whether and how CSF3 therapy adds to this process remains to be clarified.
Acknowledgments
Funding: this work was financially supported by a grant from the Center of Translational Molecular Medicine (CTMM).
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Irandoust MI, Aarts LH, Roovers O, Gits J, Erkeland SJ, Touw IP. Suppressor of cytokine signaling 3 controls lysosomal routing of G-CSF receptor. Embo J. 2007;26(7):1782–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolfler A, Irandoust M, Meenhuis A, Gits J, Roovers O, Touw IP. Site-specific ubiquitination determines lysosomal sorting and signal attenuation of the granulocyte colony-stimulating factor receptor. Traffic. 2009;10(8):1168–79 [DOI] [PubMed] [Google Scholar]
- 3.Dale DC, Bonilla MA, Davis MW, Nakanishi AM, Hammond WP, Kurtzberg J, et al. A randomized controlled phase III trial of recombinant human granulocyte colony-stimulating factor (filgrastim) for treatment of severe chronic neutropenia. Blood. 1993;81(10):2496–502 [PMC free article] [PubMed] [Google Scholar]
- 4.Dale DC, Link DC. The many causes of severe congenital neutropenia. N Engl J Med. 2009;360(1):3–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Touw IP, van de Geijn GJ. Granulocyte colony-stimulating factor and its receptor in normal myeloid cell development, leukemia and related blood cell disorders. Front Biosci. 2007;12:800–15 [DOI] [PubMed] [Google Scholar]
- 6.Germeshausen M, Skokowa J, Ballmaier M, Zeidler C, Welte K. G-CSF receptor mutations in patients with congenital neutropenia. Curr Opin Hematol. 2008;15(4):332–7 [DOI] [PubMed] [Google Scholar]
- 7.Dong F, Dale DC, Bonilla MA, Freedman M, Fasth A, Neijens HJ, et al. Mutations in the granulocyte colony-stimulating factor receptor gene in patients with severe congenital neutropenia. Leukemia. 1997;11(1):120–5 [DOI] [PubMed] [Google Scholar]
- 8.Germeshausen M, Ballmaier M, Welte K. Incidence of CSF3R mutations in severe congenital neutropenia and relevance for leukemogenesis: Results of a long-term survey. Blood. 2007;109(1):93–9 [DOI] [PubMed] [Google Scholar]
- 9.Link DC, Kunter G, Kasai Y, Zhao Y, Miner T, McLellan MD, et al. Distinct patterns of mutations occurring in de novo AML versus AML arising in the setting of severe congenital neutropenia. Blood. 2007;110(5):1648–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forbes LV, Gale RE, Pizzey A, Pouwels K, Nathwani A, Linch DC. An activating mutation in the transmembrane domain of the granulocyte colony-stimulating factor receptor in patients with acute myeloid leukemia. Oncogene. 2002;21(39):5981–9 [DOI] [PubMed] [Google Scholar]
- 11.Dong F, van Paassen M, van Buitenen C, Hoefsloot LH, Lowenberg B, Touw IP. A point mutation in the granulocyte colony-stimulating factor receptor (G-CSF-R) gene in a case of acute myeloid leukemia results in the overexpression of a novel G-CSF-R isoform. Blood. 1995;85(4):902–11 [PubMed] [Google Scholar]
- 12.Beekman R, Valkhof MG, Sanders MA, van Strien PM, Haanstra JR, Broeders L, et al. Sequential gain of mutations in severe congenital neutropenia progressing to acute myeloid leukemia. Blood. 2012;119(22):5071–7 [DOI] [PubMed] [Google Scholar]
- 13.Plo I, Zhang Y, Le Couedic JP, Nakatake M, Boulet JM, Itaya M, et al. An activating mutation in the CSF3R gene induces a hereditary chronic neutrophilia. J Exp Med. 2009;206(8):1701–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lowenberg B, van Putten WL, Touw IP, Delwel R, Santini V. Autonomous proliferation of leukemic cells in vitro as a determinant of prognosis in adult acute myeloid leukemia. N Engl J Med. 1993;328(9):614–9 [DOI] [PubMed] [Google Scholar]
- 15.Zhu QS, Xia L, Mills GB, Lowell CA, Touw IP, Corey SJ. G-CSF induced reactive oxygen species involves Lyn-PI3-kinase-Akt and contributes to myeloid cell growth. Blood. 2006;107(5):1847–56 [DOI] [PMC free article] [PubMed] [Google Scholar]