

Abstract
Background:
Muir-Torre syndrome (MTS) is an autosomal dominant syndrome characterized by neoplasms of the sebaceous gland or keratoacanthomas, in addition to visceral malignancies. Cerebral neoplasms in patients with hereditary nonpolyposis colorectal cancer (HNPCC) or familial adenomatous polyposis suffer from Turcot's syndrome. Genetic mutations in MutS homolog (MSH)-2, MutL homolog (MLH)-1, and MutS homolog (MSH)-6 DNA mismatch repair genes are the most common in MTS with MSH-2 being the most predominant. In HNPCC MLH-1 and MSH-2 mutations are approximately equal in prevalence.
Case Description:
We present the case of a 58-year-old male with a prior history of being treated for colonic adenocarcinoma and skin lesions leading to a diagnosis of MTS. The patient later developed a World Health Organization (WHO) grade 4 glioma requiring surgical resection. Pathology revealed mutations in MSH-2 and MSH-6 mismatch repair genes.
Conclusions:
This case represents the first report of Turcot's and MTS with extensive molecular testing on the cerebral neoplasm demonstrating a molecular relationship between Turcot's and MTS and only the second published report of simultaneous Turcot's and MTS.
Keywords: Glioblastoma multiforme, Muir-Torre syndrome, MutS homolog-2, Turcot's syndrome
INTRODUCTION
Muir-Torre syndrome (MTS) is a rare skin condition with an autosomal dominant inheritance pattern, which is characterized clinically by the coincident occurrence of neoplasms of the sebaceous gland or keratoacanthomas associated with one or more visceral malignancies in the absence of precipitating factors, such as, ulcerative colitis or hepatitis. Sebaceous tumors associated with MTS include sebaceous adenoma and epithelioma, basal cell epithelioma with sebaceous differentiation, and sebaceous carcinoma.[22] Colorectal and genitourinary carcinomas constitute the most commonly seen visceral tumors in patients with MTS.[1,4] Patients with MTS exhibit genetic mutations in the human MutS homolog (MSH)-2, MutL homolog (MLH)-1, and MutS homolog (MSH)-6 DNA mismatch repair genes.[14] Hereditary nonpolyposis colorectal cancer (HNPCC) and familial adenomatous polyposis (FAP) are genetic disorders, which predispose patients to colorectal cancer. When these patients subsequently develop primary brain tumors the condition is termed Turcot's Syndrome. We present the case of a 58-year-old male with a known diagnosis of MTS who subsequently developed a glioblastoma multiforme with mutations in MSH2 and MSH6 representing a case of simultaneous MTS and Turcot's Syndrome.
CASE REPORT
History
A 58-year-old male was diagnosed with colon cancer in February 1999, stage T3 N1 M0, which was treated by right hemicolectomy followed by adjuvant chemotherapy with 5-fluorouracil and leucovorin. He had no further recurrence of this tumor. In 2004, the patient started noticing “cysts” on his skin, for which he presented to a dermatologist. Biopsies of the skin lesions were taken. This prompted genetic testing on the patient's colonic adenocarcinoma, which revealed a mutation in the MSH2 gene, rendering the patient with a diagnosis of MTS. The patient presented to neurosurgical attention in 2007 with complaints of unsteady gait along with episodes of transient paresthesias involving the left arm, torso, and leg. His immediate family history included a deceased sister with a history of an astrocytoma and deceased mother with colon cancer; genetic testing was not performed on either.
Examination
The patient exhibited no neurologic deficits. Magnetic resonance imaging (MRI) studies revealed the presence of a well-demarcated, right posterior parietal mass roughly 2.2 cm in diameter. The tumor appeared as an isointense signal on the T1-weighted image with enhancement after administration of gadolinium contrast [Figure 1], and as a hyperintense signal on the T2-weighted image. Computed tomography (CT) of the chest, abdomen, and pelvis was negative for masses.
Figure 1.
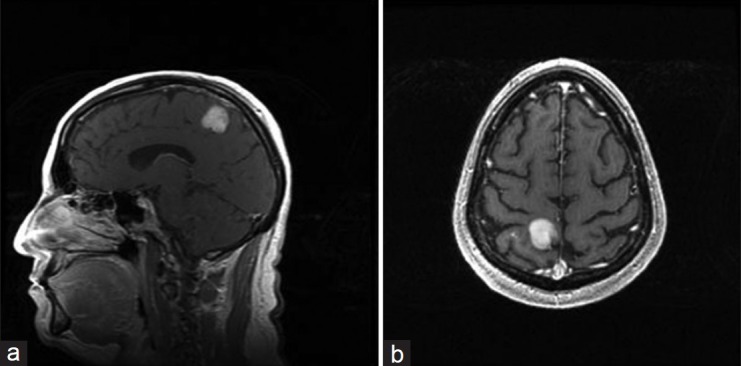
Preoperative sagittal (a) and axial (b) T1-weighted magnetic resonance images of the brain with gadolinium contrast demonstrate the presence of an enhancing mass in the right posterior parietal region
Operation/pathology/postoperative course
Given the patient's history and the need to determine whether the brain mass represented a metastatic lesion from his previous colon cancer or a primary brain tumor, the patient initially underwent an MRI-guided, frameless stereotactic biopsy of the enhancing margin of the brain lesion for histologic diagnosis. Pathology revealed scattered atypical glial cells with monosomy of chromosome 7 in some of the cells. A distinct classification was unable to be determined. Therefore, roughly 2 weeks following the biopsy, the patient underwent an image-guided, awake right parietal craniotomy for resection of the tumor. The surgery proceeded without complication and the patient was discharged to home with no new neurological deficits on postoperative day one. The pathological report from this specimen definitively proved the tumor to be a glioblastoma multiforme (World Health Organization [WHO] grade 4) with positive immunohistochemical staining for p53 and GFAP and a Ki-67 proliferative index of 25%. Immunostaining for the mismatch repair proteins MSH2, MLH1, and MSH6 revealed that the tumor lacked staining with MSH2 and MSH6 with intact staining for MLH1, suggestive of a germline mutation. Molecular analysis demonstrated a high level of microsatellite instability by detecting evidence of microsatellite instability in 5 of 10 genetic loci used as markers.
DISCUSSION
MTS was first described by Muir and Torre in the late 1960s in separate case reports. In Muir, et al.'s study, the patient's visceral malignancies included primary carcinomas of the small intestine, colon, and larynx, with concomitant facial keratoacanthomata.[16] Torre described an individual who had cancer of the ampulla of Vater along with numerous sebaceous tumors (e.g., sebaceous adenomas).[23]
Lynch, et al. initially discussed the concept of MTS representing a clinical variant of HNPCC when the authors observed sebaceous gland neoplasms among their study population of families affected by colon cancer.[11] Further studies demonstrated genetic mutations common to both, including mutations in the MSH2, MLH1, and MSH6 DNA mismatch repair genes, resulting in deficient DNA mismatch-repair proteins, and, in turn, microsatellite instability.[2,3,5,7,8,10,12–14,15,18,19] Of note, the proportion of mutations involving MSH2 and MLH1 is almost equal in HNPCC, while in MTS, mutations in the MSH2 gene is implicated in the overwhelming majority of time.[9,14] Microsatellite-stable neoplasms, however, do occur in patients with MTS, prompting researchers to postulate that a second variant of MTS exists, with its genetic basis potentially involving other gene sequences or promoter hypermethylation.[20,21]
The development of brain tumors in patients with colon cancer associated with the inherited conditions of either HNPCC or FAP is termed Turcot's syndrome. Similar to MTS, Turcot's syndrome type 1 is a clinical variant of HNPCC and specifically entails those individuals who develop brain neoplasms.[6] To our knowledge, our case represents the second published report of a brain neoplasm occurring in a patient previously diagnosed with MTS, and thus only the second documented case of a patient with concomitant diagnoses of MTS and Turcot's syndrome. Okamoto, et al.[17] described a 49-year-old male with a history of colon cancer and sebaceous adenoma who presented with a right frontal lobe mass and underwent a partial frontal lobectomy with subtotal tumor resection. After histologic examination and molecular analysis, the lesion was revealed to be consistent with an astrocytoma (WHO Grade 3) with microsatellite instability and mutation of the p53 gene.[17] Further molecular testing for mutations in the MSH2, MLH1, or MSH6 DNA mismatch repair genes was not performed in the previous study.
CONCLUSIONS
By demonstrating a mutation in the MSH2 and MSH6 mismatch repair genes from the tumor specimen, we present the first case in which genotypic changes consistent with HNPCC has been conclusively documented in a patient who has, what may be best categorized as an overlap syndrome of Muir-Torre and Turcot's syndromes.
Footnotes
Available FREE in open access from: http://www.surgicalneurologyint.com/text.asp?2013/4/1/52/110512
Contributor Information
Ramesh Grandhi, Email: grandhir@upmc.edu.
Christopher P. Deibert, Email: deibertcp@upmc.edu.
Stephen M. Pirris, Email: pirris.stephen@mayo.edu.
Barry Lembersky, Email: lemberskybc@upmc.edu.
Arlan H. Mintz, Email: pittmed@comcast.com.
REFERENCES
- 1.Akhtar S, Oza KK, Khan SA, Wright J. Muir-Torre syndrome: Case report of a patient with concurrent jejunal and ureteral cancer and a review of the literature. J Am Acad Dermatol. 1999;41:681–6. doi: 10.1016/s0190-9622(99)70001-0. [DOI] [PubMed] [Google Scholar]
- 2.Bapat B, Xia L, Madlensky L, Mitri A, Tonin P, Narod SA, et al. The genetic basis of Muir-Torre syndrome includes the hMLH1 locus ;59:736-9, Am J Hum Genet. 1996;59:736–9. [PMC free article] [PubMed] [Google Scholar]
- 3.Bocker T, Diermann J, Friedl W, Gebert J, Holinski-Feder E, Karner-Hanusch J, et al. Microsatellite instability analysis: A multicenter study for reliability and quality control. Cancer Res. 1997;57:4739–43. [PubMed] [Google Scholar]
- 4.Cohen PR, Kohn SR, Kurzrock R. Association of sebaceous gland and internal malignancy: The Muir-Torre syndrome. Am J Med. 1991;90:606–13. [PubMed] [Google Scholar]
- 5.Entius MM, Keller JJ, Drillenburg P, Kuypers KC, Giardiello FM, Offerhaus GJ. A microsatellite instability and expression of MLH-1 and hMSH2 in sebaceous gland carcinomas as markers for Muir-Torre Syndrome. Clin Cancer Res. 2000;6:1784–9. [PubMed] [Google Scholar]
- 6.Farrell CJ, Plotkin SR. Genetic causes of brain tumors: Neurofibromatosis, tuberous sclerosis, von Hippel-Lidau, and other syndromes. Neurol Clin. 2007;24:925–46. doi: 10.1016/j.ncl.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 7.Honchel R, Halling KC, Schaid DJ, Pittelkow M, Thibodeau SN. Microsatellite instability in Muir-Torre syndrome. Cancer Res. 1994;54:1159–63. [PubMed] [Google Scholar]
- 8.Kruse R, Lamberti C, Wang Y, Ruelfs C, Bruns A, Esche C, et al. Is the mismatch repair deficient type of Muir-Torre syndrome confined to mutations in the hMSH2 gene? Hum Genet. 1996;98:747–50. doi: 10.1007/s004390050298. [DOI] [PubMed] [Google Scholar]
- 9.Kruse R, Rütten A, Lamberti C, Hosseiny-Malayeri HR, Wang Y, Ruelfs C, et al. Muir-Torre phenotype has a frequency of DNA mismatch-repair-gene mutations similar to that in hereditary nonpolyposis colorectal cancer families defined by the Amsterdam criteria. Am J Hum Genet. 1998;63:63–70. doi: 10.1086/301926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kruse R, Ruzicka T. DNA mismatch repair and the significance of a sebaceous skin tumor for visceral cancer prevention. Trends Mol Med. 2004;10:136–41. doi: 10.1016/j.molmed.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 11.Lynch HT, Lynch PM, Pester J, Fusaro RM. The cancer family syndrome: Rare cutaneous phenotypic linkage of Torre's syndrome. Arch Intern Med. 1981;141:607–11. [PubMed] [Google Scholar]
- 12.Machin P, Catasus L, Pons C, Muñoz J, Conde-Zurita JM, Balmaña J, et al. Microsatellite instability and immunostaining for MSH-2 and MLH-1 in cutaneous and internal tumors from patients with the Muir-Torre syndrome. J Cutan Pathol. 2002;29:415–20. doi: 10.1034/j.1600-0560.2002.290705.x. [DOI] [PubMed] [Google Scholar]
- 13.Mangold E, Pagenstecher C, Leister M, Mathiak M, Rütten A, Friedl W, et al. A genotype-phenotype correlation in HNPCC: Strong predominance of MSH2 mutations in 41 patients with Muir-Torre syndrome. J Med Genet. 2004;41:567–72. doi: 10.1136/jmg.2003.012997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mangold E, Rahner N, Friedrichs N, Buettner R, Pagenstecher C, Aretz S, et al. MSH6 mutation in Muir-Torre syndrome: Could this be a rare finding? Br J Dermatol. 2006;156:158–62. doi: 10.1111/j.1365-2133.2006.07607.x. [DOI] [PubMed] [Google Scholar]
- 15.Mathiak M, Rütten A, Mangold E, Fischer HP, Ruzicka T, Friedl W, et al. Loss of DNA mismatch repair proteins in skin tumors from patients with Muir-Torre syndrome and MSH2 or MLH1 germline mutations. Am J Surg Pathol. 2002;26:338–43. doi: 10.1097/00000478-200203000-00007. [DOI] [PubMed] [Google Scholar]
- 16.Muir EG, Bell AJ, Barlow KA. Multiple primary carcinomata of the colon, duodenum and larynx associated with keratoacanthoma of the face. Br J Surg. 1967;54:191–5. doi: 10.1002/bjs.1800540309. [DOI] [PubMed] [Google Scholar]
- 17.Okamoto H, Mineta T, Nakahara Y, Ichinose M, Shiraishi T, Tabuchi K. Molecular analysis of astrocytoma associated with Turcot syndrome type 1: Case report. Neurol Med Chir (Tokyo) 2004;44:124–8. doi: 10.2176/nmc.44.124. [DOI] [PubMed] [Google Scholar]
- 18.Pedroni M, Sala E, Scarselli A, Borghi F, Menigatti M, Benatti P, et al. Microsatellite instability and mismatch-repair protein expression in hereditary and sporadic colorectal carcinogenesis. Cancer Res. 2001;61:896–9. [PubMed] [Google Scholar]
- 19.Pedroni M, Tamassia MG, Percesepe A, Roncucci L, Benatti P, Lanza G, Jr, et al. Microsatellite instability in multiple colorectal tumors. Int J Cancer. 1999;81:1–5. doi: 10.1002/(sici)1097-0215(19990331)81:1<1::aid-ijc1>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 20.Ponti G, Losi L, Di Gregorio C, Roncucci L, Pedroni M, Scarselli A, et al. Identification of Muir-Torre syndrome among patients with sebaceous tumors and keratoacanthomas: Role of clinical features, microsatellite instability, and immunohistochemistry. Cancer. 2005;103:1018–25. doi: 10.1002/cncr.20873. [DOI] [PubMed] [Google Scholar]
- 21.Ponti G, Ponz de Leon M. Muir-Torre syndrome. Lancet Oncol. 2005;6:980–7. doi: 10.1016/S1470-2045(05)70465-4. [DOI] [PubMed] [Google Scholar]
- 22.Schwartz RA, Torre DP. The Muir-Torre syndrome: A 25 years retrospect. J Am Acad Dermatol. 1995;33:90–104. doi: 10.1016/0190-9622(95)90017-9. [DOI] [PubMed] [Google Scholar]
- 23.Torre D. Multiple sebaceous tumors. Arch Dermatol. 1968;98:549–51. doi: 10.1001/archderm.98.5.549. [DOI] [PubMed] [Google Scholar]