

Abstract
Fatty acids such as eicosapentaenoic acid (EPA) have been shown to be beneficial for neurological function and human health. It is widely thought that oxidation products of EPA are responsible for biological activity, although the specific EPA peroxidation product(s) which exert these responses have not yet been identified. In this work we provide the first evidence that the synthesized representative cyclopentenone IsoP, 15-A3t-IsoP, serves as a potent inhibitor of lipopolysaccharide-stimulated macrophage activation. The anti-inflammatory activities of 15-A3t-IsoP were observed in response not only to lipopolysaccharide, but also to tumor necrosis factor alpha and IL-1b stimulation. Subsequently, this response blocked the ability of these compounds to stimulate nuclear factor kappa b (NFκB) activation and production of proinflammatory cytokines. The bioactivity of 15-A3t-IsoP was shown to be dependent upon an unsaturated carbonyl residue which transiently adducts to free thiols. Site directed mutagenesis of the redox sensitive C179 site of the Ikappa kinase beta subunit, blocked the biological activity of 15-A3t-IsoP and NFκB activation. The vasoprotective potential of 15-A3t-IsoP was underscored by the ability of this compound to block oxidized lipid accumulation, a critical step in foam cell transformation and atherosclerotic plaque formation. Taken together, these are the first data identifying the biological activity of a specific product of EPA peroxidation, which is formed in abundance in vivo. The clear mechanism linking 15-A3t-IsoP to redox control of NFκB transcription, and the compound's ability to block foam cell transformation suggest that 15-A3t-IsoP provides a unique and potent tool to provide vaso- and cytoprotection under conditions of oxidative stress.
Keywords: Alzheimer's disease, cytoprotection, eicosapentaenoic acid, inflammation, oxidative stress
In the last three decades, we have increasingly come to recognize that `omega' or n-3 fatty acids play essential roles in human health and development, particularly in development of the nervous system (Connor 1997; Connors et al. 2008). α-linolenic acid, eicosapentaenoic acid (EPA), and docosahexaenoic acid are all polyunsaturated fatty acids which have been at the forefront of these studies and have independently displayed cytoprotective activity. One of the primary mechanisms by which these fatty acids enhance cellular function is by the reduction of inflammation in vivo (Calder 2006). Human epidemiological and recent clinical intervention trials suggest that consumption of fish or dietary supplementation with fish oil, which is rich in EPA, reduces the incidence of cognitive decline, atherosclerosis, diabetes, and other inflammatory disorders (Kris-Etherton et al. 2002; Yokoyama and Origasa 2003; Barnham et al. 2004; Calder 2006; Mozaffarian 2007; Yokoyama et al. 2007; Saito et al. 2008). Given the unstable nature of fatty acids, identifying the bioactive metabolites and mechanism of action of n-3 fatty acids is essential to design targeted therapeutics for disorders linked to inflammatory stress. The mechanisms through which EPA exerts its biological activity are, however, poorly understood. The complexity of this problem is exacerbated by the fact that when in the cis configuration, the omega-3 fatty acids readily form dozens of potentially bioactive byproducts. Transformation events are potentiated in the presence of oxidative stress and native EPA is highly susceptible to beta-oxidation particularly in the brain (Chen et al. 2011).
We have previously determined that oxidation of EPA yields a family of prostaglandin (PG)-like molecules termed isoprostanes (IsoPs). IsoPs are formed in vitro and in vivo in settings of oxidative stress (Gao et al. 2006; Brooks et al. 2008a). One class of IsoPs shown to form in abundance is cyclopentenone IsoPs or A3/J3-IsoPs. These compounds have an analogous structure to the arachidonic acid-derived cyclopentenone prostaglandins (PGs), PGA2 and PGJ2 (Fig. 1). Even more recent work suggests that oxidative metabolites of EPA are biological active (Chaudhary et al. 2004; Mishra et al. 2004). These studies have not, however, determined which specific product(s) formed from native EPA are responsible for the biological activities reported.
Fig. 1.

Formation of cyclopentenone IsoPs through the free-radical oxidation of eicosapentaenoic acid (EPA). (a) Oxidation of EPA yields a bicyclic endoperoxide intermediate that can undergo rearrangement to form D- and E-ring IsoPs which can spontaneously dehydrate to form J- and A-ring (cyclopentenone) IsoPs. Although six regioisomers can form from oxidation of EPA, only 15-series isomers are shown for simplicity. (b) structures of the four 15-A3-IsoP stereoisomers generated from non-enzymatic peroxidation of EPA; the boxed stereoisomer was synthesized for these studies.
Oxidative stress, neuroinflammation, and atherosclerosis are closely coupled and co-occur in many neurologic diseases, in particular stroke. Pathologic activation of macrophages in the brain and neurovasculature is a major source of cerebral oxidative damage, inflammatory damage, and atherogenesis. EPA supplementation suppresses macrophage activation and subsequent neuroinflammation and pathology in several models of neurologic disease. Given that under conditions of oxidative stress, EPA-derived IsoPs form at levels 10 times higher than do IsoPs from arachidonic acid (Gao et al. 2007), we sought to isolate a single bioactive compound formed in abundance by oxidation of native EPA and determine the mechanism of bioactivity.
Experimental procedures
Materials
15-A3t-IsoP (Fig. 1) was obtained by total synthesis in a manner similar to the synthesis of A2-IsoPs (Zanoni et al. 2002). 15-A3t-IsoP was stored in ethyl acetate at −80°C until immediately before use at which time it was dried under nitrogen and resuspended in ethanol. 15-A3t-IsoP was added to culture medium immediately before its addition to cells. As compounds with α,β-unsaturated carbonyls rapidly react with albumin, serum-free medium was used for all experiments (Musiek et al. 2005). 15-A3t-IsoP was stable in this culture media for > 24 h. The IκKβ (C179A) mutant expression vector was a gift of Dr M. Karin (University of California, San Diego). Lipopolysaccharide (LPS) (from Salmonella Minnesota Re 595), arabinoside cytosine and Oil Red O stain were purchased from Sigma-Aldrich (St Louis, MO, USA). Tumor necrosis factor-α (TNFα) and interleukin-1β (IL-1β) were obtained from R & D Systems (Minneapolis, MN, USA). inhibitor of kappa b (IκBα), cyclo-oxygenase 2 (COX-2), and NFκB p65 subunit antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and inducible nitric oxide synthase (iNOS), extracellular signal-related kinase, and the glyceraldehyde 3-phosphate dehydrogenase antibodies were from Cell Signaling Technologies (Danvers, MA, USA). GW9662, T00907, and G418 sulfate were obtained from Cayman Chemical Co. (Ann Arbor, MI, USA). Fetal bovine serum was purchased from Hyclone (Logan, UT, USA). All cell culture media and supplies were from Invitrogen (Carlsbad, CA, USA) unless otherwise noted.
Cell culture
RAW267.4 murine macrophage cells were obtained from ATCC (Manassas, VA, USA). NFκB reporter macrophages were a generous gift from the laboratory of Dr Timothy Blackwell. These cells were originally obtained from the bone marrow of transgenic mice expressing a reporter plasmid containing the human immunodeficiency virus-long terminal repeat 36-bp enhancer (containing a total of eight NFκB-binding sites) upstream of the herpes simplex virus minimal thymidine kinase promoter driving expression of Photinus luciferase (Blackwell et al. 2000). All cells were grown in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 100 units/mL penicillin, and 100 mg/mL streptomycin. Cells were plated on 24- or 6-well plates at least 24 h before any experimental manipulation.
Immunofluorescence microscopy
For immunofluorescence staining, RAW264.7 cells were grown on glass coverslips. Following exposure to 15-A3t-IsoP or vehicle, cultures were fixed in 10% formaldehyde for 10 min, rinsed with phosphate-buffered saline (PBS), permeabilized with 0.1% Triton X-100, and blocked for 1 h with 8% bovine serum albumin diluted in PBS. Coverslips were then incubated overnight at 4°C in rabbit anti-p65 (1 : 100) primary antibody in 1% bovine serum albumin. Cells were washed in PBS for a total of 25 min and incubated in Cy-2-labeled secondary antibodies for 1 h. Cells were then washed again and stained with 1.4 μM 4′,6-diamidino-2-phenylindole (DAPI) for 10 min followed by further washes. Coverslips were mounted on microscope slides, and fluorescence was visualized with a Zeiss Axioplan microscope.
Cell counts
Cells were counted by fluorescence microscopy and the number of cells exhibiting nuclear localization of NFκB after treatment for the indicated time with LPS and 15-A3t-IsoP was obtained as a percentage of the whole DAPI positive population (~200 cells counted/variable). Three independent experiments were quantified by an investigator blinded to the experimental condition. Experiments quantifying the percentage of cells exhibiting nuclear accumulation of the NFκB p65 subunit were performed by immunofluorescence microscopy using a 40× objective. Data are the mean ± SD from at least 10 fields of view and represent a total of 500–750 cells.
NFκB reporter assay
Primary cells stably expressing NFκB-driven luciferase reporter were utilized to determine the direct effect on NFκB-mediated transcription. Following exposure to 15-A3t-IsoP or vehicle, cells were scraped at selected time points in Lysis Buffer (Promega, Madison, WI, USA). The cell debris was pelleted by centrifugation at 17 178 g for 5 min, and 20 μL of the supernatant was used to assay luciferase activity using a luminometer. Results were later normalized to cellular protein levels.
Measurement of nitrite
Nitrite, a stable breakdown product of nitric oxide, was measured in cell medium using the Griess reaction. Briefly, 100 μL of conditioned medium was mixed with 50 μL each of Griess reagent R1 and R2 (Cayman Chemical) in a 96-well plate, and absorbance was measured at 540 nm. Quantitation was achieved by comparison with sodium nitrite standards in Dulbecco's modified Eagle's medium.
Measurement of PGD2
PGD2, the primary product of COX-2 activation in macrophages, was measured in cell media. After 24 h of LPS stimulation, PGD2 levels were quantified in 200 μL of cell media by stable isotope dilution assays. Briefly, [4H2]-labeled PGD2 internal standard was added to samples. Samples were then purified using C18 and silica Sep-Pack extraction methods and thin layer chromatography. Samples were then analyzed as the pentafluorobenzyl ester, methoxylamine, trimethylsilyl ether derivatives via gas chromatography-negative ion chemical ionization mass spectrometry (GC-NICI-MS) and data were normalized to protein concentrations.
IκK transfection
I kappa kinase (IκK), a possible target for 15-A3t-IsoP interaction, was mutated at the cysteine 179 (C179) residue to determine the effect on 15-A3t-IsoP bioactivity. RAW264.7 cells were transfected with NFκB-luc reporter plasmid plus an empty vector, WT, or mutant (C179A) IκKβ expression vectors using the Amaxa Nucleofector System (Amaxa, Inc., Bethesda, MD, USA). Forty-eight hours after transfection, cells were pre-incubated with vehicle or 15-A3t-IsoP for 30 min, stimulated with vehicle or LPS for 4 h, and then harvested and subjected to the luciferase reporter assay. RAW264.7 cells were also transfected with the pcDNA3.1 antibiotic resistance plasmid (Invitrogen) plus WT or mutant (C179A) IκKβ expression vectors as described above. Transfected cells were selected using G418 sulfate (800 μg/mL media) for 1 month. Stably transfected cell lines were treated as described previously for PGD2 and nitrite assays, and the media was subjected to nitrite and PGD2 assessments.
Western blotting
Total protein extract was prepared, and a small aliquot of the protein suspension was removed and stored at −20°C for protein quantification using the bicinchoninic acid protein assay (Bio-Rad Laboratories, Hercules, CA, USA). Equal protein concentrations were separated using Criterion XT 10% Tris gels (Bio-Rad). Proteins were then transferred to polyvinylidene difluoride membranes and blocked for 15 minutes using methanol. Membranes were incubated overnight in primary antibody diluted in non-fat milk containing 0.1% Tween 20. The following day, membranes were washed in PBS containing 0.1% Tween 20 and incubated in horseradish peroxidase-conjugated secondary antibodies for 1 h at 22°C. Protein bands were visualized by chemiluminescence. Western blots subject to semi-quantitative analysis using the NIH ImageJ analysis program as previously described (McLaughlin et al. 2003; Brown et al. 2010).
Sodium borohydride reduction and C18 SepPak purification of synthetic 15-A3t-IsoP
15-A3t-IsoP is hypothesized to exert biological activity through the α,β-unsaturated carbonyl. To test the importance of this moiety, 100 μg of 15-A3t-IsoP was adjusted to pH 3, extracted with ethyl acetate, dried, and resuspended in 1 mL of methanol. One milliliter of 12% (w/w) sodium borohydride (NaBH4), a weak reducing agent, in water was added to the methanol and incubated on ice for 30 min. Samples were diluted in 20 mL of water and adjusted to pH 3 and then subjected to solid−phase extraction using a C18 SepPak. The SepPak column was washed with 7 mL of methanol and 10 mL of water (pH 3), and then the sample was added, followed by washes with 10 mL of water (pH 3) and 10 mL of heptane. The samples were eluted with 10 mL of 1 : 1 ethyl acetate/heptane and dried under nitrogen and resuspended in ethanol at appropriate stock concentrations.
Conjugation of 15A3t-IsoP with GSH
As another method of determining the importance of the α,β-unsaturated carbonyl of the 15-A3t-IsoP activity, 100 μg of 15-A3t-IsoP was incubated in 0.1M K3PO4 buffer (pH 6.5) in the presence of a 10-fold excess (~1 μg) of GSH and 1 mg of equine liver glutathione-S-transferase (GST) containing a mixture of GSTs at 37°C for 2 h (Chen et al. 1999). The incubation mixture was then acidified to pH 3 and extracted with 2 volumes of CH2Cl2. followed by C18 silica Sep-Pak extraction (Fam et al. 2002). The samples were then dried under nitrogen and resuspended in ethanol at appropriate stock concentrations.
Oil Red O staining
The accumulation of oxidized low-density lipoprotein (OxLDL) is an important step in the formation of foam cells. Lipid accumulation and foam cell formation can be visualized using Oil Red O staining (Mori et al. 2001). RAW264.7 cells were treated with the mitotic inhibitor cytosine arabinoside and grown on coverslips. Following cellular exposure to OxLDL (100 μg/mL) and 15-A3t-IsoP (10 μM) for 48 h, cultures were fixed for 40 min at 22°C with 4% paraformaldehyde and stained with 0.5% Oil Red O stain for 45 min. Coverslips were subsequently washed and dehydrated with 70% EtOH. Coverslips were mounted on microscope slides, and imagining was performed with a Zeiss Axioplan microscope.
F2-Isoprostane assay
To measure the level of 15-A3t-IsoP-induced oxidative stress in cells, total F2-IsoPs were measured in combined medium and cell pellet as described previously (Morrow and Roberts 1999). Briefly, cells were scraped directly into their conditioned medium and then collected by centrifugation. The cell pellet was resuspended in 0.5 mL of methanol containing 0.005% butylated hydroxytoluene, sonicated, and subjected to chemical saponification using 15% potassium hydroxide to hydrolyze bound F2-IsoPs. The conditioned medium was then added back to the cell lysate and adjusted to pH 3 followed by addition of [4H2]-labeled 15-F2t-IsoP internal standard. Free F2-IsoPs were then purified by C18 and silica Sep-Pak extraction and thin layer chromatography and then analyzed as the pentafluorobenzyl ester, trimethylsilyl ether derivatives via GC-NICI-MS.
Statistics
Except where otherwise noted, data were summarized and are represented as mean ± SEM. The statistical significance of differences between means was assessed using one-way analysis of variance (anova) at the 95% level (p < 0.05), followed by the Tukey's multiple comparison tests using GraphPad Prism software.
Results
15-A3t-IsoP inhibits LPS-induced iNOS and COX-2 expression and activity in RAW264.7 macrophages
When oxidized in vivo, native EPA produces cyclopentenone isoprostanes which are structurally similar to anti-inflammatory cyclopentenone PGs (Fig. 1). These products are formed in abundance both in vivo and in vitro (Brooks et al. 2008a). We obtained a single regio- and stereoisomer formed from the endogenous oxidation of EPA–15-A3t–IsoP-and used this to study the biological activities of the cyclopentenone IsoPs as a class.
Dose–response curves using MTT assays revealed no cytotoxicity in RAW264.7 cells when incubated for 24 h in 25 μM 15-A3t-IsoP or elevation in oxidative stress as measured by free and bound 15-F2t-IsoP (control levels 137 ± 9 and 15-A3t-IsoP 161 ± 9 pg F2t-IsoP/gram protein; n = 5, p = 0.19).
We did, however, find that that 15-A3t-IsoP potently suppressed LPS-induced PGD2 and nitric oxide production as measured by accumulation of PGD2 and nitrite in cell media and normalized to protein concentration with an approximate IC50 of 20 μM (Fig. 2a and b). 15-A3t-IsoP also inhibited the expression of the LPS-stimulated pro-inflammatory proteins iNOS and cyclooxygenase-2 (COX-2) (Fig. 2c and d). Time-matched incubations in 15-A3t-IsoP alone did not alter the expression or activity of iNOS, COX-2 or their biochemical products (data not shown). Densitometric analysis of 15-A3t-IsoP effects on LPS stimulated COX-2 expression revealed a 17 ± 4% decrease in protein levels when cells were harvested 6 h after LPS stimulation and a 93 ± 2% decrease in iNOS expression when cells were harvested 9 h after LPS stimulation.
Fig. 2.

15-A3t-IsoP inhibits LPS-induced production of inflammatory mediators in RAW264.7 macrophages. RAW264.7 macrophages were pre-incubated with varying concentrations of 15-A3t-IsoP for 30 min and then stimulated with LPS (1 μg/mL) for 24 h in the presence of 15-A3t-IsoP. A, 15-A3t-IsoP inhibits LPS-induced PGD2 production, the primary product of COX-2 activity in macrophages, as quantified by GC/MS. B, 15-A3t-IsoP inhibits LPS-induced production of nitrite, a stable metabolite of iNOS activity, as quantified by Griess assay. Assays were performed 24 h after the initiation of the pre-incubation period. Statistical analysis of PDG2 (a) and nitrate formation (b) were performed using a two-tailed between group anova and demonstrated a significant treatment effect with p < 0.01. Post hoc analysis using Tukey's HSD post hoc testing revealed control versus LPS p < 0.001 for both Fig. 2A and B. Asterisks are used to denote statistically significant effects of 15-A3t-IsoP + LPS compared to LPS alone. All data represent mean ± SD of four separate experiments carried out in triplicate where data were normalized to the protein concentrations of samples. (c, d) 15-A3t-IsoP (5, 10, and 25 μM) inhibits LPS-induced expression of the proteins COX-2 after 6 h of LPS stimulation (c) and iNOS after 9 h of LPS stimulation (d) as analyzed by western blot. GAPDH and ERK blots are shown as loading controls. Blots in panels (c) and (d) are representative of four independent experiments. Semi-quantitative densitometric analysis of the blots was performed using Image J and data shown are the optical densities (OD) for each condition ± SD where the first lane is taken as a baseline value of 100%.
15-A3t-IsoP Acts as an Inhibitor of the NFκB pathway
As the expression of iNOS and COX-2 enzymes is known to be regulated by the transcription factor NFκB (Chawla et al. 2001; Chun and Surh 2004), we examined the effects of 15-A3t-IsoP on NFκB signaling. NFκB transcription activity was assessed using immortalized mouse macrophage cell line expressing an NFκB-activation responsive luciferase reporter. Luciferase measurements revealed a dose-dependent decrease in NFκB-mediated transcription with 25 μM dropping LPS-stimulated activation by more than 70% (Fig. 3a). 15-A3t-IsoP had no effect on NFκB transcription or the degradation of IκB even when incubations were continued for 24 h (data not shown but see Brooks et al. 2008b; Cox et al. 2009). We next sought to characterize the method by which 15-A3t-IsoP inhibitedNFκB signaling.
Fig. 3.

15-A3t-IsoP inhibits LPS-induced NFκB nuclear translocation and activation. (a) NFκB reporter macrophages were pre-treated with varying concentrations of 15-A3t-IsoP for 30 min and then stimulated with LPS (1 μg/mL) for 4 h. Luciferase assays were then performed and data represent mean ± SD of five separate experiments; data were normalized to protein concentration expressed as % LPS-induced luciferase activity increase. Statistical analysis of were performed using a one-tailed anova and demonstrated a significant treatment effect with p < 0.01. Post hoc analysis revealed control versus LPS p < 0.001. Asterisks are used to denote statistically significant effects of various concentrations of 15-A3t-IsoP + LPS compared with LPS alone. (b) RAW264.7 cells were pre-treated with vehicle or 15-A3t-IsoP (25 μM) and then stimulated with vehicle or LPS (1 μg/mL) for 1 h. Cells were then subjected to immunofluorescent microscopy following staining of the NFκB p65 subunit (red) and nucleus (DAPI staining, blue).
NFκB is sequestered in the cytoplasm of macrophage cells and, upon inflammatory stimulation, the p50/p65 NFκB heterodimers translocate from the cytosol to the nucleus. Immunostaining of macrophages demonstrated that the NFκB p65 subunit was confined primarily to the cytoplasm in vehicle-treated cells whereas LPS stimulation resulted in intense nuclear p65 accumulation and colocalization with the nucleic acid stain DAPI after 1 h. Pre-treatment of macrophages with 15-A3t-IsoP abrogated LPS-induced p65 nuclear translocation (Fig. 3b). Cell counts demonstrated a significant change in p65-nuclear staining between LPS-stimulated and 15-A3t-IsoP pre-treated cells, indicating that cyclopentenone IsoPs have the ability to prevent NFκB nuclear translocation in the presence of an inflammatory mediator. Over 90% of the 500+ cells counted in the LPS stimulated condition had colocalization of DAPi and p65 compared to less than 13% in cells exposed to 15-A3t-IsoP and LPS.
Inhibition of NFκB Signaling by 15-A3t-IsoP occurs at the level of IκK function
We next sought to characterize the mechanism by which 15-A3t-IsoP prevented the translocation of NFκB to the nucleus. LPS-induced NFκB translocation of p65 is dependent on the phosphorylation and degradation of IκBα, the chaperone protein associating with the NFκB heterodimers which maintain the transcription factor in the cytoplasm (de Winther et al. 2005; Gloire et al. 2006).
Western blot analysis demonstrated that LPS-induced stress resulted in degradation of the vast majority of the IκBα after 15 min of stimulation (Fig. 4a) and that this decrease was maintained for at least 30 min (Fig. 4b). 15-A3t-IsoP blocked the LPS-stimulated IκBα degradation both at 15 min as well as at 30 min (Fig. 4a and b).
Fig. 4.
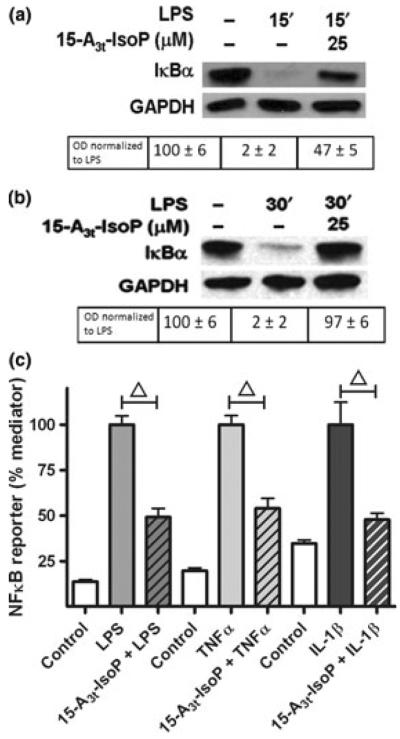
15-A3t-IsoP inhibits LPS-induced inflammation in macrophages at the level of the IκK. (a, b) RAW264.7 macrophages were pre-treated with varying concentrations of 15-A3t-IsoP for 30 min, stimulated with LPS (1 μg/mL), and harvested at 15 (a) or 30 min (b). Protein lysates were subjected to western blot analysis with the anti-IκBα-antibody and results are representative of four independent experiments. GAPDH blots are shown as loading controls. Semi-quantitative analysis of optical densities is shown below each group with average ± SD expressed for all analyses. Data in panels (a) and (b) represent four independent experiments. (c) NFκB reporter macrophages were pre-treated with vehicle or 15-A3t-IsoP for 30 min and then stimulated with LPS (1 μg/mL), TNFα (10 ng/mL), or IL-1β (20 ng/mL) for 4 h. Data expressed as % inflammatory mediator-stimulated luciferase activity. Statistical analysis of was performed using a two-tailed between group anova for each treatment (TNFα, LPS or IL1β) and demonstrated a significant treatment effect of each drug group with p < 0.01. Post hoc analysis using Tukey's HSD testing revealed significance of 15-A3t-IsoP addition compared with treatment alone with a p < 0.01 denoted by a bracketed △ representing a p < 0.01.
These data suggest that either 15-A3t-IsoP modifies inflammatory stress signals which promote their degradation or that 15-A3t-IsoP might work in a manner that would solely impact the signaling of LPS-induced NFκB stabilization. To address this question, we measured the ability of 15-A3t-IsoP to alter the activation of other IκBα-dependent pathways including TNFα, and IL-1β induced inflammation. Each of these stimuli act via distinct receptors and signaling cascades but converge on the IκB kinase (IκK) complex to activate NFκB (Karin and Ben-Neriah 2000).
We found that 15-A3t-IsoP blocked the activation of NFκB triggered by all three pro-inflammatory stimuli as assessed by the NFκB-mediated reporter system (Fig. 4c). This suggests that 15-A3t-IsoP-mediated inhibition of NFκB signaling does not occur at a receptor level but likely at the common point in these three pathways.
Bioactivity of 15-A3t-IsoP is independent of PPARγ and is abrogated by chemical reduction or conjugation of the cyclopentenone ring
Several reports have indicated that either direct or indirect activation of the peroxisome proliferator activator receptor (PPAR)γ nuclear receptor by native EPA as well as cyclopentenone-containing molecules and mediate the anti-inflammatory properties of these compounds (Chawla et al. 2001; Chene et al. 2007). To assess the role of PPARγ in mediating the effects of 15-A3t-IsoP, we used two structurally unrelated receptor antagonists, GW9662 and T0070907. Neither compound blocked the anti-inflammatory activity measured by NFκB reporter assays 4 h after LPS stimulation. Notably, both PPARγ antagonists were used at > 10-fold higher levels than required to potentiate inflammation suggesting that modification of PPARγ is not crucially involved to the anti-inflammatory properties of 15-A3t-IsoP (Fig. 5a).
Fig. 5.
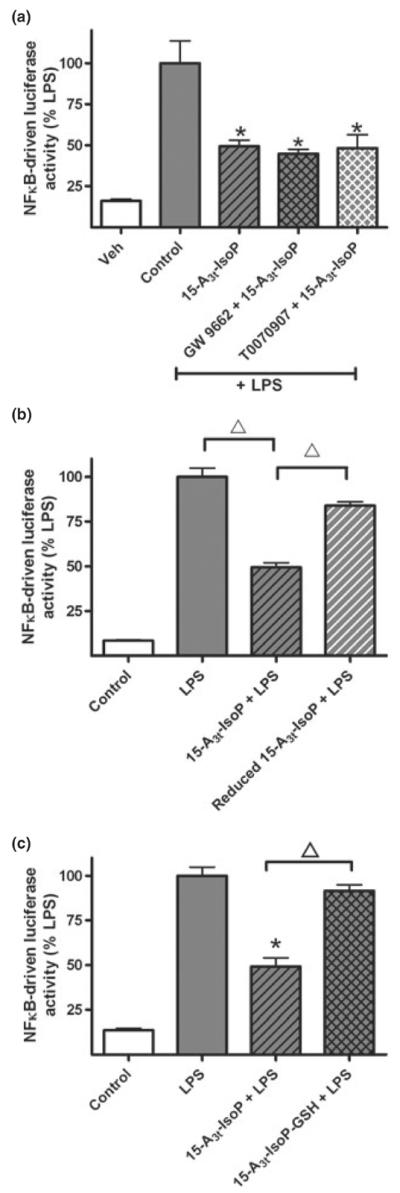
The anti-inflammatory effects of 15-A3t-IsoP are independent of PPARγ activation and are dependent on the α,β-unsaturated carbonyl moiety. (a) NFκB reporter cells were pre-treated with vehicle or one of two PPARγ antagonists (GW9662, 1 μM, or T0070907, 500 nM) for 2 h, exposed to 15-A3t-IsoP (25 μM) for 30 min, and then stimulated with LPS (1 μg/mL) as in previous experiments. Data expressed as % LPS-induced luciferase activity increase. Statistical analysis of was performed using a two-tailed between group anova and demonstrated a significant treatment. Post hoc analysis revealed that LPS only control (gray bar) was significantly different from control (white bar) (a). Additional comparisons revealed 15-A3t-IsoP, 15-A3t-IsoP with GW9662 and 15-A3t-IsoP with T0070907 were significantly different from LPS alone but not from one another as denoted by a * representing a p < 0.01 compared with LPS alone. (b, c) 15-A3t-IsoP was subjected to reduction with NaBH4 for 30 min (b) or incubated with GST and GSH for 2 h (c) and purified by C18 SepPak extraction before being applied to NFκB reporter cells as in previous experiments. C18 SepPak extraction removes residual GST, GSH, and NaBH4. LPS-stimulated NFκB reporter activity was significantly different from control in both panels (b) and (c) and 15-A3t-IsoP's effect on NFκB was significantly diminished when using the reduced form of the compound or the GSH adduct as denoted by bracketed △ representing a p < 0.01 compared with 15-A3t-IsoP + LPS. Data are shown as ± SD from four independent experiments carried out in triplicate.
In evaluating structural modifications which may account for the anti-inflammatory action of 15-A3t-IsoP, we next evaluated thiol-rich candidate molecules whose biological activity may be compromised by Michael adduction with cyclopentenone-containing compounds (Perez-Sala et al. 2002). In the first set of experiments, 15-A3t-IsoP was incubated with NaBH4, an agent whose weak reducing properties transform the carbonyl moiety on the cyclopentenone ring to a non-reactive alcohol (Brooks et al. 2008a; Cox et al. 2008, 2009), resulting in complete inhibition of the anti-inflammatory effect of 15-A3t-IsoP. When 15-A3t-IsoP was subjected to the identical incubation and purification steps without NaBH4, the compound maintained its biological activity as seen in NFκB-reporter assays (Fig. 5b). This suggested an essential role for the reactive cyclopentenone ring in mediating the bioactivity of 15-A3t-IsoP.
Cyclopentenone-containing eicosanoids can be metabolized via conjugation with GSH by GST (Atsmon et al. 1990; Milne et al. 2004). Glutathione is the most abundant cellular antioxidant. Our group has shown previously shown that EPA-derived A3/J3-IsoPs are susceptible to GSH conjugation (Brooks et al. 2008a) and the modification of GSH and GST by cyclopentenone isoprostanes yields GSH conjugates that no longer possess anti-inflammatory activities (Musiek et al. 2008). Similarly, 15-A3t-IsoP incubated with GSH and equine liver GST substantially inhibited its anti-inflammatory effects (Fig. 5c). It should be noted that residual GSH, GST, and NaBH4 were removed from these experiments by C18 column purification prior to cell treatment using previously stated methods (Chen et al. 1999). Taken together this suggests that thiol modification of the reactive cyclopentenone ring abrogates the anti-inflammatory activity of 15-A3t-IsoP.
Mutation of the IκKβ cysteine 179 impairs the anti-inflammatory effects of 15-A3t-IsoP
Given the importance of the cyclopentenone ring structure to the biological effects of 15-A3t-IsoP, we sought to determine whether this compound may inhibit NFκB function through adduction specific thiols within the IκK complex. Previous studies demonstrated that the cysteine 179 (C179) residue contains a thiol group susceptible to Michael adduction. It was also shown that cyclopentenone PGs, as well as their electrophilic metabolites, are known to inhibit NFκB signaling via adduction to this residue (Rossi et al. 2000). Thus, we examined the impact of a mutation of the IκKβ C179 to an alanine (C179A), which would prevent Michael adduction to that site but otherwise not affect the activity of the IκKβ. As shown in Fig. 6a, over-expression of WT or mutant C179A IκKβ caused a marked increase in NFκB-luciferase reporter activity in transiently transfected RAW264.7 cells in the absence of LPS signaling. 15-A3t-IsoP suppressed this increase significantly in WT IκKβ over-expressing cells but was unable to abrogate the inflammatory effect of LPS in the C179A mutation expressing cells. 15-A3t-IsoP therefore exerts some of its anti-inflammatory activity through adduction to this particular cysteine residue. Other studies demonstrated that 15-A3t-IsoP was able to significantly inhibit both PGD2 (Fig. 6b) and nitrite (data not shown) production in cells stably transfected with the WT construct. 15-A3t-IsoP, however, was less successful in inhibiting LPS-induced inflammation in cells stably transfected with the mutant C179A construct. Thus a specific residue on the IκK complex was determined to be a primary site of 15-A3t-IsoP biological activity, and, for the first time, the pathway of a specific product of non-enzymatic EPA peroxidation has been explicitly demonstrated.
Fig. 6.

Mutation of cysteine 179 on the IκKb subunit inhibits 15-A3t-IsoP activity in inflamed macrophages. (a) RAW264.7 cells were transiently transfected with an NFκB reporter luciferase reporter plasmid as well as either wild-type (left portion of graph) or mutant (C179A, right portion of graph) IκKβ expression vectors. Twenty-four hours post-transfection, cells were pre-treated with 15-A3t-IsoP (25 μM) for 30 min and stimulated with LPS (1 μg/mL) for 4 h. Cells were then assayed for luciferase activity. (b) RAW264.7 cells were transfected with either wild-type (left portion of graph) or mutant (C179A, right portion of graph) IκKβ expression vectors as well as a pcDNA3.1 antibiotic selection vector. Selection was carried out utilizing G418 sulfate as an antibiotic. After selection, cells were pre-treated with 15-A3t-IsoP (25 μM) for 30 min and stimulated with LPS (1 μg/ mL) for 24 h. PGD2 levels from the media were then assayed. Statistical analysis was performed using a two-tailed between groups anova and Tukey's HSD post hoc analysis. Paired analysis revealed that LPS was significantly different from control as denoted with an *p < 0.001 but that the only difference between LPS and LPS + 15-A3t-IsoP was in WT cells as denoted by * representing a p < 0.01. Data represented as mean ± SD and represents either five (a) or four (b) independent experiments.
15-A3t-IsoP Affects lipid accumulation in macrophages in the presence of oxidized LDL
Increased activation of NFκB signaling has been shown to correlate with atherosclerosis (Brand et al. 1996; Wilson et al. 2000). Inhibition of the NFκB signaling pathway, however, has been offered as a possible method for prevention of foam cell formation because inhibition of this pathway decreases lipid loading in macrophages treated with OxLDL (Ferreira et al. 2007). To study effects that 15-A3t-IsoP might exert on lipid loading, RAW264.7 macrophages were treated for 48 hours with vehicle, OxLDL (100 μg/mL) or OxLDL and 15-A3t-IsoP (10 μM). Forty-eight hours of treatment with either OxLDL or 15-A3t-IsoP did not have an effect on cellular viability (data not shown). Vehicle-treated cells demonstrated low levels of intracellular lipid accumulation as determined by Oil Red O staining. (Fig. 7a). Oil Red O stains neutral lipids, and cells demonstrating more than five intracellular Oil Red O droplets were considered to be positive for intracellular lipid accumulation. Treatment of cells with OxLDL significantly increased the number of stain-positive cells. Incubation of cells with 15-A3t-IsoP along with OxLDL, however, significantly decreased lipid loading, returning to near control levels (Fig. 7b). Thus, treatment of RAW264.7 cells with 15-A3t-IsoP revealed a decrease in OxLDL-induced lipid loading in cells with inhibited NFκB signaling. These data point to an underlying mechanism for the anti-atherosclerotic effects associated with EPA supplementation.
Fig. 7.
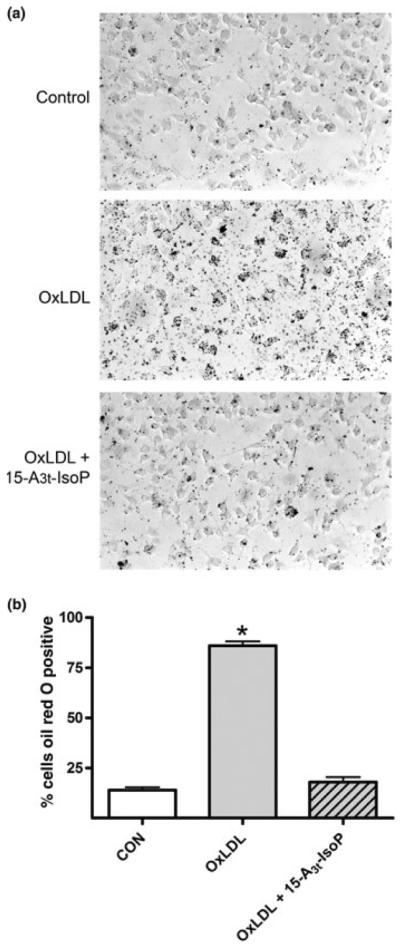
Incubation of cells with 15-A3t-IsoP decreases lipid accumulation in cells treated with oxidized LDL. (a) RAW264.7 cells were pre-treated with vehicle or 15-A3t-IsoP (10 μM) and then incubated with OxLDL (1 μg/mL) for 48 h. Cells were then subjected to light microscopy at 20× magnification after Oil Red O staining to measure intracellular lipid accumulation. (b) Quantification of lipid loading by macrophages demonstrating the percentage ± SD of cells positive for Oil Red O stain from three independent experiments. Statistical analysis was performed using a two-tailed between groups anova and Tukey's HSD post hoc analysis. Paired analysis revealed that OxLDL was significantly different from control as denoted with an *p < 0.001 OxLDL + 15-A3t-IsoP was not significantly different from control.
Discussion
The CNS vasculature endures tremendous physical and biochemical stress ensuring the energetic demands of the brain are met while serving as an essential barrier to prevent the accumulation/promote the removal of toxins. Plaque formation within the vasculature is a major cause of ischemic stroke as leukocytes enters damaged tissue through blood vessel endothelium where they undergo a series of changes to become a macrophage. Neuroinflammatory stress has been implicated in acute diseases such as stroke and traumatic brain injury as well as chronic diseases including Alzheimer's, multiple sclerosis and Parkinson's disease amongst others (Bazan et al. 2002; Phillis et al. 2006; Glass et al. 2010). Moreover, the use of fish oil, a rich source of the omega-3 or n-3 fatty acids has been shown to be beneficial in the prevention of important human diseases such as atherosclerosis and sudden cardiac death, and various inflammatory disorders. Although the mechanism(s) by which the n-3 fatty acids exert their biological activity is unknown, an important anti-atherogenic and anti-inflammatory mechanism of EPA action is its interference with the arachidonic acid cascade that generates pro-inflammatory eicosanoids via the cyclooxygenases and lipo-oxygenases
Oxidative stress is an essential component of macrophage transformation and it promotes the production of highly potent lipid peroxidation products formed from arachidonic acid and other polyunsaturated fatty acids. Oxidative stress and vascular dysfunction has been implicated not only in acute disorders including stroke and traumatic brain injury, but also in chronic `peripheral' conditions which impact the CNS including diabetes, hypertension and vasculitis. Additional complications of vascular dysfunction have also been associated with neurodegenerative diseases and oncogenic transformation. A number of clinical trials have now shown improvements in learning and memory, vascular integrity, neuronal survival and human health when subjects were given dietary supplementation with the `omega' or n-3 fatty acids docosahexaenoic acid (DHA) and EPA (De Caterina and Zampolli 2001; Simopoulos 2002; Cleland et al. 2006; Mozaffarian 2007; Calder 2008; Saito et al. 2008).
Although many studies have focused on the beneficial aspects of native EPA and its enzymatically derived metabolites, there is a growing consensus that nonenzymatic oxidation products of EPA exert potent, beneficial biological activities. Vallve et al. (2002) reported that non-enzymatic aldehyde oxidation products of EPA decrease the expression of the CD36 receptor in macrophages, a receptor linked to atherosclerosis. Moreover, free-radical catalyzed oxidation of EPA is a major consequence of an oxidizing environment as demonstrated by the formation of A3/J3-IsoPs in subjects fed an EPA-enhanced diet (Gao et al. 2006; Brooks et al. 2008a). Recent work confirmed that these EPA oxidation compounds are formed in vivo in micromolar quantities in settings of oxidative stress (Brooks et al. 2008a). We believe the current work uses EPA products likely in the pathophysiologically relevant range given that A3/J3-IsoPs from EPA-fed mice exposed to oxidative stress was 102 ng/g in liver. As the MW of A3/J3-IsoP is 353 ng/nmol, this equals 0.289 nmol/g. Assuming liver tissue has a density similar to water, then this is 0.289 nmol/mL = 289 nM.
Levels of A3/J3-IsoPs have not been quantified in brain tissue. However, EPA is enriched in brain tissue, suggesting that low micromolar quantities of A3/J3-IsoP could conceivably be achieved in brain tissue in vivo. Increasing concentrations of native EPA in vivo induces a sharp decrease in the level of arachidonic-acid derived IsoPs and a dramatic increase in EPA-derived IsoPs (Davis et al. 2006). EPA-derived IsoPs form at levels ten times higher in settings of oxidative stress than do IsoPs from arachidonic acid (Gao et al. 2007). By conducting a total synthesis of a specific regio- and stereoisomer of an endogenously formed EPA-derived cyclopentenone IsoP, 15-A3t-IsoP, we can begin to study activities of specific oxidation products of EPA that forms in vivo in micromolar quantities in settings of oxidative stress.
The enzymatic and chemical oxidation of fatty acids plays a central role in regulating inflammation. Prostaglandins, thromboxanes, IsoPs, and leukotrienes initiate and propagate signals essential for inflammatory stress via direct activation of G-protein-coupled activation, nuclear receptor activation, and in some cases by post-translational protein modification. Exposure to native EPA and DHA has been linked specifically to activation of the PPARγ receptor (Li et al. 2005), inhibition of toll-like receptor activity (Lee et al. 2003), alterations of lipid raft composition in T-cells (Li et al. 2006), and inhibition of the production of cytokines such as IL-1β and TNFα in monocytes (Babcock et al. 2002) and IL-6 and IL-8 in endothelial cells (De Caterina et al. 1994; Khalfoun et al. 1997).
In evaluating the mechanism of action of 15-A3t-IsoP, we found that unlike 15-A2t-IsoP, that is formed by oxidation of arachidonic acid, and exacerbates oxidative stress in RAW264.7 cells (Musiek et al. 2005), 15-A3t-IsoP was not cytotoxic and did not increase oxidative stress in macrophages. This underscores the importance of the fatty acid backbone (from which oxidized lipids are derived) in determining the biological activities of these compounds.
We found that similar to the activity of the cyclopentenone PGs, cyclopentenone IsoPs exert potent immunomodulatory effects. When administered to LPS-stimulated macrophages, 15-A3t-IsoP inhibited NFκB activation via impairment of IκBα degradation and IκK activity. Furthermore, 15-A3t-IsoP attenuated LPS-induced expression of the NFκB-responsive proteins iNOS and COX-2 while rapidly blocking nitric oxide and PG production. The cyclopentenone ring on these compounds of 15-A3t-IsoP is necessary for their bioactivity, and their ability to undergo a Michael adduction to a specific cysteine residue on the IκK complex was important for inhibition of LPS-induced inflammation.
The proposed pathway of EPA-derived cyclopentenone IsoP anti-inflammatory action can be found in Fig. 8. These data demonstrate that the mechanism of action of A3t-IsoP is similar to other α,β-unsaturated carbonyl-containing compounds such as PGA2 and A2/J2-IsoPs (Rossi et al. 2000; Musiek et al. 2005). These effects were also shown to participate in a PPARγ-independent manner. Therefore, the cytoprotective potential of EPA-derived 15-A3t-IsoP lies not only in the anti-inflammatory effects we observed but also in the ability of this compound to diminish total lipid oxidation in macrophages. Based on these data, one might expect that the beneficial effects of supplementation with EPA may be closely associated with formation of A3-IsoP levels at concentrations enabling anti-inflammatory effects while still avoiding the unwanted effects that occur with complete NFκB inhibition. Indeed, this study lays the groundwork defining a broader pleotropic beneficial action of EPA supplementation much in the same way as has been demonstrated for DHA (Horrocks and Yeo 1999; Montine et al. 2004) and offer an excellent biological metric of efficacy given the correlation between A3t-IsoP levels and anti-inflammatory activity of EPA-derived cyclopentenone IsoPs.
Fig. 8.

Model of NFκB signaling inhibition by EPA-derived cyclopentenone IsoPs. Based on our findings, we suggest that 15-A3t-IsoP is formed in cellular membranes and can adduct to exposed cysteine residues on the β subunit of the IκK complex, hindering the complex's ability to activate NFκB signaling. This results in decreased iNOS and provides a beneficial effect on vascular integrity.
In this study, we report that in addition to identifying a redox sensitive site on IκK which is modified in response to A3t-IsoP treatment, we found that A3t-IsoP is able to inhibit a major step in atherosclerosis – the formation of foam cells in response to treatment with oxidized lipids. Macrophages have been shown to internalize OxLDL, and this increase in lipid loading was determined to be an important first-step in the progression of atherosclerosis (Hansson et al. 2006). 15-A3t-IsoP decreased the accumulation of cytosolic droplets, and, consequently, could delay the formation of foam cells from macrophages interacting with OxLDL in vivo.
We believe this work warrants further study to determine the nature and extent of the anti-atherosclerotic activity of 15-A3t-IsoP. Additional studies would determine if this effect is caused by prevention of cholesterol accumulation or via other mechanisms, such as alteration of NFκB-driven migration molecules necessary for deposition of foam cells (Brach et al. 1991; Bond et al. 1998) or the inhibition of NFκB-controlled inflammatory mediators (Gu et al. 1998; Burleigh et al. 2002; Zhao and Funk 2004). Given that the A3-IsoPs are not cytotoxic and do not increase the formation of other lipid-derived peroxidation products, this specific EPA product may be more useful than native EPA in eliciting anti-inflammatory and cytoprotective responses in cells.
Acknowledgements
Research was supported by NIH grants NS050396 (BM), GM15431 (JDM), DK48831 (JDM), CA77839 (JDM) and training grants from PhRMA (ESM.) and the Vanderbilt Brain Institute (JNS). Statistical and graphical support was provided by P30HD15052 (Vanderbilt Kennedy Center). We thank Dr. Michael Karin for generously providing the IκK wild-type and C179A plasmids, Dr. Timothy Blackwell for generously providing the murine NFκB reporter cells, and Dr. Pat Levitt for generously providing the pcDNA3.1 plasmid. Technical assistance on this work was graciously provided by Stephanie Sanchez and Jacquelynn Brown and editorial assistance by Jessica Cohen and Lauren Koenig. We very much appreciate the thoughtful suggestions on experimental design provided by Dr. Gregg Stanwood.
Abbreviations used
- BCA
bicinchoninic acid
- CH2Cl2
dicholoromethane
- COX-2
cyclo-oxygenase 2
- DAPI
4′,6-diamidino-2-phenylindole
- DHA
docosahexaenoic acid
- EPA
eicosapentaenoic acid
- GC-NICI-MS
gas chromatography-negative ion chemical ionization mass spectrometry
- GST
glutathione-S-transferase
- IL-1β
interleukin-1 beta
- iNOS
inducible nitric oxide synthase
- IsoP
isoprostane
- IκBα
inhibitor of kappa b
- IκK
I kappa kinase
- K3PO4
potassium phosphate
- LPS
lipopolysaccharide
- LT
leukotriene
- NaBH4
sodium borohydride
- NFκB
nuclear factor kappa b
- OxLDL
oxidized low-density lipoprotein
- PBS
phosphate-buffered saline
- PG
prostaglandin
- PGD2
prostaglandin D2
- PPAR
peroxisome proliferator activator receptor
- PUFA
polyunsaturated fatty acids
- TLR
toll-like receptor
- TNFα
tumor necrosis factor alpha
Footnotes
The authors of this work declare they have no financial conflicts of interest in this work.
References
- Atsmon J, Freeman ML, Meredith MJ, Sweetman BJ, Roberts LJ., 2nd Conjugation of 9-deoxy-delta 9,delta 12(E)-prostaglandin D2 with intracellular glutathione and enhancement of its antiproliferative activity by glutathione depletion. Cancer Res. 1990;50:1879–1885. [PubMed] [Google Scholar]
- Babcock TA, Novak T, Ong E, Jho DH, Helton WS, Espat NJ. Modulation of lipopolysaccharide-stimulated macrophage tumor necrosis factor-alpha production by omega-3 fatty acid is associated with differential cyclooxygenase-2 protein expression and is independent of interleukin-10. J. Surg. Res. 2002;107:135–139. doi: 10.1006/jsre.2002.6498. [DOI] [PubMed] [Google Scholar]
- Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004;3:205–214. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- Bazan NG, Colangelo V, Lukiw WJ. Prostaglandins and other lipid mediators in Alzheimer's disease. Prostaglandins Other Lipid Mediators. 2002;68-9:197–210. doi: 10.1016/s0090-6980(02)00031-x. [DOI] [PubMed] [Google Scholar]
- Blackwell TS, Yull FE, Chen CL, Venkatakrishnan A, Blackwell TR, Hicks DJ, Lancaster LH, Christman JW, Kerr LD. Multiorgan nuclear factor kappa B activation in a transgenic mouse model of systemic inflammation. Am. J. Respir. Crit. Care Med. 2000;162:1095–1101. doi: 10.1164/ajrccm.162.3.9906129. [DOI] [PubMed] [Google Scholar]
- Bond M, Fabunmi RP, Baker AH, Newby AC. Synergistic upregulation of metalloproteinase-9 by growth factors and inflammatory cytokines: an absolute requirement for transcription factor NF-kappa B. FEBS Lett. 1998;435:29–34. doi: 10.1016/s0014-5793(98)01034-5. [DOI] [PubMed] [Google Scholar]
- Brach MA, Henschler R, Mertelsmann RH, Herrmann F. Regulation of M-CSF expression by M-CSF: role of protein kinase C and transcription factor NF kappa B. Pathobiology. 1991;59:284–288. doi: 10.1159/000163664. [DOI] [PubMed] [Google Scholar]
- Brand K, Page S, Rogler G, et al. Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J. Clin. Invest. 1996;97:1715–1722. doi: 10.1172/JCI118598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks JD, Milne GL, Yin H, Sanchez SC, Porter NA, Morrow JD. Formation of highly reactive cyclopentenone isoprostane compounds (A3/J3-isoprostanes) in vivo from eicosapentaenoic acid. J. Biol. Chem. 2008a;283:12043–12055. doi: 10.1074/jbc.M800122200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks JD, Sanchez SC, Milne GL, McLaughlin BA, Morrow JD. Highly reactive cyclopentenone isoprostanes derived from eicosapentaenoic acid exert potent anti-inflammatory effects through inhibition of NF kappa B signaling. Free Radic. Biol. Med. 2008b;45:S133–S133. [Google Scholar]
- Brown JE, Zeiger SLH, Hettinger JC, Brooks JD, Holt B, Morrow JD, Musiek ES, Milne G, McLaughlin B. Essential role of the redox-sensitive kinase p66shc in determining energetic and oxidative status and cell fate in neuronal preconditioning. J. Neurosci. 2010;30:5242–5252. doi: 10.1523/JNEUROSCI.6366-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burleigh ME, Babaev VR, Oates JA, et al. Cyclooxygenase-2 promotes early atherosclerotic lesion formation in LDL receptor-deficient mice. Circulation. 2002;105:1816–1823. doi: 10.1161/01.cir.0000014927.74465.7f. [DOI] [PubMed] [Google Scholar]
- Calder PC. Use of fish oil in parenteral nutrition: Rationale and reality. Proc. Nutr. Soc. 2006;65:264–277. doi: 10.1079/pns2006500. [DOI] [PubMed] [Google Scholar]
- Calder PC. Polyunsaturated fatty acids, inflammatory processes and inflammatory bowel diseases. Mol. Nutr. Food Res. 2008;52:885–897. doi: 10.1002/mnfr.200700289. [DOI] [PubMed] [Google Scholar]
- Chaudhary A, Mishra A, Sethi S. Oxidized omega-3 fatty acids inhibit pro-inflammatory responses in glomerular endothelial cells. Nephron. Exp. Nephrol. 2004;97:e136–e145. doi: 10.1159/000079178. [DOI] [PubMed] [Google Scholar]
- Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat. Med. 2001;7:48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- Chen Y, Morrow JD, Roberts LJ., II Formation of reactive cyclopentenone compounds in vivo as products of the isoprostane pathway. J. Biol. Chem. 1999;274:10863–10868. doi: 10.1074/jbc.274.16.10863. [DOI] [PubMed] [Google Scholar]
- Chen CT, Liu Z, Bazinet RP. Rapid de-esterification and loss of eicosapentaenoic acid from rat brain phospholipids: an intracerebroventricular study. J. Neurochem. 2011;116:363–373. doi: 10.1111/j.1471-4159.2010.07116.x. [DOI] [PubMed] [Google Scholar]
- Chene G, Dubourdeau M, Balard P, et al. n-3 and n-6 polyunsaturated fatty acids induce the expression of COX-2 via PPARgamma activation in human keratinocyte HaCaT cells. Biochim. Biophys. Acta. 2007;1771:576–589. doi: 10.1016/j.bbalip.2007.02.014. [DOI] [PubMed] [Google Scholar]
- Chun KS, Surh YJ. Signal transduction pathways regulating cyclooxygenase-2 expression: potential molecular targets for chemoprevention. Biochem. Pharmacol. 2004;68:1089–1100. doi: 10.1016/j.bcp.2004.05.031. [DOI] [PubMed] [Google Scholar]
- Cleland JG, Freemantle N, Coletta AP, Clark AL. Clinical trials update from the American Heart Association: REPAIR-AMI, ASTAMI, JELIS, MEGA, REVIVE-II, SURVIVE, and PROACTIVE. Eur. J. Heart Fail. 2006;8:105–110. doi: 10.1016/j.ejheart.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Connor WE. The beneficial effects of omega-3 fatty acids: cardiovascular disease and neurodevelopment. Curr. Opin. Lipidol. 1997;8:1–3. doi: 10.1097/00041433-199702000-00001. [DOI] [PubMed] [Google Scholar]
- Connors SL, Levitt P, Matthews SG, Slotkin TA, Johnston MV, Kinney HC, Johnson WG, Dailey RM, Zimmerman AW. Fetal mechanisms in neurodevelopmental disorders. Pediatr. Neurol. 2008;38:163–176. doi: 10.1016/j.pediatrneurol.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Cox BE, Milne GL, Brooks JD, Sanchez SC, Yin HY, Morrow JD. Formation of novel D-ring and E-ring isoprostanes-like compounds in vivo and in vitro from the oxidation of eicosapentaenoic acid. Free Radic. Biol. Med. 2008;45:S69–S69. [Google Scholar]
- Cox BE, Brooks JD, Pfister SL, Morrow JD, Yin HY, Milne GL, Roberts LJ. Comparison studies on the biological activity of 3-series isoprostanes derived from eicosapentaenoic acid and 2-series isoprostanes derived from arachidonic acid. Free Radic. Biol. Med. 2009;47:S72–S72. [Google Scholar]
- Davis TA, Gao L, Yin H, Morrow JD, Porter NA. In vivo and in vitro lipid peroxidation of arachidonate esters: the effect of fish oil omega-3 lipids on product distribution. J. Am. Chem. Soc. 2006;128:14897–14904. doi: 10.1021/ja064399o. [DOI] [PubMed] [Google Scholar]
- De Caterina R, Zampolli A. n-3 fatty acids: antiatherosclerotic effects. Lipids. 2001;36(Suppl):S69–S78. doi: 10.1007/s11745-001-0685-9. [DOI] [PubMed] [Google Scholar]
- De Caterina R, Cybulsky MI, Clinton SK, Gimbrone MA, Jr, Libby P. The omega-3 fatty acid docosahexaenoate reduces cytokine-induced expression of proatherogenic and proinflammatory proteins in human endothelial cells. Arterioscler. Thromb. 1994;14:1829–1836. doi: 10.1161/01.atv.14.11.1829. [DOI] [PubMed] [Google Scholar]
- Fam SS, Murphey LJ, Terry ES, et al. Formation of highly reactive A-ring and J-ring isoprostane-like compounds (A4/J4-neuroprostanes) in vivo from docosahexaenoic acid. J. Biol. Chem. 2002;277:36076–36084. doi: 10.1074/jbc.M205638200. [DOI] [PubMed] [Google Scholar]
- Ferreira V, van Dijk KW, Groen AK, Vos RM, van der Kaa J, Gijbels MJ, Havekes LM, Pannekoek H. Macrophage-specific inhibition of NF-kappaB activation reduces foam-cell formation. Atherosclerosis. 2007;192:283–290. doi: 10.1016/j.atherosclerosis.2006.07.018. [DOI] [PubMed] [Google Scholar]
- Gao L, Yin H, Milne GL, Porter NA, Morrow JD. Formation of F-ring isoprostane-like compounds (F3-isoprostanes) in vivo from eicosapentaenoic acid. J. Biol. Chem. 2006;281:14092–14099. doi: 10.1074/jbc.M601035200. [DOI] [PubMed] [Google Scholar]
- Gao L, Wang J, Sekhar KR, et al. Novel n-3 fatty acid oxidation products activate Nrf2 by destabilizing the association between Keap1 and Cullin3. J. Biol. Chem. 2007;282:2529–2537. doi: 10.1074/jbc.M607622200. [DOI] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem. Pharmacol. 2006;72:1493–1505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, Rollins BJ. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell. 1998;2:275–281. doi: 10.1016/s1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- Hansson GK, Robertson AK, Soderberg-Naucler C. Inflammation and atherosclerosis. Annu. Rev. Pathol. 2006;1:297–329. doi: 10.1146/annurev.pathol.1.110304.100100. [DOI] [PubMed] [Google Scholar]
- Horrocks LA, Yeo YK. Health benefits of docosahexaenoic acid (DHA) Pharmacol. Res. 1999;40:211–225. doi: 10.1006/phrs.1999.0495. [DOI] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- Khalfoun B, Thibault F, Watier H, Bardos P, Lebranchu Y. Docosahexaenoic and eicosapentaenoic acids inhibit in vitro human endothelial cell production of interleukin-6. Adv. Exp. Med. Biol. 1997;400B:589–597. [PubMed] [Google Scholar]
- Kris-Etherton PM, Harris WS, Appel LJ. Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation. 2002;106:2747–2757. doi: 10.1161/01.cir.0000038493.65177.94. [DOI] [PubMed] [Google Scholar]
- Lee JY, Plakidas A, Lee WH, Heikkinen A, Chanmugam P, Bray G, Hwang DH. Differential modulation of Toll-like receptors by fatty acids: preferential inhibition by n-3 polyunsaturated fatty acids. J. Lipid Res. 2003;44:479–486. doi: 10.1194/jlr.M200361-JLR200. [DOI] [PubMed] [Google Scholar]
- Li H, Ruan XZ, Powis SH, Fernando R, Mon WY, Wheeler DC, Moorhead JF, Varghese Z. EPA and DHA reduce LPS-induced inflammation responses in HK-2 cells: evidence for a PPAR-gamma-dependent mechanism. Kidney Int. 2005;67:867–874. doi: 10.1111/j.1523-1755.2005.00151.x. [DOI] [PubMed] [Google Scholar]
- Li Q, Tan L, Wang C, Li N, Li Y, Xu G, Li J. Polyun-saturated eicosapentaenoic acid changes lipid composition in lipid rafts. Eur. J. Nutr. 2006;45:144–151. doi: 10.1007/s00394-005-0574-7. [DOI] [PubMed] [Google Scholar]
- McLaughlin BA, Hartnett KA, Erhardt JA, Legos JJ, White RF, Barone FC, Aizenman E. Caspase 3 activation is essential for neuroprotection in ischemic preconditioning. Proc. Natl Acad. Sci. USA. 2003;100:715–720. doi: 10.1073/pnas.0232966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne GL, Zanoni G, Porta A, Sasi S, Vidari G, Musiek ES, Freeman ML, Morrow JD. The cyclopentenone product of lipid peroxidation, 15-A2t-isoprostane, is efficiently metabolized by HepG2 cells via conjugation with glutathione. Chem. Res. Toxicol. 2004;17:17–25. doi: 10.1021/tx034213o. [DOI] [PubMed] [Google Scholar]
- Mishra A, Chaudhary A, Sethi S. Oxidized omega-3 fatty acids inhibit NF-kappaB activation via a PPARalpha-dependent pathway. Arterioscler. Thromb. Vasc. Biol. 2004;24:1621–1627. doi: 10.1161/01.ATV.0000137191.02577.86. [DOI] [PubMed] [Google Scholar]
- Montine KS, Quinn JF, Zhang J, Fessel JP, Roberts LJ, Morrow JD, Montine TJ. Isoprostanes and related products of lipid peroxidation in neurodegenerative diseases. Chem. Phys. Lipids. 2004;128:117–124. doi: 10.1016/j.chemphyslip.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Mori M, Itabe H, Higashi Y, Fujimoto Y, Shiomi M, Yoshizumi M, Ouchi Y, Takano T. Foam cell formation containing lipid droplets enriched with free cholesterol by hyperlipidemic serum. J. Lipid Res. 2001;42:1771–1781. [PubMed] [Google Scholar]
- Morrow JD, Roberts LJ., II Mass spectrometric quantification of F2-isoprostanes in biological fluids and tissues as measure of oxidant stress. Methods Enzymol. 1999;300:3–12. doi: 10.1016/s0076-6879(99)00106-8. [DOI] [PubMed] [Google Scholar]
- Mozaffarian D. JELIS, fish oil, and cardiac events. Lancet. 2007;369:1062–1063. doi: 10.1016/S0140-6736(07)60504-2. [DOI] [PubMed] [Google Scholar]
- Musiek ES, Gao L, Milne GL, et al. Cyclopentenone isoprostanes inhibit the inflammatory response in macrophages. J. Biol. Chem. 2005;280:35562–35570. doi: 10.1074/jbc.M504785200. [DOI] [PubMed] [Google Scholar]
- Musiek ES, Brooks JD, Joo M, et al. Electrophilic cyclopentenone neuroprostanes are anti-inflammatory mediators formed from the peroxidation of the omega-3 polyunsaturated fatty acid docosahexaenoic acid. J. Biol. Chem. 2008;283:19927–19935. doi: 10.1074/jbc.M803625200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Sala D, Cernuda-Morollon E, Pineda-Molina E, Canada FJ. Contribution of covalent protein modification to the anti-inflammatory effects of cyclopentenone prostaglandins. Ann. N Y Acad. Sci. 2002;973:533–536. doi: 10.1111/j.1749-6632.2002.tb04695.x. [DOI] [PubMed] [Google Scholar]
- Phillis JW, Horrocks LA, Farooqui AA. Cyclooxygenases, lipoxygenases, and epoxygenases in CNS: Their role and involvement in neurological disorders. Brain Res. Rev. 2006;52:201–243. doi: 10.1016/j.brainresrev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- Saito Y, Yokoyama M, Origasa H, et al. Effects of EPA on coronary artery disease in hypercholesterolemic patients with multiple risk factors: Sub-analysis of primary prevention cases from the Japan EPA Lipid Intervention Study (JELIS) Atherosclerosis. 2008;300:135–140. doi: 10.1016/j.atherosclerosis.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Simopoulos AP. Omega-3 fatty acids in inflammation and autoimmune diseases. J. Am. Coll. Nutr. 2002;21:495–505. doi: 10.1080/07315724.2002.10719248. [DOI] [PubMed] [Google Scholar]
- Vallve JC, Uliaque K, Girona J, Cabre A, Ribalta J, Heras M, Masana L. Unsaturated fatty acids and their oxidation products stimulate CD36 gene expression in human macrophages. Atherosclerosis. 2002;164:45–56. doi: 10.1016/s0021-9150(02)00046-1. [DOI] [PubMed] [Google Scholar]
- Wilson SH, Caplice NM, Simari RD, Holmes DR, Jr, Carlson PJ, Lerman A. Activated nuclear factor-kappaB is present in the coronary vasculature in experimental hypercholes-terolemia. Atherosclerosis. 2000;148:23–30. doi: 10.1016/s0021-9150(99)00211-7. [DOI] [PubMed] [Google Scholar]
- de Winther MP, Kanters E, Kraal G, Hofker MH. Nuclear factor kappaB signaling in atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2005;25:904–914. doi: 10.1161/01.ATV.0000160340.72641.87. [DOI] [PubMed] [Google Scholar]
- Yokoyama M, Origasa H. Effects of eicosapentaenoic acid on cardiovascular events in Japanese patients with hypercholes-terolemia: rationale, design, and baseline characteristics of the Japan EPA Lipid Intervention Study (JELIS) Am. Heart J. 2003;146:613–620. doi: 10.1016/S0002-8703(03)00367-3. [DOI] [PubMed] [Google Scholar]
- Yokoyama M, Origasa H, Matsuzaki M, et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet. 2007;369:1090–1098. doi: 10.1016/S0140-6736(07)60527-3. [DOI] [PubMed] [Google Scholar]
- Zanoni G, Porta A, Vidari G. First total synthesis of A(2) isoprostane. J. Org. Chem. 2002;67:4346–4351. doi: 10.1021/jo025652f. [DOI] [PubMed] [Google Scholar]
- Zhao L, Funk CD. Lipoxygenase pathways in atherogenesis. Trends Cardiovasc. Med. 2004;14:191–195. doi: 10.1016/j.tcm.2004.04.003. [DOI] [PubMed] [Google Scholar]