

Abstract
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) confer adaptive immunity to prokaryotes through a small RNA-mediated mechanism. Specific endoribonucleases are required by all CRISPR-bearing organisms to process CRISPR RNAs into small RNA that serve as guides for defensive effector complexes. The molecular mechanism of how the endoribonucleases process the class of CRISPR RNA containing no secondary structural features remains largely elusive. Here we report cocrystal structures of a processing endoribonuclease bound with a noncleavable RNA substrate and its product-like fragment derived from a nonpalindramic repeat. The enzyme stabilizes a short RNA stem-loop structure near the cleavage site and cleaves the phosphodiester bond using an active site comprised of arginine and lysine residues. The distinct RNA binding and cleavage mechanisms underline the diversity in CRISPR RNA processing.
Introduction
Many bacteria and archaea employ a strategy based on Clustered Regularly Interspaced Palindromic Repeats (CRISPRs) to defend themselves against harmful nucleic acids (Barrangou et al., 2007; Brouns et al., 2008). Knowledge of this mechanism has fueled studies of RNA-mediated gene regulation and silencing and is believed to have potential applications in areas including development of new strategies for controlling antibiotics resistance of bacterial pathogens (Deveau et al., 2010; Horvath and Barrangou, 2010; Karginov and Hannon, 2010; Marraffini and Sontheimer, 2010; Sorek et al., 2008; van der Oost et al., 2009). CRISPRs are genetic loci of identical repeats interspaced by unique spacer sequences that are derived from past infections (Mojica et al., 2005; Pourcel et al., 2005). CRISPR loci often include adjacent sets of protein encoding genes (CRISPR-associated or cas genes) (Haft et al., 2005; Makarova et al., 2006). The repeat-spacer array and the Cas proteins instigate three functional phases of this remarkable immunity mechanism: spacer incorporation, CRISPR RNA (crRNA) biogenesis, and interference (Barrangou and Horvath, 2012; Bhaya et al., 2011; Terns and Terns, 2011; Wiedenheft et al., 2012). In many organisms, the step of crRNA biogenesis critically depends on the activity of the endonuribonuclease Cas6 that recognizes and excises the repeat-spacer array transcript (Brouns et al., 2008; Carte et al., 2008; Haurwitz et al., 2010; Richter et al., 2012; Zhang et al., 2012).
Cas6 cleaves within the repeat region of the repeat-spacer array transcript to yield individual spacer RNA flanked by portions of the repeat (Fig. 1A). Mature crRNA subsequently assemble with other Cas proteins into effector complexes. These complexes target invading DNA or RNA for destruction. A processed crRNA typically contains the last 8 nucleotides (nts) of the repeat (5′-handle), a spacer sequence (guide) (Brouns et al., 2008; Haurwitz et al., 2010; Lintner et al., 2011) and, in some cases, a variable 3′ end comprised of the 5′ portion of the repeat (Hale et al., 2008; Richter et al., 2012; Zhang et al., 2012) (Fig. 1A). The variability of the 3′ flanking repeat appears to be effector complex-specific (Hale et al., 2009; Lintner et al., 2011; Zhang et al., 2012) and its further processing is believed to be carried out by unknown exonuclease activities (Hale et al., 2008). The 5′-handle of the crRNA plays critical roles in the specific assembly of effector complexes while the guide facilitates selection of the target DNA or RNA in the destruction process (Hale et al., 2009; Wiedenheft et al., 2011a; Wiedenheft et al., 2011b; Zhang et al., 2012). The unprocessed 3′-flanking repeat is used to anchor Cas6 onto the effector complex, thereby coupling crRNA processing to effector assembly (Haurwitz et al., 2012; Wiedenheft et al., 2011a).
Fig. 1.
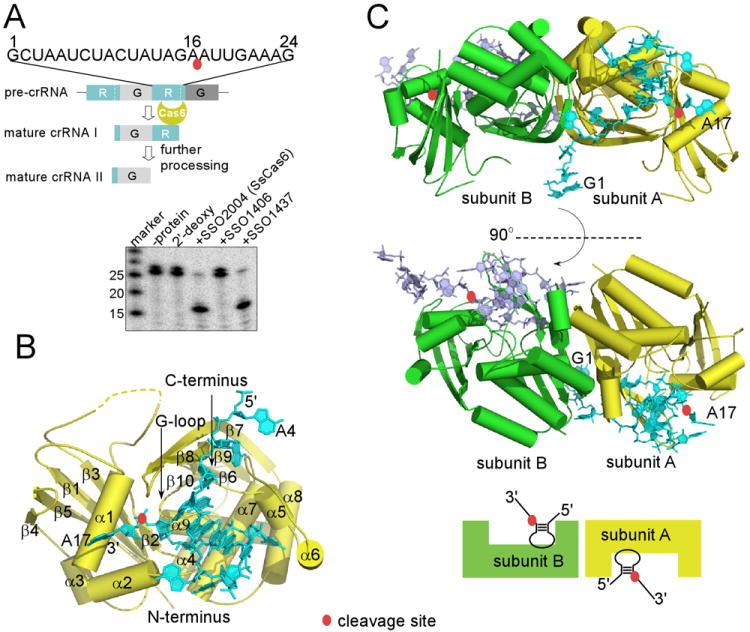
Overview of CRISPR RNA processing activities and structures of SsCas6 bound with a repeat RNA. The scissile phosphate group is indicated by a red oval. (A). Sulfolobus sulfotaricus P2 (Ss) CRISPR RNA processing activities of Ss proteins SSO2004, SSO1437, and SSO1406. The repeat RNA from Ss CRISPR locus F was cleaved by the two close homologs SSO2004 and SSO1437 but not SSO1406 whose sequence clearly distinguishes itself from the other two. “2′-deoxy” denotes the cleavage result with SSO2004 and the same 24mer repeat RNA containing 2′-deoxy modification at position 16. Markers were used to provide relative positions of non-related RNA oligomers. (B). Overview of SsCas6 structure and its complex with the repeat RNA. The structure of SsCas6 in isolation is similar to that bound with RNA and is thus not shown separately here. Secondary structure elements of SsCas6 and the RNA nucleotides are labeled. (C). Two orthogonal views of the dimeric structure of SsCas6 bound with the 24mer noncleavable RNA. The monomeric units are distinguished by different colors.
Whereas currently known Cas6 likely share a structural fold and a similar 3′ cleavage product (i.e., 5′-handle), they appear to employ different methods for recognition of crRNA, due largely to the highly variable structures of the repeat RNA. Cas6 proteins belong to the Repeat Associated Mysterious Protein (RAMP) family of proteins that are characterized by, in most cases, a tandem ferredoxin fold (Makarova et al., 2011). In contrast, repeat RNA of even closely related organisms share little sequence homology except for a prominent palindromic feature in many organisms (Kunin et al., 2007). The lack of sequence and structural similarities in repeat RNAs raises the question how structurally different RNAs are recognized by Cas6 proteins. Crystal structures of Cas6-RNA complexes determined so far suggest at least two distinct methods of recognition. Both E. coli and Pseudomonas aeruginosa (Pa) CRISPR repeats have clear palindromic features. Consistently, the structure of Thermus thermophilus Cas6, Cas6e (also named CasE or Cse3) bound with its repeat RNA and that of Pa Cas6, Cas6f (also named Csy4), bound with its repeat RNA revealed a critical importance of the stem-loop structure of the RNA in their recognition and cleavage (Gesner et al., 2011; Haurwitz et al., 2012; Haurwitz et al., 2010; Sashital et al., 2011). On the other hand, Pyroccucus fulgidus (Pf) RNAs lack this secondary motif and the structure of a PfCas6-RNA complex captured only the 5′ region of the repeat RNA in a single stranded conformation with its cleavage site region disordered (Wang et al., 2011), suggesting a binding model consistent with recognition of single stranded region. However, structure data on recognition of the cleavage sites of the nonpalindromic repeats remain missing, leaving a gap in our understanding of crRNA processing mechanisms.
Sulfolobus sulfotaricus P2 (Ss) provides a rich source of crRNA that result from its six CRISPR loci A-F (Lillestol et al., 2009; Lintner et al., 2011; Zhang et al., 2012). Ss repeats do not appear to exhibit conserved palindromic features and are thus categorized into a cluster of unstructured repeats (Kunin et al., 2007). Among the five annotated Ss Cas6 proteins (SSO1381, SSO1406, SSO1422, SSO1437, and SSO2004), SSO2004 was the first to be shown to possess specific processing activity on Ss repeat RNAs (Lintner et al., 2011). To understand how Cas6 processes RNAs with no detectable secondary structures, we studied crystal structures of SSO2004 (SsCas6 herein), both in absence and presence of a repeat RNA substrate. Our structural and complementary biochemical analyses show that SsCas6 is able to bind and cleave the nonstructured RNA by stabilizing an otherwise unstable duplex of merely two base pairs near the cleavage site, leading to an inline conformation around the scissile phosphate necessary for its breakage.
Results
Overview of SsCas6-RNA Complex Structures
We first confirmed that a recombinant C-terminally histidine-tagged SsCas6 purified from E. coli was able to cleave repeat RNAs. SsCas6 was incubated with a 5′-radiolabeled 24mer repeat RNA derived from Ss CRISPR locus F at 45°C and a 16mer product was observed (Fig. 1A). The same RNA substrate bearing 2′-deoxy modification at position 16 was not cleaved, suggesting a cleavage mechanism similar to that of RNase A (Fig. 1A). Due to small perturbation to RNA structure by the 2’-deoxy modification, the modified RNA is expected to be a competitive inhibitor of the enzyme. The closest homolog of SsCas6, SSO1437, also exhibited a similar activity while SSO1406, whose sequence clearly belongs to a different subgroup than SsCas6 and SSO1437, did not process the same RNA (Fig. 1A). The cleaved 3′ product matches the sequence of the 5′-handle previously determined by deep sequencing of the crRNA associated with the Ss Cascade or the Cmr effector complex (Lintner et al., 2011; Zhang et al., 2012).
We solved three different crystal structures of SsCas6: that of SsCas6 in isolation at 3.0 Å (SsCas6), that of SsCas6 bound with the 2′-deoxy modified 24mer at 2.5 Å (24mer noncleavage complex), and that of SsCas6 bound with a 16mer RNA mimicking the 5′-cleavage product without the 2′,3′ cyclic phosphate at 2.8 Å (16mer product-like complex). The first two structures were determined by a single wavelength anomalous diffraction (SAD) method using crystals containing selenomethionine-substituted SsCas6 and the last was determined by a molecular replacement (MR) method. The register of the protein backbone was confirmed by positions of selenomethionine residues and the position of U8 was confirmed by 5-bromouridine (Figure S1). In all three models, SsCas6 lacks residues 63-77 due to disorder. In addition, SsCas6 bound with 24mer RNA also lacks residues 223-230. The 24mer RNA structure includes nucleotides 1-17 in one but 3-17 and 19-23 in the other protomer. The 16mer RNA structure contains nucleotides 1-16 in all eight protomers. Data collection, phasing, and refinement statistics for each complex are listed in Table 1.
Table 1.
Crystallographic data collection and refinement statistics*
| SsCas6-24mer noncleavable complex (4ILL) | SsCas6-16mer Product-like complex (4ILM) | SsCas6 (4ILR) | |
|---|---|---|---|
| Data Collection Statistics | |||
| Space group | C2 | P21 | P6522 |
| a | 159.7 | 79.0 | 58.4 |
| b | 68.5 | 154.6 | 58.4 |
| c | 79.8 | 130.8 | 471.0 |
| α | 90.0 | 90.0 | 90.0 |
| β | 119.1 | 93.6 | 90.0 |
| γ | 90.0 | 90.0 | 120.0 |
| Resolution range (Å) | 50.0-2.5 (2.6-2.5) | 50.0-3.0 (3.1-3.0) | 50.0-3.0 (3.1-3.0) |
| No. of observed unique reflections | 24962 (988) | 57752 (2099) | 8986 (171) |
| Redundancy | 9.4 (3.8) | 6.7 (3.5) | 28.2 (15.6) |
| Completeness (%) | 93.8 (54.4) | 95.9 (70.6) | 89.1 (34.2) |
| <I>/<σ(I)> | 41.4 (2.2) | 24.9 (1.7) | 42.8 (3.5) |
| Rsym(%) | 9.3 (51.1) | 8.8 (62.3) | 8.4 (42.7) |
| Refinement statistics | |||
| Resolution range (Å) | 50.0-2.5 (2.6-2.5) | 50.0-3.0 (3.1-3.0) | 50.0-3.0 (3.1-3.0) |
| Rwork(%) | 20.9 | 22.9 | 25.2 |
| Rfree(%) | 24.6 | 28.3 | 29.0 |
| Model information | |||
| No. of protein/RNA complexes | 2/2 | 8/8 | 1/0 |
| No. of amino acids/nucleotides/water | 560/37/14 | 2248/128/4 | 280/0/0 |
| Root-mean-square deviations (rmsd) | |||
| Bond length (Å) | 0.009 | 0.010 | 0.011 |
| Bond angle (°) | 1.268 | 1.221 | 1.350 |
| Ramachandran plot of protein residues | |||
| Preferred regions (%) | 96.0 | 91.3 | 92.3 |
| Allowed regions (%) | 4.0 | 8.7 | 7.7 |
| Disallowed region (%) | 0.0 | 0.0 | 0.0 |
Values in parentheses are for the last resolution shell. PDBid for each structure is indicated after each title.
SsCas6 comprises a tandem ferredoxin fold arranged side-by-side and displays two distinct surfaces: one of β-sheets and one of α-helices (Fig. 1B). Like other RAMP proteins, both ferredoxin domains are interrupted by insertions to the canonical βαββαβ arrangement (Figure S2). The most notable is a three-helix insertion (α5-α7) following the first β-strand (β6) of the C-terminal ferredoxin domain. Together with another helix insertion to the N-terminal ferredoxin fold (α2), these insertions form an important binding platform for the repeat RNA (Fig. 1B).
The overall structure of SsCas6-RNA is a 2:2 (protein:RNA) homodimer. There are two protein-RNA complexes in the asymmetric units of the 24mer noncleavable complex crystals while there are eight protein-RNA complexes in the16mer product-like complex crystal. However, the eight noncrystallographic symmetry-related protomers comprise only four pairs of the same homodimer as that of the 24mer crystal. Although the isolated SsCas6 crystal has one protomer in its asymmetric unit, it again forms the same homodimer via crystallographic symmetry interactions. Thus, in all structures, SsCas6 or SsCas6-RNA complexes form the same homodimer mediated by protein residue interactions. The dimerization interface has a relatively extensive buried surface solvent accessible area (1289 Å2) that is suggestive of dimerization in solution and is made primarily of the helices (α5-α8) and β7 of its C-terminal ferredoxin domain (Figure S3). Cas6 dimerization has been previously observed in a non-catalytic Cas6 protein from Pyrococcus horikoshii (Ph) bound with its repeat RNA that, however, is mediated by the bound RNA molecules (Wang et al., 2012). The other three Cas6 do not seem to form dimers (Gesner et al., 2011; Haurwitz et al., 2012; Haurwitz et al., 2010; Sashital et al., 2011; Wang et al., 2011). Thus, dimerization is likely an inherent property of the protein unique to SsCas6. Structure superimposition revealed few differences in SsCas6 among the three structures with an exception for the β7-β8 hairpin loop (discussed below). Structural differences among protomers or between the 24mer and the 16mer complexes are observed only in two termini of the bound RNA due to different packing interactions, suggesting that the observed core enzyme-RNA structure is independent of crystal packing interactions.
SsCas6 Stabilizes a Short RNA Stem-loop Motif near the Cleavage Site
The bound 24mer RNA forms two single-stranded regions (nucleotides 1-5 and 17-23) and a central stem-loop (nucleotides 6-16) (Fig. 2). Of the five loop nucleotides of the stem-loop, three extrude out of helical stacking (A9, U11, and U13) and two stack on the three base-pair stem (C10 and A12) (Fig. 2). The short RNA stem-loop interacts extensively with SsCas6 while the single stranded regions have only limited contacts with the enzyme. The terminal nucleotides that do not contact SsCas5 (G1, C2, G21, A22, A23, and A24) are either disordered or are stabilized by crystal packing interactions. Thus the stem-loop is the primary structure recognized by SsCas6 (Fig. 2A).
Fig. 2.
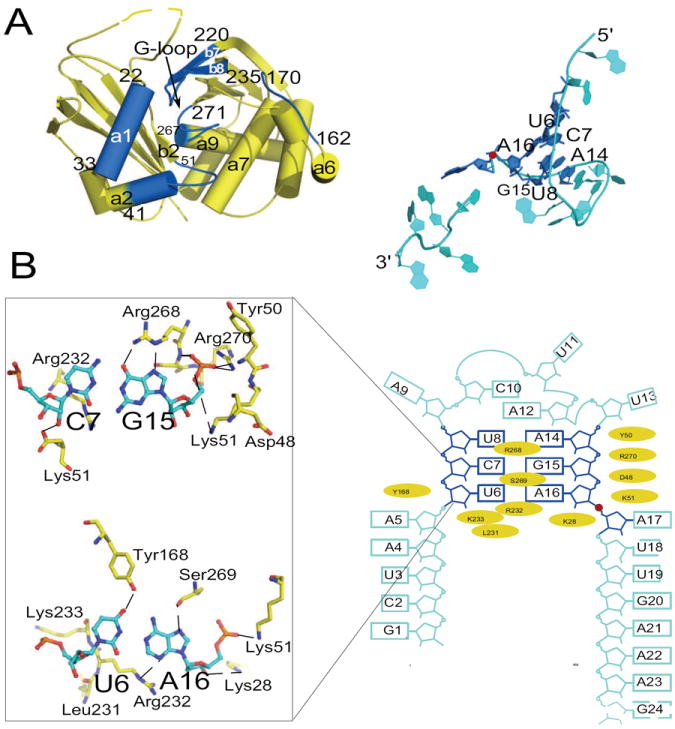
Detailed views of SsCas6-RNA interactions and the RNA secondary structure. Red sphere indicates the scissile phosphate group. (A). Regions on SsCas6 and on the 24mer RNA that form the protein-RNA interface are highlighted in blue and are shown in two separately panels. (B). Schematic of RNA secondary structure and residues of SsCas6 that interact with the RNA. The interaction residues were identified by their solvent accessible surface area changes upon RNA binding (> 5 Å2). Structures of two base pairs that interact extensively with the protein residues are depicted in details.
SsCas6 uses a narrow surface to interact with the RNA. It comprises α1, α2, loop α2β2, the G-loop and loop β7β8 (Fig. 2A). SsCas6 forms both non-specific and specific interactions with the RNA. The most extensive interaction with the RNA phosphate sugar backbone is formed with the short stem, in particular, the 3′ side of the stem (nucleotides 14-16). The backbone of nucleotides 14-16 lies against a positively charged surface of SsCas6 comprised primarily of loop α2β2 and the G-loop (Fig. 2A and 2B). The amino group of Lys51 contacts the non-bridging oxygen atoms of both G15 and A16. The guanidinium group of Arg270 of the G-loop further stabilizes the negative charge of nonbridging oxygen of G15. In addition, the side chain atoms of Ser148 on α5 and Tyr180 on α7 contact the nonbridging oxygen atoms of A14. With the exception of Arg270 that is strictly conserved, Lys51, Ser148 and Tyr180 may be substituted by other residue in SsCas6 close homologs (Figure S3), suggesting a shape-specific interaction in this region.
In addition to electrostatic and hydrogen-bonding interactions with the phosphate backbone, SsCas6 forms base-specific interactions with the short stem. For the U6-A16 base pair, O4 of U6 and N6 of A16 are within hydrogen bonding distances from the hydroxyl groups of Tyr168 of loop α6α7 and Ser269 of the G-loop, respectively (Fig. 2B). Ser269 is well conserved while Tyr168 is often replaced by phenylalanine in SsCas6 homologs (Figure S3). A16 is further stabilized by a close contact between its N3 atom and the guanidinium group of Arg232 (Fig. 2B). The G15C7 pair involves the most extensive interactions. Arg268 of the G-loop lies near the Watson-Crick edge of G15 and forms base-specific interactions with the G15C7 base pair (Fig. 2B). Its guanidinium group simultaneously contacts O6 and N7 of G15 (Fig. 2B). The fact that Arg268 is often replaced by lysine in other SsCas6 homolog suggests a possibility of coevolution between Cas6 and its substrates (Figure S3).
Although the penta-nucleotide loop contacts SsCas6, it does so through its two extruded nucleotides, U11 and U13, in a manner independent of bases. Protein residues (Lys15, Lys49, and Tyr50) stabilize the phosphate sugar back of both U11 and U13. Although the exocyclic oxygen atoms of U11 contact Asp48 side chain atoms, Asp48 is not well conserved, suggesting that the protein may be able to accommodate other nucleotides in the loop. The fact that there is no contact between the protein and the rest of the loop nucleotides suggests that both the size and the base identity of the loop are not critical to RNA recognition.
Two Specific Base Pairs Comprise the Minimal Recognition Motif of SsCas6
Formation of the stem-loop structure in SsCas6-bound repeat RNA is unexpected given the instability of this structure in isolation (ΔG0 > 0 by mfold, http://mfold.rna.albany.edu/?q=mfold) (Zuker, 2003) and the nearly flat thermal melting profile (data not shown). We thus tested the requirement of the stem loop structure in RNA cleavage by SsCas6. Deletion of a significant portion of the single stranded region at both 5′ and 3′ ends did not prevent the enzyme from cleaving the RNA (Fig. 3). In contrast, disruption of either U6-A16 or C7-G15 base pair (U6A and C7G) nearly abolished enzyme activity. Furthermore, compensatory mutations (U6A/A16U and C7G/G15C) to restore base pairing at these two positions did not rescue the activity (Fig. 3), confirming the observed specific recognition of these two base pairs. Interestingly, mutation of U8 to adenosine, which disrupted the third base pair, or substitution of the AU by GC pair had only a minor effect on enzyme activity (Fig. 3), suggesting a less stringent dependence on this base pair. We also tested importance of the size of the penta-loop by either deleting two non-interacting A9C10 or by inserting two adenosine residues between A9 and C10 (Fig. 3). While deletion of A9 and C10 significantly reduced RNA cleavage activity, insertion of two adenosine nucleotides had little impact on RNA cleavage activity, suggesting a relaxed requirement for the number of nucleotides in the loop (Fig. 3). These results define an essential core structure required for recognition by SsCas6 that comprises two specific base pairs upstream the cleavage site of the repeat RNA.
Fig. 3.

Activity assay results for RNA structure requirement for its processing. (A) Nomenclatures for RNA variants used in the activity assay. Nucleotide changes are indicated by arrows and the resulting mutant RNAs are represented by a single letter. The cleavage site is indicated by a red oval. (B) Radiographs of RNA cleavage reactions with the wild-type and variants of the repeat RNA incubated with 1 μM SsCas6 at 45°C (see Materials and Methods for details).
The Structure of the Active Site
Cas6 proteins reported so far, including SsCas6, do not require metal ions for cleavage and are believed to employ a general acid and base mechanism similar to those of RNase A (Raines, 1998) and tRNA splicing endonuclease (Xue et al., 2006) for the RNA cleavage reaction. Like RNase A and tRNA splicing endonuclease, a number of Cas6 proteins contain a critical histidine residue in their putative active sites and, in the case of Cas6f, was assigned to be the general base (Carte et al., 2010; Haurwitz et al., 2012; Haurwitz et al., 2010; Sashital et al., 2011; Wang et al., 2011). An important serine and tyrosine was also found crucial to RNA cleavage activity of Cas6f (Haurwitz et al., 2012; Haurwitz et al., 2010) and Cas6e (Gesner et al., 2011; Sashital et al., 2011) respectively. In contrast, SsCas6 does not contain any conserved histidine or tyrosine in its sequence (Figure S3 and S4). Furthermore, although several serine residues are conserved, they are found in positions nonequivalent to that in Cas6f (Figure S3). Examination of the SsCas6-24mer and SsCas6-16mer structures identified, surprisingly, four positively charged residues in its active site. These include Lys25 and Lys28 of α1 where the critical histidine of Cas6f and tyrosine of Cas6e are located, Lys51 on the loop connecting α2-β2, and Arg232 on the long β-hairpin loop where the critical serine of Cas6f resides. Observed separately in the 24mer noncleavable and 16mer product-like complex structures, the three lysine residues are within 3.5 Å to phosphate groups (Lys51 to A16, both Lys25 and Lys28 to A17) and Arg232 is close to 2′-OH of A16 (2.6 Å) (Fig. 4A). In the 24mer noncleavable complex structure where the nucleophile 2′-OH group is missing, Arg232 is further away from the active site than that in the 16mer product-like complex, suggesting its specific stabilization role for the 2′-OH group of A16 (Fig. 4A). Strikingly, both Lys28 and Arg232 are strictly and Lys25 and Lys51 are well conserved in close homologs of SsCas6 (Figure S3).
Fig. 4.

Active site geometry and mutational study results. The scissile bond is between nucleotides 16 and 17. (A). Close-up view of the structure near the RNA cleavage site of the 24mer noncleavable complex (upper panel) and that of the 16mer product-like complex (lower panel). The scissile phosphate position is indicated by a red oval. Protein residues within 3.6 Å of any of the atoms of A16 and A17 are displayed and those within 3.2 Å are indicated by dashed lines to RNA. (B). RNA cleavage activity assay results indicate critical roles of four positively charged residues in catalysis. Trace amount of 5′-radiolabeled 24mer repeat RNA was incubated with 1 μM SsCas6 (WT) and various mutants (indicated by single letter amino acid codes) for 10 minutes at 45°C. The reaction products were separated on a denaturing polyacrylamide gel and visualized by phosphorimaging. The lane labeled with “-” contains RNA and the reaction buffer. (C) Progression curves of enzyme activities under a single turnover condition for the wild-type and four mutants of SsCas6. The single-turnover rates for the wild-type and the mutants are labeled near the respective curves.
In addition to the placement of four positively charged residues at the active site, an RNA structure suggestive of the “inline attack” conformation at the cleavage site is also observed. Similar to the conformation previously observed in Cas6e- or Cas6f-bound RNA, the leaving nucleotide, A17, is flipped out of the helical stack, leading to a near inline geometry among the three atoms participating in bond breakage. An earlier computational study on tRNA splicing endonuclease provided quantitative support to a moderate stabilization effect (by ~12 fold) of the surrounding protein surface on the otherwise unstable inline conformation in solution (Min et al., 2007), suggesting that SsCas6 may similarly employ conformational trapping as a strategy for rate enhancement. To assess the importance of the active site and surrounding residues to the RNA cleavage reaction, we carried out site-directed mutations to the surrounding residues and tested their effects on RNA cleavage. We found that Arg232Ala, Lys25Ala, Lys28Ala, or Lys51Ala had significant impact on activity (Fig. 4B) while Ser46Ala, His57Ala, Glu225Ala or Asp226Ala did not (Fig. 4B). To quantify contributions of the four positively charged residues to the RNA cleavage activity, we compared the single turnover rate of the wild-type enzyme to those of the four mutants (Fig. 4C). Both Lys51Ala and Arg232Ala had significant reduction in activity (2.6×102-fold and 1.5×102-fold reduction, respectively), supporting their importance in catalysis and/or stabilizing the RNA conformation.
Discussion
Many CRISPR repeats contain no detectable palindromic feature, nor do they display conserved sequences. This raises the question of how CRISPR processing endonucleases recognize and cleave RNA transcribed from this group of CRISPR repeats. Here we show by both crystallographic and biochemical studies that a CRISPR repeat RNA derived from a nonpalindromic repeat forms a short stem-loop motif, comprising minimally of two base pairs, near the site of cleavage. The endonuclease that cleaves this CRISPR repeat RNA, SsCas6, stabilizes and specifically recognizes the stem-loop motif. This mode of protein-RNA interaction results in a conformation of the scissile phosphate bond that enables its breakage. Mutations of SsCas6 residues that are predicted to stabilize this conformation are found to greatly reduce the RNA cleavage activity of SsCas6.
The SsCas6-RNA complex structure suggests that extensive base pairing in RNA is not required for recognition by this class of processing endoribonucleases. The minimal two-base pair stem-loop formed near the cleavage site can be easily satisfied by many CRISPR repeat RNA devoid of stable secondary structures. Given the favorable conformation of the scissile phosphate bond for cleavage, we suggest that the observed SsCas6-RNA interactions near the cleavage site serve as a model for the class of endoribonucleases that recognize nonstructured CRISPR RNA. Individual Cas6 proteins may further fine-tune the specificity by recognizing base pairs and/or RNA nucleotides within and beyond the stem-loop motif. For instance, the previously studied PfCas6 endoribonuclease specifically recognizes the first eight nucleotides in addition to the cleavage site around nucleotide 22 (the wrap-around model) (Wang et al., 2011; Wang et al., 2012). Although the structure of the RNA cleavage site bound to PfCas6 is not observed, it is tantalizing to imagine that this region also forms a short stem-loop. If so, a general recognition model of nonstructured RNA may comprise both individually recognized peripherals and the cleavage site short stem-loop. Significantly, the short stem loop resembles the structure at the cleavage site of the CRISPR RNA derived from panlindromic repeats. This common motif near the RNA cleavage site establishes a mechanistic link in the catalytic process of the endonuribonucleases that process two different types of RNA.
The discovery of an arginine residue in SsCas6 at the position typically occupied by the general base in metal-independent endoribonucleases is surprising. Although arginine has a favorable electrostatic property for stabilizing the developing charge in the pentavalent transition state during phosphodiester bond cleavage reaction, the naturally high pKa of its guanidinium group argues against its role as a general base. However, we note that the functional groups tentatively assigned as the general base in several ribozymes also have naturally high pKa (Cochrane and Strobel, 2008). In these cases, it has been proposed that active site environment may lower the pKa values of these groups (Cochrane and Strobel, 2008). Whether this is the case for the arginine of SsCas6 awaits additional careful dissection of the catalytic mechanism.
Materials and Methods
Protein preparation and crystallization
The gene encoding SSO2004 protein was cloned into pET28a with a 6-histidine tag at the C-terminus and the protein was expressed in Escherichia coli BL21 RIPL cells (Agilent technologies, Santa Clara, CA). Cell pellets were resuspended in a lysis buffer (25mM sodium phosphate pH 7.5, 1.0M NaCl, 10 % (v/v) glycerol, 5.0mM β-mercaptoethanol, and 0.2mM phenylmethylsulfonyl fluoride). The cells were lysed by sonication and their debris was cleared by centrifugation. The supernatant was loaded onto a Ni-NTA column equilibrated with the lysis buffer supplemented with 5mM imidazole. The column was washed with the lysis buffer containing 25mM imidazole and the protein was eluted by increasing imidazole to 350mM. Fractions containing protein were pooled and loaded onto a Superdex 200 (Hiload 26/60, GE Healthcare) size-exclusion chromatography column that had been equilibrated with 20mM Tris-HCl pH 7.4, 500mM NaCl, 5 % (v/v) glycerol, and 5 mM β-mercaptoethanol. Fractions corresponding to the SSO2004 protein were pooled and concentrated. The L-selenomethionine (SeMet) labeled protein was prepared by a similar procedure. Mutant SSO2004 proteins were individually purified by heating and a single-step Ni-NTA affinity column followed by overnight dialysis in order to avoid cross contamination with the wild-type protein.
The SSO repeat RNA derived from Ss CRISPR locus F consisting of nucleotides 1-24, or 1-16 and those containing bromide substitutions were ordered from Integrated DNA technology (Coralville, IA). To crystallize the noncleavable complex, the 24mer repeat RNA containing 2′-deoxy modification at position 16 was mixed with SSO2004 protein at 1.2:1 molar ratio. For the product-like complex, the 16mer was used in place of the 24mer. In addition, a 24mer RNA containing 5-bromourdine at position 8 was used in cocrystallization in order to aid RNA tracing. All protein and protein-RNA complexes were crystallized at 30°C by using the vapor diffusion hanging-drop method. The SSO2004 protein was mixed in 1:1 volume ratio with reservoir solution containing 200mM MgCl2, 100mM Tris-Cl (pH 7.6), 32% PEG 400. Crystals grew to full size (0.3×0.4×0.1 mm3 in 3 days. The SSO2004 protein or the seleno-methionine-labeled protein bound with 24mer was mixed in 1:1 volume ratio with the reservoir solution containing 5mM MgCl2, 100mM KCl, 25% MPD, 100mM Tris-Cl (pH 7.9). Crystals(0.1×0.04×0.04 mm3) were obtained in 8 days. Lastly, The SSO2004 protein bound with 16mer RNA was mixed with the reservoir solution containing 15%(v/v) isopropanol, 1mM spermine, 10mM Co(NH4)6Cl3, 50mM Tris-HCl (pH 7.0) and crystals (0.08×0.05×0.05 mm3) were formed in 4 days.
Data collection and structure determination
The space groups and the cell dimensions of the three crystals are listed in Table 1. Crystals of the SsCas6 in isolation was directly mounted on a goniometer head from its mother liquor. Its structure was determined by single-wavelength anomalous dispersion (SAD) using data collected from a crystal containing selenomethionine-labeled proteins. Crystals of the 24mer noncleavable complex were cryo-protected in a buffer containing 400mM KCl, 20% MPD, 10% glucose, 100mM Tris-HCl (pH 7.9) prior to being mounted on a goniometer head. Crystals of the 16mer product-like complex were cryo-protected in its mother liquor containing 30% isopropanol. X-ray diffraction data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22ID or 22BM. Data were indexed, integrated, and scaled using the HKL2000 software package (Otwinowski and Minor, 1997). Phases of the 24mer crystals were obtained from a highly redundant SAD data set from crystals of selenomethionine-labeled SsCas6 protein and the 24mer RNA. The 16mer product-like complex structure was determined by the maximum-likelihood molecular replacement method using a 24mer noncleavable complex monomer structure as the search model. Structure determination, iterative model building and structure refinement were carried out using the PHENIX (Adams et al., 2011) and COOT programs (Emsley and Cowtan, 2004). The Maximum likelihood refinement protocol including experimental phase constraints was applied to complexes solved by SAD methods during initial stages of refinement. In order to reduce the data to degrees-of-freedom radio, non-crystallographic symmetry restraints were applied for both the 24mer noncleavable and the 16mer product-like complexes throughout refinement. The refinement and model statistics indicate satisfactory models for the three structures and are presented in Table 1.
RNA-cleavage reactions
In vitro RNA cleavage assay was carried out similarly as previously described (Wang et al., 2011). Briefly, SsCas6 (1 μM) was incubated with 16 nM 5′-32P-labeled 24mer repeat RNA or its variants for 30 min at 45°C in the cleavage buffer containing 20 mM Tris-HCl (pH 7.0) and 400 mM KCl. The reaction was quenched by the addition of 96% formamide dye. The cleavage products were resolved on a 15% polyacrylamide denaturing gel and visualized by phosphorimaging. For single turnover rate measurements, cleavage reactions were carried out at 45°C in a volume of 20 μl containing the same buffer, 1μM SsCas6 and 25nM RNA substrate. RNA cleavage products at various time points were separated on a 15% denaturing polyacrylamide gel and quantified by the Imagequant Software (Molecular Dynamics) of the phosphoimaging instrument. The reaction rate, kobs, for each reaction was obtained by fitting the faction of cleavage versus time to a single exponential function using SigmaPlot. Error bars were deviations from the mean of triplicate measurements.
Supplementary Material
Highlight.
A large proportion of CRISPR repeat RNAs contains no conserved primary or secondary structures. How is this group of RNA recognized and processed by CRISPR processing endoribonucleases? This study reveals an essential recognition motif near the RNA cleavage site comprised of a short stem.
Acknowledgments
This work was supported by National Institutes of Health grant R01 GM099604 to H.L. X-ray diffraction data were collected from the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions for APS beamlines may be found at http://necat.chem.cornell.edu/ and http://www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U. S. Department of Energy Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Footnotes
Author Contributions: YS performed all experiments. YS and HL designed the experiments and wrote the manuscript.
ACCESSION NUMBERS:
Coordinates and structure factors have been deposited in the Protein Data Bank with accession numbers 4ILL (SsCas6-24mer complex), 4ILM (SsCas6-16mer complex), and 4ILR (SsCas6).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams P, Afonine P, Bunkóczi Gb, Chen V, Echols N, Headd J, Hung L-W, Jain S, Kapral G, Grosse Kunstleve R, et al. The Phenix software for automated determination of macromolecular structures. Methods (San Diego, Calif) 2011;55:94–106. doi: 10.1016/j.ymeth.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau Hln, Richards M, Boyaval P, Moineau S, Romero D, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science (New York N Y) 2007;315:1709–1721. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- Barrangou R, Horvath P. CRISPR: new horizons in phage resistance and strain identification. Annual Review of Food Science and Technology. 2012;3:143–162. doi: 10.1146/annurev-food-022811-101134. [DOI] [PubMed] [Google Scholar]
- Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annual review of Genetics. 2011;45:273–297. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, Dickman MJ, Makarova KS, Koonin EV, Oost Jvd. Small CRISPR RNAs Guide Antiviral Defense in Prokaryotes. Science. 2008;321 doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carte J, Pfister NT, Compton MM, Terns RM, Terns MP. Binding and cleavage of CRISPR RNA by Cas6. RNA. 2010;16:2181–2188. doi: 10.1261/rna.2230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carte J, Wang R, Li H, Terns RM, Terns MP. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008;22:3489–3496. doi: 10.1101/gad.1742908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane JC, Strobel SA. Catalytic strategies of self-cleaving ribozymes. Acc Chem Res. 2008;41:1027–1035. doi: 10.1021/ar800050c. [DOI] [PubMed] [Google Scholar]
- Deveau Hln, Garneau J, Moineau S. CRISPR/Cas system and its role in phage-bacteria interactions. Annual Review of Microbiology. 2010;64:475–493. doi: 10.1146/annurev.micro.112408.134123. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Gesner EM, Schellenberg MJ, Garside EL, George MM, Macmillan AM. Recognition and maturation of effector RNAs in a CRISPR interference pathway. Nat Struct Mol Biol. 2011;18:688–692. doi: 10.1038/nsmb.2042. [DOI] [PubMed] [Google Scholar]
- Haft DH, Selengut J, Mongodin EF, Nelson KE. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol. 2005;1:e60. doi: 10.1371/journal.pcbi.0010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale C, Kleppe K, Terns RM, Terns MP. Prokaryotic silencing (psi)RNAs in Pyrococcus furiosus. RNA. 2008;14:2572–2579. doi: 10.1261/rna.1246808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, Terns MP. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139:945–956. doi: 10.1016/j.cell.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haurwitz R, Sternberg S, Doudna J. Csy4 relies on an unusual catalytic dyad to position and cleave CRISPR RNA. The EMBO Journal. 2012;31:2824–2832. doi: 10.1038/emboj.2012.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science. 2010;329:1355–1358. doi: 10.1126/science.1192272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- Karginov FV, Hannon GJ. The CRISPR system: small RNA-guided defense in bacteria and archaea. Mol Cell. 2010;37:7–19. doi: 10.1016/j.molcel.2009.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunin V, Sorek R, Hugenholtz P. Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol. 2007;8:R61. doi: 10.1186/gb-2007-8-4-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillestol R, Shah S, Brugger K, Redder P, Phan H, Christiansen J, Garrett R. CRISPR families of the crenarchaeal genus Sulfolobus: bidirectional transcription and dynamic properties. Molecular Microbiology. 2009;72:259–272. doi: 10.1111/j.1365-2958.2009.06641.x. [DOI] [PubMed] [Google Scholar]
- Lintner N, Kerou M, Brumfield S, Graham S, Liu H, Naismith J, Sdano M, Peng N, She Q, Copié Vr, et al. Structural and functional characterization of an archaeal clustered regularly interspaced short palindromic repeat (CRISPR)-associated complex for antiviral defense (CASCADE) The Journal of Biological Chemistry. 2011;286:21643–21699. doi: 10.1074/jbc.M111.238485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova K, Grishin N, Shabalina S, Wolf Y, Koonin E. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biology Direct. 2006;1:7. doi: 10.1186/1745-6150-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Aravind L, Wolf YI, Koonin EV. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol Direct. 2011;6:38. doi: 10.1186/1745-6150-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, Sontheimer EJ. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet. 2010;11:181–190. doi: 10.1038/nrg2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min D, Xue S, Li H, Yang W. ‘In-line attack’ conformational effect plays a modest role in an enzyme-catalyzed RNA cleavage: a free energy simulation study. Nucleic Acids Res. 2007;35:4001–4006. doi: 10.1093/nar/gkm394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60:174–182. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode in Methods in Enzymology. In: Carter CW Jr, Sweet RM, editors. Macromolecular Crystallography, Part A. Vol. 276. Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Pourcel C, Salvignol G, Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology. 2005;151:653–663. doi: 10.1099/mic.0.27437-0. [DOI] [PubMed] [Google Scholar]
- Raines R. Ribonuclease A. Chem Rev. 1998;98:1045–1065. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- Richter H, Zoephel J, Schermuly J, Maticzka D, Backofen R, Randau L. Characterization of CRISPR RNA processing in Clostridium thermocellum and Methanococcus maripaludis. Nucleic Acids Research. 2012 doi: 10.1093/nar/gks737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sashital DG, Jinek M, Doudna JA. An RNA-induced conformational change required for CRISPR RNA cleavage by the endoribonuclease Cse3. Nat Struct Mol Biol. 2011;18:680–687. doi: 10.1038/nsmb.2043. [DOI] [PubMed] [Google Scholar]
- Sorek R, Kunin V, Hugenholtz P. CRISPR--a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat Rev Microbiol. 2008;6:181–186. doi: 10.1038/nrmicro1793. [DOI] [PubMed] [Google Scholar]
- Terns M, Terns R. CRISPR-based adaptive immune systems. Current Opinion in Microbiology. 2011;14:321–327. doi: 10.1016/j.mib.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Oost J, Jore M, Westra E, Lundgren M, Brouns SJ. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends in Biochemical Sciences. 2009;34:401–407. doi: 10.1016/j.tibs.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Wang R, Preamplume G, Terns MP, Terns RM, Li H. Interaction of the Cas6 Riboendonuclease with CRISPR RNAs: Recognition and Cleavage. Structure. 2011;19:257–264. doi: 10.1016/j.str.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Zheng H, Preamplume G, Shao Y, Li H. The impact of CRISPR repeat sequence on structures of a Cas6 protein-RNA complex. Protein Science : a publication of the Protein Society. 2012;21:405–422. doi: 10.1002/pro.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenheft B, Lander GC, Zhou K, Jore MM, Brouns SJ, van der Oost J, Doudna JA, Nogales E. Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature. 2011a;477:486–489. doi: 10.1038/nature10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenheft B, Sternberg S, Doudna J. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- Wiedenheft B, van Duijn E, Bultema J, Bultema J, Waghmare S, Waghmare S, Zhou K, Barendregt A, Westphal W, Heck A, et al. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proceedings of the National Academy of Sciences of the United States of America. 2011b;108:10092–10097. doi: 10.1073/pnas.1102716108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue S, Calvin K, Li H. RNA recognition and cleavage by a splicing endonuclease. Science. 2006;312:906–910. doi: 10.1126/science.1126629. [DOI] [PubMed] [Google Scholar]
- Zhang J, Rouillon C, Kerou M, Reeks J, Brugger K, Graham S, Reimann J, Cannone G, Liu H, Albers S-V, et al. Structure and mechanism of the CMR complex for CRISPR-mediated antiviral immunity. Molecular Cell. 2012;45:303–316. doi: 10.1016/j.molcel.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.