

Abstract
Niemann-Pick disease type C (NPC disease) is an incurable, neurodegenerative, autosomal recessive disease caused by mutations in either the NPC1 or NPC2 gene. These mutations affect the intracellular trafficking of lipids and cholesterol resulting in the intralysosomal accumulation of unesterified cholesterol and glycosphingolipids, and clinical signs of ataxia and impaired motor and intellectual development with death frequently occurring in adolescence. The incidence of peripheral neuropathy in NPC patients is not known. We investigated peripheral nerves in the naturally-occurring feline model of NPC disease which has proven critical for understanding both disease pathogenesis and for evaluating experimental therapies. Electrodiagnostic studies revealed significantly slowed motor and sensory nerve conduction velocities in affected cats in the absence of altered M-wave amplitude. Histology showed demyelination characterized by thin myelin sheaths, membranous debris, myelin figures, lipid vacuolization of Schwann cell cytoplasm, and expanded paranodal areas. Axonal degeneration was not identified. A shift to small myelinated fibers was noted in affected cats and significant decreases in fiber diameter, axonal diameter, and myelin thickness was found. These changes were similar to those described in the murine model as well as in the rare patient in which nerve biopsy was performed. Characterization of the demyelinating neuropathy is necessary in NPC disease due to the clinical trial in patients scheduled for 2013 where cyclodextrin will be delivered into the cerebral ventricles in an attempt to control the CNS aspects of this disease.
INTRODUCTION
Niemann-Pick disease type C (NPC disease) is caused by mutations in either the NPC1 or NPC2 gene which result in abnormal cellular lipid and cholesterol trafficking(1). The accumulation of unesterified cholesterol and glycosphingolipids in the endosomal/lysosomal compartments of cells in the CNS and in visceral organs is associated with progressive neurological dysfunction, hepatosplenomegaly, and early death(1–3). Although the disease manifests in patients at varying ages and with varying deficits, affected individuals typically develop cerebellar ataxia, dysarthria, dysphagia, vertical supranuclear gaze palsy, seizures, and dementia, and die in adolescence(1, 3, 4). Brain histology reveals widespread neuronal cytoplasmic vacuolization, neuroaxonal dystrophy and neuronal loss most severely affecting Purkinje cells, cortical pyramidal neuron meganeurite formation and ectopic dendritogenesis, gliosis, and inflammation.(5–7) Although there is no effective cure, recent experimental work in the murine and feline model show that the administration of cyclodextrins prevent or delay CNS dysfunction and greatly extend lifespan (8, 9). These studies have resulted in a clinical trial in affected patients scheduled to start in 2013.
The incidence of clinical or sub-clinical neuropathy in NPC patients is not known. Peripheral neuropathy has only rarely been described in affected children and electrodiagnostic testing is not commonly performed (1, 4, 10, 11). One 3.5-year-old boy presented with CNS signs typical of NPC disease but also with diminished tendon reflexes and sensation (10). A mild demyelinating polyneuropathy was diagnosed following electrodiagnostic testing. Additionally, a 3-year-old boy developed muscular atrophy and decreased tendon reflexes in the lower extremities; no electrodiagnostics were described (12). Finally, a 4-year-old girl was diagnosed with electrophysiological evidence of a demyelinating motor and sensory neuropathy which was confirmed by nerve biopsy(11).
The purpose of the present study was to evaluate peripheral nerve electrodiagnostic findings and histopathology in a cohort of cats with NPC disease compared to age-matched unaffected cats. Naturally occurring feline NPC disease is due to a missense mutation in NPC1 (p.C955S; c.2864G<C) with clinical, neuropathological, and biochemical abnormalities similar to those present in juvenile-onset patients, the most common form of the disease seen in human patients (5, 6, 13–17). The feline model has been critical for identifying the late endosomal/lysosomal accumulation of gangliosides (GM2 and GM3) and unesterified cholesterol(7, 18), for evaluating the association of ganglioside storage with meganeurite formation and ectopic dendritogenesis(18), for correlating neuroaxonal dystrophy with neurological dysfunction(6), and for evaluating efficacy of experimental therapies(8, 18–21). Here we describe clinical, electrodiagnostic and pathological changes that demonstrate peripheral nerve involvement in the feline model of NPC disease.
MATERIALS AND METHODS
Animals
Cats were raised in the animal colony of the School of Veterinary Medicine, of the University of Pennsylvania, under National Institutes of Health and USDA guidelines for the care and use of animals in research. The experimental protocol was approved by the University of Pennsylvania Institutional Animal Care and Use Committee. Mature cats from the same line were bred to produce cats with autosomal recessive Niemann-Pick type C (NPC) disease. The cats were housed in 21°C with ad libitum food and water, 12-hour light cycles, with 12–15 air changes per hour. Peripheral blood leukocytes from all the cats were tested at one day of age for the NPC1 missense mutation using a PCR-based DNA test(16). Cats with no copies of the mutation were classified as control cats, while cats homozygous for the mutant allele were classified as affected. Physical and neurological examinations were performed weekly.
Electrodiagnostic Testing
Nerve conduction velocity (NCV) and compound muscle action potential (CMAP) amplitude were measured on cats under anesthesia using intravenous propofol (up to 6 mg/kg) for induction, followed by endotracheal intubation, and anesthetic maintenance with isoflurane. Data were obtained at 8 and 24 weeks of age using a Nicolet Viking Quest machine (Nicolet Biomedical, Madison, WI). 12-mm, 27-gauge subdermal recording electrodes were placed in the interosseous muscle to determine motor NCV (MNCV) in the tibial, sciatic, and ulnar nerves of the right pelvic and thoracic limbs. The sciatic nerve was stimulated at the stifle and at the level of the femoral head, and the tibial nerve was stimulated at the tarsus and stifle. The ulnar nerve was stimulated at the carpus and the elbow. 26-gauge monopolar stimulating electrodes were placed subcutaneously to stimulate each nerve. Stimulation duration was 100 μs, filter settings were 20 Hz and 2 kHz, gain was 2 mV/cm, and analysis time was 20ms. For sensory nerve conduction velocity (SNCV) recording electrodes were placed subcutaneously lateral to the radial nerve at the level of the elbow. Subcutaneous monopolar stimulating electrodes were placed in the skin over the dorsum of the paw. A 100-μs stimulus duration, band pass of 20 Hz to 2 kHz, gain of 2 μV/cm, and analysis times of 10 ms were used. Motor nerve conduction velocity was determined by dividing the distance between the stimulating electrodes by the difference in onset latency between the two recorded evoked potentials. Sensory nerve conduction velocity was determined by dividing the distance between stimulating and recording electrodes by the latency to the first peak. Amplitude was measured from peak to trough of the evoked response. Electromyography (EMG) was recorded using a 25-mm, 26-gauge concentric needle electrode inserted into the muscle. Amplifier filters were 5 Hz to 5 kHz; responses were recorded at a sensitivity of 100 uV/cm and a sweep speed of 10 ms/cm. Ten NPC disease cats and 10 control cats were compared at 8 weeks of age. Six NPC cats and 15 control cats were compared at 24 weeks of age.
Post mortem examination
Cats were euthanized using an overdose of barbiturates in accordance with the American Veterinary Medical Association guidelines. All NPC disease cats were euthanized when they were no longer able to maintain sternal recumbency without assistance which occurs at a mean age of 20.5 +/− 4.8 weeks (17). Control cats were euthanized between 20 and 29 wk of age for histological comparison. Immediately before sacrifice, each cat was given 0.5 mL of heparin (1000 units/mL) i.v. After sacrifice, the cats were perfused with 500 mL of 0.9% cold saline and samples of brain, liver, spleen, and lung as well as peripheral nerves (radial, ulnar, sciatic, tibial, and peroneal nerves) were collected for histological and biochemical analyses.
Light and Electron Microscopy
Nerve specimens as necropsy samples were collected immediately following euthanasia and saline perfusion. Specimens were either immersion-fixed in 10% neutral buffered formalin or placed on a tongue depressor, wrapped in a saline dampened gauze sponge, placed into a watertight container, and kept chilled during shipping. Upon receipt, fixed nerves were transferred to 2.5% glutaraldehyde, post-fixed in 1% aqueous osmium tetroxide and processed to araldite resin blocks. Thick sections (1 μm) were cut and stained with toluidine blue prior to light microscopic examination. Thin sections (60–90 nm) were cut with a diamond knife and stained with uranyl acetate and lead citrate prior to electron microscopic examination.
Teased fibers from the ulnar nerve were processed as above except that the araldite resin used for overnight infiltration lacked hardener. After infiltration, excess araldite resin was wiped off and the nerve subsequently teased in Epon 812 resin without hardener before cover slipping and examination by light microscopy.
Morphometry
For quantitative assessment, high quality nerve specimens adequately fixed and free of artifact were chosen. Using point counting techniques(22) and a grid with a magnified distance of 0.08 mm between intersection points, fascicular area, defined as the number of points falling on the endoneurium of nerve fascicles, was determined. The total number of myelinated fibers, the number of fibers with inappropriately thin myelin sheaths relative to their axon diameter as well as those with myelin splitting and ballooning, and the number of probable regenerative clusters (defined as two or more closely apposed myelinated fibers were assessed in each nerve specimen and normalized to fascicular area(23). As previously described(24, 25), only those fibers with clear, faintly staining space evident between separated myelin with an asymmetrical profile are considered to have splitting and ballooning, while profiles containing paranodal regions or Schmidt-Lanterman clefts were not counted. In addition, computer-assisted analyses of axonal size-frequency distributions of myelinated fibers, nerve fiber diameter, axonal diameter, myelin thickness and G ratio (structural index of axonal myelination defined as axonal diameter/fiber diameter based on measurements by perimeter) were determined using SPOT™ Advanced Imaging software and modifications of Adobe Photoshop(26).
Statistical Analysis
Differences between nerve conduction velocity, and morphometric parameters of affected and control cats were tested using two-tailed, unpaired t-tests after determining that variances were not significantly different. When variances were unequal, an unpaired t-test with Welch's correction was used.
RESULTS
Clinical Examinations
All NPC cats developed progressive intention tremors and cerebellar ataxia eventually resulting in their inability to walk, however, none developed appendicular muscle flaccidity, hyporeflexia or evidence of diminished sensation. All affected cats were euthanized when they were no longer able to maintain sternal recumbency without support at a mean age of 22.4 +/− 3.7 weeks. Unaffected cats developed no neurologic deficits and were euthanized to serve as age-matched controls.
Peripheral Nerve Electrodiagnostic Testing
Significantly diminished sciatic MNCV was found in NPC disease cats at both 8 and 24 weeks of age. Tibial MNCV was significantly slower in affected cats at 24 weeks of age (Fig. 1). Radial SNCV was also significantly slower in affected cats compared to control cats at both 8 and 24 weeks of age. In control cats, the tibial, ulnar, and radial nerves showed increasing NCV over time (8 to 24 weeks of age) and reached statistical significance (p < 0.05). Individual nerves in affected cats at both ages all showed increasing NCV over time, but only the radial and sciatic nerves reached statistical significance (p < 0.05). No significant differences in amplitudes of motor or sensory evoked potentials were found between affected and control cats at either age (data not shown). No EMG abnormalities were identified in any cats evaluated.
Fig. 1.
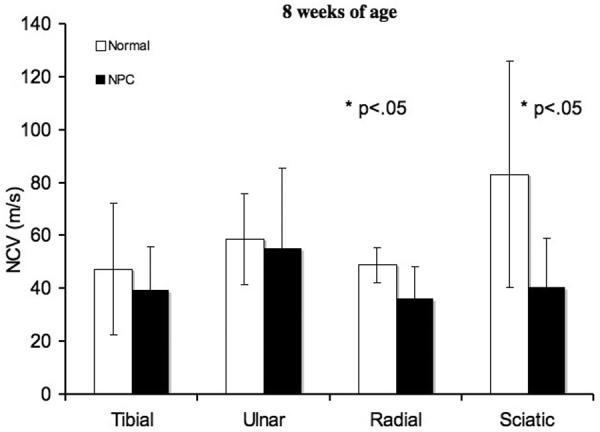
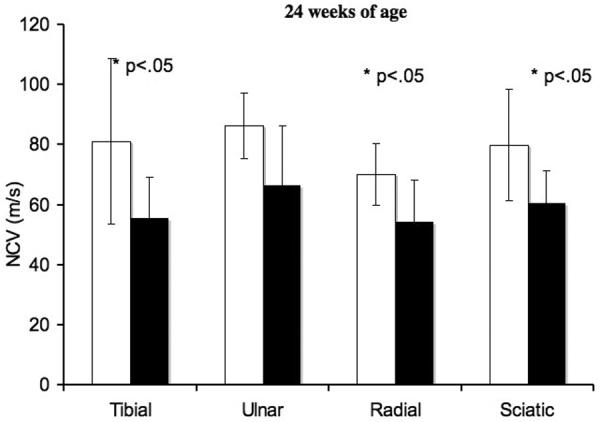
Nerve conduction velocities of peripheral nerves. In the radial and sciatic nerves, at 8 weeks of age, there were significant differences (*p < 0.05) in NCV between normal and affected cats. Significant differences (*p < 0.05) were found in the tibial, radial, and sciatic nerves at 24 weeks of age. Means and standard deviations are presented for normal and affected cats at each age range.
Light and Electron Microscopy
At the light microscopic level, toluidine blue stained resin embedded sections (1 μm) from the radial, ulnar, sciatic, tibial and peroneal nerves were evaluated from control and affected cats. Similar pathologic changes were identified in all nerves of affected cats and representative changes are illustrated for the ulnar nerve (Fig. 2). Compared to control ulnar nerve (Fig. 2a), the density of myelinated fibers was subjectively appropriate. However, thickness of myelin sheaths varied considerably between nerve fibers with many showing inappropriately thin myelin sheaths (Fig. 2b,c,d). Scattered nerve fibers also contained membranous debris and lipid substrates (Fig. 2d). Axonal degeneration was not found. Electron microscopic changes were consistent with those observed at the light level (Fig. 3). Demyelinated nerve fibers were evident with membranous debris, myelin figures and vacuoles containing lipid substrates within the Schwann cell cytoplasm (Fig. 3a,b). Schwann cell nuclei and axons were normal in appearance. Specifically, axons showed no evidence of neurofilament abnormalities and no axoplasmic dense bodies. We also performed a survey of high quality plastic thick sections of all nerves for evidence of darkly staining axoplasm which was not identified. Nerve fibers with inappropriately thin myelin sheaths (Fig. 3c) and fibers with appropriate myelin sheaths but containing large vacuoles and myelin debris in the Schwann cell cytoplasm (Fig. 2d) were observed. Variability in thickness of the myelin sheath and expanded paranodal areas containing lipid substrates and debris were also noted in teased fibers from the ulnar nerve of affected cats compared to unaffected controls (Fig. 4).
Fig. 2.
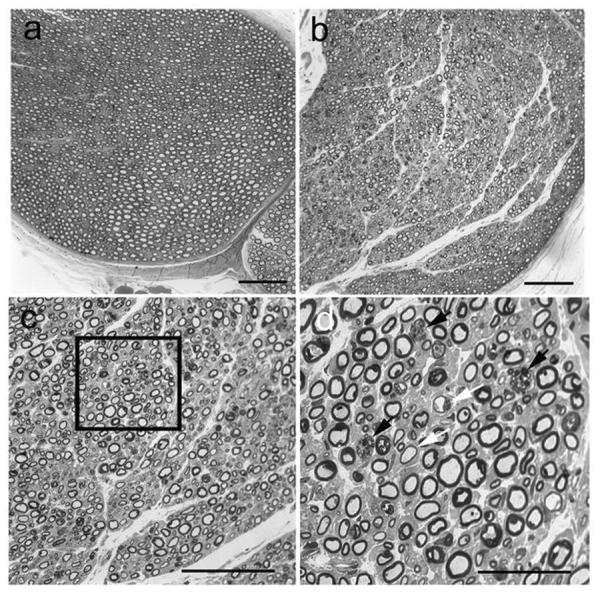
Pathological changes affecting myelin were evident in peripheral nerve biopsies from NPC affected cats compared to unaffected cats. Compared to control ulnar nerve (a), variability in thickness of the myelin sheath were noted (b,c). Insert in c is expanded in d and shows inappropriately thin myelin sheaths (white arrows) and Schwann cell cytoplasm containing myelin debris and vacuoles (black arrows). Bars = 50 μm for a–c and 25 μm for d.
Fig. 3.
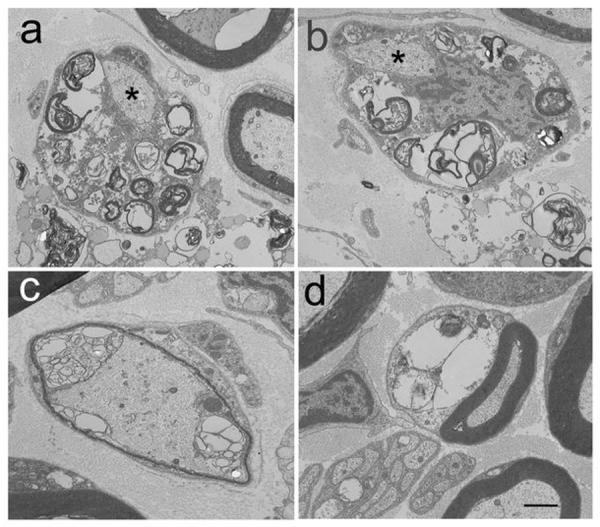
Electron micrographs of the ulnar nerve from an NPC affected cat confirmed the light microscopic findings. Myelin debris and vacuoles were noted in Schwann cell cytoplasm (a,b) with normal appearing axons (a,b asterisks) and Schwann cell nuclei (b). Inappropriately thin myelin sheaths (c) and large vacuoles containing lipid substrates (d) were noted. Bar in d = 0.87 μm for a and b, and 0.95 μm for c and d.
Fig. 4.
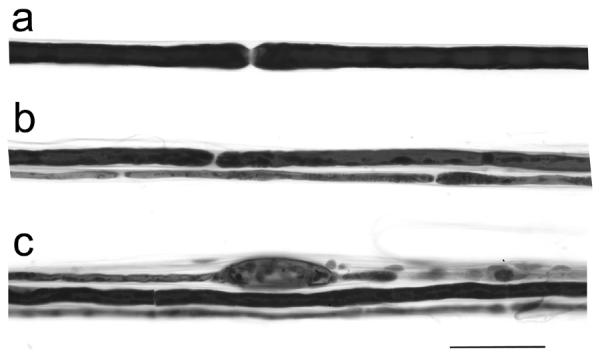
Teased fiber preparations from the ulnar nerve of a control (a) and NPC affected (b,c) cat showed variability in nerve fiber thickness (b) and paranodal swellings containing myelin debris (c). Bar = 50 μm for all figures.
Morphometry
Axonal size-frequency analysis was performed on the ulnar nerve of cats affected with NPC disease and compared to unaffected cats (Fig. 5). A shift in the axonal size-frequency distribution from large myelinated fibers >5 μm to small myelinated fibers ≤5 μm was noted compared to controls. A statistically significant decrease in fiber diameter (p=0.03), axonal diameter (p=0.04) and myelin thickness (p=0.02) was also noted (Table 1) with a non-significant trend toward an increased myelinated fiber density.
Fig. 5.
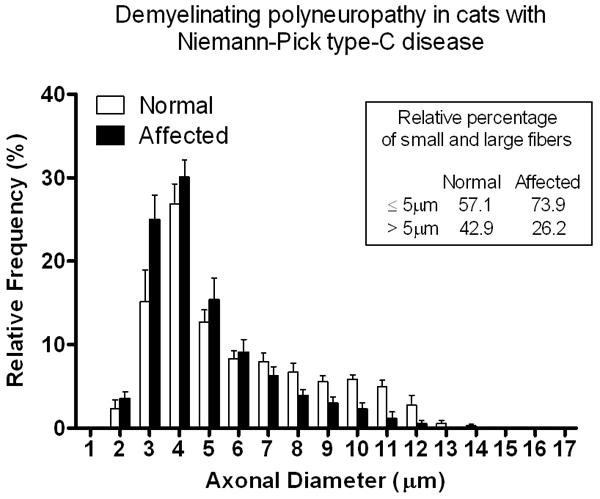
Histogram showing the axonal size-frequency distribution in the ulnar nerves of NPC affected cats compared to unaffected cats. There was a significant shift to smaller caliber fibers (p= 0.03) in NPC affected cats compared to unaffected cats. s
Table 1.
Morphometric analysis of the ulnar nerve in cats affected with Niemann-Pick Disease compared to control cats
| Normal (n=6) | Affected (n=8) | P Value | |
|---|---|---|---|
| MF Density | 10826±1140 (8721–11980) | 13007±4876 (7211–22200) | ns |
| Split MF Density | 40±31 (5–80) | 78±61 (13–79) | ns |
| % Split MF | 0.4±0.3 (0.0–0.8) | 0.6±0.4 (0.1–1.2) | ns |
| Thin MF Density | 871±291 (297–1360) | 1162±721 (467–2564) | ns |
| % Thin MF | 7.8±3.1 (3.4–11.7) | 8.4±2.6 (6.2–12.4) | ns |
| Cluster Density | 1±1 (0–2) | 27±27 (0–70) | 0.03 |
| # Myelinated Sprouts/Cluster | 1±1 (0–3) | 2±1 (0–3) | ns |
| Fiber diameter (μm) | 8.82±1.53 (7.27–11.73) | 6.91±0.82 (6.20–8.76) | 0.03 |
| Axonal diameter (μm) | 5.40±0.86 (4.43–6.90) | 4.38±0.60 (3.08–5.58) | 0.04 |
| G ratio | 0.61±0.02 (0.59–0.65) | 0.63±0.03 (0.59–0.68) | ns |
| Myelin thickness (μm) | 1.73±0.35 (1.42–2.42) | 1.26±0.17 (1.04–1.59) | 0.02 |
Data are presented as mean±/SD (range) and were analyzed by a two-tailed, unpaired t-test. When variances were unequal, a t-test with Welch's correction was used. MF= myelinated fiber; split MF=fibers with split myelin sheaths including ballooned myelin sheaths; thin MF=fiber with myelin sheaths that are thin by comparison with those fibers with similar axonal diameter; G ratio=structural index of axonal myelination defined as axonal diameter/fiber diameter based on measurements by perimeter; ns-not significant
DISCUSSION
Brain from NPC disease patients reveals widespread neuronal and glial cytoplasmic vacuolization, neuronal loss most severely affecting the Purkinje cells, neuroaxonal dystrophy, cerebrocortical meganeurite formation, ectopic dendritogenesis, and gliosis (1, 3, 7, 27). Cerebral white matter is generally normal although demyelination with perivascular accumulation of macrophages has been reported (28). The histologic abnormalities contribute to the phenotype of ataxia, impaired mental development, seizures, dysphagia, dysarthria, and dementia. Unlike the CNS, however, the peripheral nervous system shows more subtle clinical and morphologic changes. Diminished tendon reflexes and superficial sensation have been described in a small number of patients and a demyelinating motor and sensory polyneuropathy has been identified through electrodiagnostic testing and nerve biopsy (10–12). Although the incidence of clinical polyneuropathy in NPC disease appears low (4), the incidence of subclinical neuropathy is not known.
The feline model has clinical, neuropathological, and biochemical CNS abnormalities similar to those seen in juvenile-onset patients and exhibits Purkinje cell death, ectopic dendritogenesis, neuroaxonal dystrophy, and central myelin deficits (7, 15, 17). No signs of peripheral nervous system involvement are present on clinical examination. However, electrodiagnostic studies show clear evidence of slow nerve conduction velocity in both motor and sensory nerves in the absence of changes in evoked potential amplitude and in the absence of electromyographic abnormalities. These changes are consistent with a demyelinating polyneuropathy which was confirmed on histology. Our findings in the feline model are similar to what was described in the mouse model(29). The lipid inclusions within Schwann cells evident by light microscopy, the many myelin figures observed by electron microscopy, and the paranodal swellings corresponding to sites of inclusion accumulations observed with teased nerve fibers, are similar between species. There was no evidence of segmental demyelination or axonal degeneration. Further, histograms (Fig. 5) showed a reduced percentage of large myelinated fibers in the NPC cats compared to unaffected cats and an increased population of small fibers. Morphometric analysis demonstrated statistically significant differences in cluster density, fiber diameter, axonal diameter and myelin thickness between normal and NPC affected cats (Table 1). The increased population of small caliber nerve fibers may reflect the statistically significant increase in cluster density in addition to the decreased myelin thickness. Similarly, a reduced percentage of large myelinated fibers with an increased population of small caliber fibers was also noted in the mouse model (29). Unlike the mouse model, however, distended axons containing peripherally located, massive accumulations of small membrane bound electron dense bodies, were not observed in the cat model. In fact, statistically significant evidence for a decreased axonal diameter was found in NPC cats (Table 1). Such electron dense bodies in the mouse model were identical to those found in dystrophic axons in the CNS(29). Although axonal spheroids packed with electron-dense material have been described in the CNS of affected cats (13), they are absent in peripheral nerves. In one human patient examined, electron dense bodies were identified within some PNS axonal spheroids but also not to the extent described in the murine model(11). The absence of axonal electron dense bodies in peripheral nerves of the NPC cat may reflect a species variation, or alternatively, an age-related change that could develop if the cats were allowed to reach a more advanced age, although the latter is less likely since affected mice were examined at 60 days of age.
The cause of the demyelinating polyneuropathy associated in NPC disease is not well understood. The light and electromicroscopic findings in the NPC mouse were postulated to indicate defective myelin turnover and a defect in the utilization of cholesterol was proposed as the cause (29). In support of this hypothesis, trauma to the sciatic nerve in NPC mice resulted in delayed myelination compared to normal mice, and reutilization of cholesterol, as assayed by labeling with radioactive acetate, was reduced (30). The cause for defective myelination in the CNS in NPC is also poorly understood. In the CNS, expression of myelin basic protein (MBP) in oligodendrocytes is decreased in the NPC1 mouse model compared to normal mice(31, 32). No increase in oligodendrocyte apoptosis and no defect in differentiation of oligodendrocyte progenitor cells into premyelinating oligodendrocytes was seen in affected mice. The mechanisms of the defect in myelination was proposed to be associated with an observed increase in the polysialylated-neural cell adhesion molecule (PSA-NCAM), a negative regulator of myelination, and/or a defect in differentiation of premyelinating oligodendrocytes into myelinating oligodendrocytes(32). The later hypothesis is supported by the findings of increased numbers of premyelinating oligodendrocytes in the cerebral cortex of NPC mice and a decrease in myelin gene regulatory factor, a transcription factor which promotes formation of myelin proteins, and is necessary for the formation of myelinating oligodendrocytes(33).
In conclusion, our findings provide evidence for a subclinical motor and sensory polyneuropathy in the feline NPC disease model which is similar to what has been described in some human patients as well as in the murine model. As experimental therapies are evaluated for NPC disease, it will be necessary to identify additional surrogate markers of disease progression and severity which can be quantified and evaluated over time to give evidence of therapeutic effect. It is also necessary to identify subclinical disease which may manifest with new clinical signs when the more dominant CNS dysfunction is ameliorated.
Acknowledgments
Sources of support: NIH R01-NS073661-01, NCRR02512, Ara Parseghian Medical Research Foundation, Dana's Angels Research Trust, and National Niemann Pick Disease Foundation (CHV)
References
- 1.Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis. 2010e;5:16. doi: 10.1186/1750-1172-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ory DS. Niemann-Pick type C: a disorder of cellular cholesterol trafficking. Biochim Biophys Acta. 2000e;1529:331–9. doi: 10.1016/s1388-1981(00)00158-x. [DOI] [PubMed] [Google Scholar]
- 3.Patterson MC, Vanier MT, Suzuki K, Morris JA, Carstea E, Neufeld EB, Blanchette-Mackie EJ, Pentchev PG. Niemann-Pick disease type C: a lipid trafficking disorder. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, editors. Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; NY: 2001. [Google Scholar]
- 4.Sevin M, Lesca G, Baumann N, Millat G, Lyon-Caen O, Vanier MT, Sedel F. The adult form of Niemann-Pick disease type C. Brain. 2007e;130:120–33. doi: 10.1093/brain/awl260. [DOI] [PubMed] [Google Scholar]
- 5.March PA, Thrall MA, Brown DE, Mitchell TW, Lowenthal AC, Walkley SU. GABAergic neuroaxonal dystrophy and other cytopathological alterations in feline Niemann-Pick disease type C. Acta Neuropathol. 1997e;94:164–72. doi: 10.1007/s004010050689. [DOI] [PubMed] [Google Scholar]
- 6.Zervas M, Dobrenis K, Walkley SU. Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J Neuropathol Exp Neurol. 2001e;60:49–64. doi: 10.1093/jnen/60.1.49. [DOI] [PubMed] [Google Scholar]
- 7.Walkley SU, Suzuki K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim Biophys Acta. 2004e;1685:48–62. doi: 10.1016/j.bbalip.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 8.Liu B. Therapeutic potential of cyclodextrins in the treatment of Niemann-Pick type C disease. Clinical Lipidology. 2012e;7:289–301. doi: 10.2217/clp.12.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aqul A, Liu B, Ramirez CM, Pieper AA, Estill SJ, Burns DK, Repa JJ, Turley SD, Dietschy JM. Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment. J Neurosci. 31:9404–13. doi: 10.1523/JNEUROSCI.1317-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zafeiriou DI, Triantafyllou P, Gombakis NP, Vargiami E, Tsantali C, Michelakaki E. Niemann-Pick type C disease associated with peripheral neuropathy. Pediatr Neurol. 2003e;29:242–4. doi: 10.1016/s0887-8994(03)00219-4. [DOI] [PubMed] [Google Scholar]
- 11.Hahn AF, Gilbert JJ, Kwarciak C, Gillett J, Bolton CF, Rupar CA, Callahan JW. Nerve biopsy findings in Niemann-Pick type II (NPC) Acta Neuropathol. 1994e;87:149–54. doi: 10.1007/BF00296184. [DOI] [PubMed] [Google Scholar]
- 12.Alvelius G, Hjalmarson O, Griffiths WJ, Bjorkhem I, Sjovall J. Identification of unusual 7-oxygenated bile acid sulfates in a patient with Niemann-Pick disease, type C. J Lipid Res. 2001e;42:1571–7. [PubMed] [Google Scholar]
- 13.Lowenthal AC, Cummings JF, Wenger DA, Thrall MA, Wood PA, de Lahunta A. Feline sphingolipidosis resembling Niemann-Pick disease type C. Acta Neuropathol. 1990e;81:189–97. doi: 10.1007/BF00334507. [DOI] [PubMed] [Google Scholar]
- 14.Brown DE, Thrall MA, Walkley SU, Wenger DA, Mitchell TW, Smith MO, Royals KL, March PA, Allison RW. Feline Niemann-Pick disease type C. Am J Pathol. 1994e;144:1412–5. [PMC free article] [PubMed] [Google Scholar]
- 15.Munana KR, Luttgen PJ, Thrall MA, Mitchell TW, Wenger DA. Neurological manifestations of Niemann-Pick disease type C in cats. J Vet Intern Med. 1994e;8:117–21. doi: 10.1111/j.1939-1676.1994.tb03208.x. [DOI] [PubMed] [Google Scholar]
- 16.Somers KL, Royals MA, Carstea ED, Rafi MA, Wenger DA, Thrall MA. Mutation analysis of feline Niemann-Pick C1 disease. Mol Genet Metab. 2003e;79:99–103. doi: 10.1016/s1096-7192(03)00074-x. [DOI] [PubMed] [Google Scholar]
- 17.Vite CH, Ding W, Bryan C, O'Donnell P, Cullen K, Aleman D, Haskins ME, Van Winkle T. Clinical, electrophysiological, and serum biochemical measures of progressive neurological and hepatic dysfunction in feline Niemann-Pick type C disease. Pediatr Res. 2008e;64:544–9. doi: 10.1203/PDR.0b013e318184d2ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zervas M, Somers KL, Thrall MA, Walkley SU. Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr Biol. 2001e;11:1283–7. doi: 10.1016/s0960-9822(01)00396-7. [DOI] [PubMed] [Google Scholar]
- 19.Somers KL, Brown DE, Fulton R, Schultheiss PC, Hamar D, Smith MO, Allison R, Connally HE, Just C, Mitchell TW, Wenger DA, Thrall MA. Effects of dietary cholesterol restriction in a feline model of Niemann-Pick type C disease. J Inherit Metab Dis. 2001e;24:427–36. doi: 10.1023/a:1010588112003. [DOI] [PubMed] [Google Scholar]
- 20.Ward S, O'Donnell P, Fernandez S, Vite CH. 2-hydroxypropyl-beta-cyclodextrin raises hearing threshold in normal cats and in cats with Niemann-Pick type C disease. Pediatr Res. 2010e;68:52–6. doi: 10.1203/PDR.0b013e3181df4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stein VM, Crooks A, Ding W, Prociuk M, O'Donnell P, Bryan C, Sikora T, Dingemanse J, Vanier MT, Walkley SU, Vite CH. Miglustat improves purkinje cell survival and alters microglial phenotype in feline Niemann-Pick disease type C. J Neuropathol Exp Neurol. 71:434–48. doi: 10.1097/NEN.0b013e31825414a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weibel ER. Stereological methods. Academic Press; New Yrok: 2007. [Google Scholar]
- 23.Bradley WG, Good P, Rasool CG, Adelman LS. Morphometric and biochemical studies of peripheral nerves in amyotrophic lateral sclerosis. Ann Neurol. 1983e;14:267–77. doi: 10.1002/ana.410140304. [DOI] [PubMed] [Google Scholar]
- 24.Mizisin AP, Kalichman MW, Bache M, Dines KC, DiStefano PS. NT-3 attenuates functional and structural disorders in sensory nerves of galactose-fed rats. J Neuropathol Exp Neurol. 1998e;57:803–13. doi: 10.1097/00005072-199809000-00001. [DOI] [PubMed] [Google Scholar]
- 25.Mizisin AP, Shelton GD, Burgers ML, Powell HC, Cuddon PA. Neurological complications associated with spontaneously occurring feline diabetes mellitus. J Neuropathol Exp Neurol. 2002e;61:872–84. doi: 10.1093/jnen/61.10.872. [DOI] [PubMed] [Google Scholar]
- 26.Mizisin AP, Nelson RW, Sturges BK, Vernau KM, Lecouteur RA, Williams DC, Burgers ML, Shelton GD. Comparable myelinated nerve pathology in feline and human diabetes mellitus. Acta Neuropathol. 2007e;113:431–42. doi: 10.1007/s00401-006-0163-8. [DOI] [PubMed] [Google Scholar]
- 27.Vanier MT, Millat G. Niemann-Pick disease type C. Clin Genet. 2003e;64:269–81. doi: 10.1034/j.1399-0004.2003.00147.x. [DOI] [PubMed] [Google Scholar]
- 28.Elleder M, Jirasek A, Smid F, Ledvinova J, Besley GT. Niemann-Pick disease type C. Study on the nature of the cerebral storage process. Acta Neuropathol. 1985e;66:325–36. doi: 10.1007/BF00690966. [DOI] [PubMed] [Google Scholar]
- 29.Higashi Y, Murayama S, Pentchev PG, Suzuki K. Peripheral nerve pathology in Niemann-Pick type C mouse. Acta Neuropathol. 1995e;90:158–63. doi: 10.1007/BF00294315. [DOI] [PubMed] [Google Scholar]
- 30.Goodrum JF, Pentchev PG. Cholesterol reutilization during myelination of regenerating PNS axons is impaired in Niemann-Pick disease type C mice. J Neurosci Res. 1997e;49:389–92. doi: 10.1002/(sici)1097-4547(19970801)49:3<389::aid-jnr14>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 31.Takikita S, Fukuda T, Mohri I, Yagi T, Suzuki K. Perturbed myelination process of premyelinating oligodendrocyte in Niemann-Pick type C mouse. J Neuropathol Exp Neurol. 2004e;63:660–73. doi: 10.1093/jnen/63.6.660. [DOI] [PubMed] [Google Scholar]
- 32.Yan X, Lukas J, Witt M, Wree A, Hubner R, Frech M, Kohling R, Rolfs A, Luo J. Decreased expression of myelin gene regulatory factor in Niemann-Pick type C 1 mouse. Metab Brain Dis. 26:299–306. doi: 10.1007/s11011-011-9263-9. [DOI] [PubMed] [Google Scholar]
- 33.Emery B, Agalliu D, Cahoy JD, Watkins TA, Dugas JC, Mulinyawe SB, Ibrahim A, Ligon KL, Rowitch DH, Barres BA. Myelin Gene Regulatory Factor Is a Critical Transcriptional Regulator Required for CNS Myelination. Cell. 2009e;138:172–85. doi: 10.1016/j.cell.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]