

Summary
Metabolomic profiling of obese versus lean humans reveals a branched-chain amino acid (BCAA)-related metabolite signature that is suggestive of increased catabolism of BCAA and correlated with insulin resistance. To test its impact on metabolic homeostasis, we fed rats on high-fat (HF), HF with supplemented BCAA (HF/BCAA) or standard chow (SC) diets. Despite having reduced food intake and weight gain equivalent to the SC group, HF/BCAA rats were equally insulin resistant as HF rats. Pair-feeding of HF diet to match the HF/BCAA animals or BCAA addition to SC diet did not cause insulin resistance. Insulin resistance induced by HF/BCAA feeding was accompanied by chronic phosphorylation of mTOR, JNK, and IRS1(ser307), accumulation of multiple acylcarnitines in muscle, and was reversed by the mTOR inhibitor, rapamycin. Our findings show that in the context of a poor dietary pattern that includes high fat consumption, BCAA contributes to development of obesity-associated insulin resistance.
Keywords: Obesity, branched-chain amino acids, organic acids, acylcarnitines, hormones, fatty acids, metabolomics, body composition, energy expenditure
Introduction
Obesity has reached epidemic proportions in many countries around the world, and is strongly linked to a number of chronic diseases, including diabetes, hypertension and cardiovascular disease (Hedley et al., 2004; Sowers et al., 2003). Whereas many comparisons of obese and lean subjects exist in the literature, these are often focused on one or a small group of experimental variables. Here we sought to apply comprehensive metabolic profiling tools to gain a complete understanding of metabolic, endocrine, inflammatory, and physiologic differences between obese and lean subjects. Included were measurements of nineteen hormones of energy balance and fuel homeostasis, four pro- and anti-inflammatory cytokines, physiological variables such as insulin sensitivity, body composition, and resting metabolic rate, ten metabolites assayed by conventional enzyme assays, and as a novel feature, ninety-eight intermediary metabolites in six chemical groups measured by targeted mass spectrometry (MS) (Haqq et al., 2005).
This comprehensive metabolic profiling was performed on 74 obese (median BMI of 36.6 kg/m2) and 67 lean (median BMI of 23.2 kg/m2) subjects. We selected “healthy” obese subjects free of diabetes or other serious illness. Not surprisingly, we found the obese participants to be more insulin resistant on average than the lean controls. We also found a novel metabolic “signature” related to branched-chain amino acid (BCAA) catabolism in obese subjects. Animal studies based on these findings demonstrate that supplementation of a high fat (HF) diet with BCAA (HF/BCAA) reduces food intake and body weight, but causes the animals to be equally insulin resistant as heavier animals fed on a non-supplemented HF diet. Importantly, animals fed on HF diet at a rate matched to that of the HF/BCAA-fed animals do not become insulin resistant, and the HF/BCAA-induced insulin resistance is selectively reversed by the mTOR inhibitor rapamycin. From these findings, we propose a pathway by which dysregulated BCAA metabolism makes an independent contribution to development of insulin resistance and glucose intolerance, ultimately leading to type 2 diabetes.
Results
Demographics and Clinical Characteristics
73 obese and 67 lean subjects underwent baseline evaluation. The obese subjects were comprised of 70% women and 41% African Americans, whereas the lean subjects were 57% women and 45% African Americans. Median age of the obese subjects was 52 years, and their median body mass index (BMI) was 36.6 kg/m2, compared to 50 years and 23.2 kg/m2 for the lean controls. Additional demographic and clinical data is provided in Table 1.
Table 1. Baseline Characteristics and Physiologic Measures.
| All results presented as medians (25th, 75th percentile) or N (percent). | |||
|---|---|---|---|
| Obese | Lean | p-value | |
| Baseline Characteristics | N=74 | N=67 | |
| Age (years) | 52.0(46.0,60.0) | 50.0(38.0,60.0) | 0.2391 |
| African-American | 30(40.5%) | 30(44.8%) | 0.6115 |
| Female | 52(70.3%) | 38(56.72%) | 0.0944 |
| BMI | 36.6(32.7,40.7) | 23.2(21.8,23.8) | <0.0001 |
| Lean, BMI<25 kg/m2 | 0(0.0%) | 67(100.0%) | <0.0001 |
| Obese, stage I (30-34.9kg/m2) | 28(37.8%) | 0(0.0%) | |
| Obese, stage II (35-39.9 kg/m2) | 24(32.4%) | 0(0.0%) | |
| Obese, stage III (≥40 kg/m2) | 22(29.7%) | 0(0.0%) | |
| Waist circumference (cm)* | 112.6(103.9,117.7) | 84.1(78.0,88.7) | <0.0001 |
| Taking lipid medication | 29(39.2%) | 9(13.4%) | 0.0006 |
| Taking blood pressure medication | 50(67.6%) | 16(23.9%) | <0.0001 |
| Dietary intake (% of kcal) | N=66 | N=66 | |
| Fat | 42.2(37.1,46.8) | 35.7(31.1,39.7) | <0.0001 |
| Carbohydrate | 43.4(37.6,48.7) | 50.0(41.9,55.0) | 0.0005 |
| Protein | 15.5(13.5,18.2) | 14.3(13.0,16.9) | 0.0719 |
| Physical activity (METs)** | 1431(459,3510) | 2126(1386,3816) | 0.0957 |
| Physiologic Measures*** | |||
| Body composition | |||
| Fat mass (kg) | 36.45(31.92,43.38) | 18.65(13.66,21.38) | <0.0001 |
| Lean mass (kg) | 55.09(49.60,68.43) | 45.45(41.28,58.20) | <0.0001 |
| Subcutaneous fat (area) | 37903.00(34958.00,46235.0) | 16176.00(12186.00,22042.00) | <0.0001 |
| Visceral fat (area) | 20971.00(14004.00,26325.00) | 6712.50(4068.50,9865.00) | <0.0001 |
| Resting metabolic rate | |||
| REE/LBM | 28.02(25.98,30.41) | 29.32(26.84,32.06) | 0.027 |
| RER(RQ) | 0.85(0.79,0.90) | 0.81(0.78,0.84) | 0.0038 |
| Insulin resistance | |||
| HOMA | 5.73(3.88,8.29) | 2.51(2.01,3.32) | <0.0001 |
| Si | 2.12(1.27,2.99) | 4.44(3.66,6.30) | <0.0001 |
| AIRg | 599.00(329.00,924.00) | 398.00(223.00,569.00) | 0.0491 |
N for mea surement of waist circumference = 68 for obese and 62 for lean.
N for measurement of physical activity = 47 for obese and 59 for lean.
N varies due to variations in completion of data collection.
N for fat and lean mass = 48 for obese and 64 for lean.
N for subcutaneous fat = 41 and 60 and for visceral fat = 47 and 60 for obese and lean, respectively.
N for REE/LBM = 45 obese and 62 for lean; N for RER = 53 and 62 for obese and lean.
N for HOMA = 74 for obese and 67 for lean.
N for Si = 26 for obese and 25 for lean and AIRg = 26 for both obese and lean, respectively.
Based on self-administered Block Food Frequency Questionnaire (Harlan and Block, 1990), obese subjects had a higher dietary intake of fat (p < 0.0001), a lower intake of carbohydrate (p = 0.0005), and a trend towards an increase in protein consumption (p = 0.072). Physical activity measured by the International Physical Activity Questionnaire (IPAQ) trended lower in obese compared to lean subjects (p = 0.0957).
Physiologic measures
Obese subjects had twice as much total fat mass as lean controls, but only a 17% increase in lean mass (Table 1). The higher total fat mass in obese subjects was accompanied by a 2.3-fold increase in subcutaneous fat mass and a 3.1-fold increase in visceral fat mass (p < 0.0001 in both cases). Respiratory exchange rate (RER), also known as respiratory quotient (RQ), was significantly higher in obese subjects (p = 0.0038). Thus, obese subjects appear to have a preference for oxidation of non-lipid fuels (glucose and amino acids) compared to lean controls (as indicated by the higher RQ), even though they have substantially larger total, subcutaneous, and visceral fat depots.
Obese subjects were less insulin sensitive than the lean controls, by two independent criteria (Table 1). First, the homeostasis model assessment (HOMA) index was 2.3-fold higher in obese subjects than in lean controls (p < 0.0001). Second, in a subset of 26 obese and 26 lean subjects underwent intravenous glucose tolerance testing, the insulin sensitivity index, Si, was 2.1-fold higher in lean subjects (p < 0.001). The obese subjects that underwent IVGTT showed a trend toward higher acute insulin response to glucose (AIRG; p = .049), consistent with obesity-related insulin resistance.
Hormones and Cytokines
Among a total of 19 hormones of energy balance and metabolic regulation measured in this study, only 5 were not significantly different in obese versus lean subjects—amylin, glucagon-like peptide-1 (GLP-1), resistin, pancreatic polypeptide, and insulin-like growth factor binding protein-3 (IGFBP-3).
Table 2 demonstrates that pancreatic islet peptides other than amylin (insulin, C-peptide, and glucagon) were all significantly elevated in serum of obese versus lean subjects (p < 0.0001 in all cases). Levels of the incretin hormone GIP were also higher (p = 0.001), in accord with the higher insulin levels in obese versus lean subjects.
Table 2. Hormones, Cytokines, and Metabolites.
| All results presented as medians (25 thth, 75th percentile). | ||||
|---|---|---|---|---|
| Obese | Lean | p-value | ||
| Hormones | N=74 | N=67 | ||
| Insulin (μUI/mL) | 22.01(15.71,30.14) | 9.99(8.36,13.51) | <0.0001 | |
| C-peptide (ng/mL) | 3.62(2.71,4.58) | 1.90(1.59,2.34) | <0.0001 | |
| Glucagon (pg/mL) | 91.18(79.63,120.72) | 68.92(56.08,89.58) | <0.0001 | |
| GIP (pg/mL) | 43.99(26.28,62.25) | 29.58 (20.39,44.90) | 0.0011 | |
| Leptin (ng/mL) | 29.67(19.71,44.08) | 7.83(3.67,14.77) | >0.0001 | |
| Adiponectin (μg/mL) | 6.06(4.00,8.94) | 9.62(6.96,14.85) | <0.0001 | |
| Ghrelin (pg/mL) | 674.09(504.85,881.23) | 965.75(636.23,1269.37) | <0.0001 | |
| NPY (pM) | 51.18(43.74,61.72) | 44.47(35.05,52.50) | 0.0006 | |
| PYY (pg/mL) | 141.88(112.55,168.19) | 103.36(88.74,126.14) | <0.0001 | |
| Resistin (ng/mL) | 3.84(3.3,4.8) | 3.64(2.9,4.9) | 0.34 | |
| Amylin (pM)§ | 4.78(0.00,16.03) | 3.94(0.00,31.52) | 0.81 | |
| GLP-1 (pg/mL)* | 24.21(17.62,35.71) | 28.36(15.75,92.69) | 0.42 | |
| PP (pM) | 24.26(14.46,44.99) | 24.46(11.83,52.38) | 0.63 | |
| Growth hormone axis (ng/mL) | ||||
| Growth hormone | 0.08(0.00,0.46) | 0.30(0.01,1.64) | 0.0394 | |
| Total IGF-1 | 134.26(93.26,184.64) | 250.20(153.34,325.16) | <0.0001 | |
| IGFBP-1 | 8.25(3.47,17.21) | 19.75(12.21,28.53) | <0.0001 | |
| IGFBP-2 | 244.27(66.66,362.42) | 603.57(384.00,960.54) | <0.0001 | |
| IGFBP-3 | 3290.0(2920.8,3804.4) | 3301.0(2771.9,4041.0) | 0.89 | |
| Free IGF** | 0.4595(0.3365,0.713) | 0.558(0.406,0.926) | 0.01 | |
| Cytokines | N=74 | N=67 | ||
| Interleukin-6 (pg/mL) | 7.62(4.31,11.35) | 8.18(5.80,10.89) | 0.32 | |
| TNF-α (pg/mL) | 9.61(6.73,14.89) | 6.93(5.07,14.1) | 0.19 | |
| C-reactive protein (mg/L) | 4.48(2.35,7.55) | 0.85(0.45,1.35) | <0.0001 | |
| Interleukin-10 (pg/mL)§ | 0.91(0.01,2.87) | 1.85(0.25,3.11) | 0.0251 | |
| Conventional Metabolites | N=74 | N=67 | ||
| Fasting glucose (mg/dL) | 105.0(97.5,116.0) | 100.5(92.5,106.0) | 0.005 | |
| Free fatty acid (mmol/L) | 0.58(0.45,0.72) | 0.43(0.33,0.68) | 0.004 | |
| Ketones (μmol/L) | 82.5(46.5,120.0) | 55.5(39.5,140.5) | 0.28 | |
| Cholesterol (mg/dL) | 191.0(174.5,213.0) | 188.5(161.5,223.0) | 0.67 | |
| LDL-cholesterol (mg/dL) | 119.3(103.0,139.7) | 105.8(86.2,124.8) | 0.02 | |
| Triglyceride (mg/dL) | 106.5(77.0,163.0) | 63.5(46.0,89.0) | <0.0001 | |
| HDL-cholesterol (mg/dL) | 49.9(38.7,58.7) | 62.2(54.5,77.6) | <0.0001 | |
| Plasma lactic acid (mM) | 1.3(1.2,1.6) | 1.1(0.86,1.4) | 0.0006 | |
| Plasma pyruvic acid (mM) | 0.12(0.10,0.15) | 0.09(0.07,0.11) | <0.0001 | |
| β-Hydeoxybutyrate (μmol/L) | 47.8(27.5,73.0) | 35.0(22.5,100.5) | 0.35 | |
| Amino Acids (μM) | N=74 | N=67 | ||
| Valine | 281.4(249.2,332.9) | 235.3(204.1,257.0) | <0.0001 | |
| Leucine/Isoleucine | 170.0(150.2,200.8) | 149.0(132.5,176.6) | <0.0001 | |
| Glutamate/Glutamine | 118.4(91.4,143.7) | 81.2(66.7,95.2) | <0.0001 | |
| Glycine | 282.6(245.6,319.6) | 328.4(265.6,403.0) | 0.0007 | |
| Alanine | 433.4(394.5,492.3) | 367.3(297.1,420.0) | <0.0001 | |
| Phenylalanine | 72.6(66.3,78.9) | 61.6(55.1,68.8) | <0.0001 | |
| Tyrosine | 79.5(68.5,90.0) | 67.1(56.7,73.5) | <0.0001 | |
| Aspartate/Asparagine | 20.1(17.3,23.8) | 16.5(13.5,19.7) | <0.0001 | |
| Arginine | 135.2(116.5,148.5) | 115.3(101.6,137.0) | 0.0007 | |
| Citrulline | 32.0(27.9,40.3) | 36.3(30.5,40.7) | 0.04 | |
| Histidine | 81.6(73.5,88.9) | 81.9(71.9,91.6) | 0.57 | |
| Methionine | 27.5(25.1,30.7) | 27.6(24.2,30.9) | 0.68 | |
| Ornithine | 69.6(55.3,80.8) | 64.8(53.3,71.8) | 0.18 | |
| Proline | 176.4(155.4,231.5) | 158.1(138.4,201.7) | 0.02 | |
| Serine | 115.6(100.7,131.9) | 116.7(102.2,138.6) | 0.69 | |
| Acylcarnitines (μM) | N=74 | N=67 | ||
| Proprionyl (C3) | 0.45(0.35,0.54) | 0.38(0.28,0.48) | 0.0034 | |
| Isovaleryl/2-methylbutyryl (C5) | 0.12(0.10,0.16) | 0.09(0.07,0.13) | 0.0004 | |
| Hexanoyl (C6)§ | 0.10(0.04,0.16) | 0.06(0.00,0.11) | 0.0089 | |
| Octenoyl (C8:1) | 0.28(0.19,0.35) | 0.18(0.16,0.27) | 0.0006 | |
| Organic Acids (mmol/mol) | N=70 | N=67 | ||
| Ethyl malonate | 3.03(2.41,4.02) | 2.61(2.28,2.95) | 0.0054 | |
| Isobutyryl glycine | 0.40(0.26,0.58) | 0.53(0.40,0.86) | 0.001 | |
| Isovaleryl glycine | 0.62(0.42,0.95) | 0.92(0.53,1.33) | 0.0031 | |
| α-Ketoglutarate | 4.72(3.37,7.26) | 8.19(4.21,12.75) | 0.0005 | |
| Free Fatty Acids (μM) | N=74 | N=67 | ||
| Octanoic | C8:0 | Nd | Nd | Nd |
| Decanioc | C10:0 | Nd | Nd | Nd |
| Lauri | C12:0 | Nd | Nd | Nd |
| Myristic | C14:0 | 9.98(7.9,12.7) | 7.45(5.8,11.5) | 0.01 |
| Myristoleic | C14:1 | Nd | Nd | Nd |
| Palmitic | C16:0 | 141.3(103.4,176.5) | 101.3(73.3,150.5) | 0.0004 |
| Palmitoleic | C16:1 | 21.5(13.4,35.0) | 14.6(8.0,25.4) | 0.001 |
| Stearic | C18:0 | 42.4(32.2,55.0) | 36.9(27.5,53.2) | 0.3 |
| Oleic | C18:1 | 206.4(156.2,270.6) | 157.9(103.7,241.2) | 0.002 |
| Linoleic | C18:2 | 81.3(62.9,99.7) | 75.8(46.3,94.3) | 0.087 |
| α-Linolenic | C18:3 | 6.87(4.8,9.7) | 5.68(3.9,8.5) | 0.048 |
| Arachidic | C20:0 | Nd | Nd | Nd |
| Gondioc | C20:1 | Nd | Nd | Nd |
| Eicosadienioc | C20:2 | Nd | Nd | Nd |
| Dihomogamma-linolenic | C20:3 | Nd | Nd | Nd |
| Arachidonic | C20:4 | 5.98(4.3,7.8) | 4.6(3.3,6.8) | 0.006 |
| Docosahexaenioc | C20:6 | Nd | Nd | Nd |
| Total Fatty Acids (μM) | N=73 | N=67 | ||
| Octanoic*** | C8:0 | 29.70(26.23,39.90) | 36.66(27.47,43.77) | 0.073 |
| Decanioc | C10:0 | 5.30(4.43,6.58) | 4.96(4.05,6.43) | 0.079 |
| Lauri | C12:0 | 10.12(7.62,16.85) | 8.59(6.33,13.79) | 0.05 |
| Myristic | C14:0 | 102.3(77.4,155.7) | 71.9(50.2,111.2) | <0.0001 |
| Myristoleic | C14:1 | 7.2(5.3,10.8) | 4.7(3.1,7.2) | <0.0001 |
| Palmitic | C16:0 | 1744.9(1398.3,2169.6) | 1234.8(982.1,1591.5) | <0.0001 |
| Palmitoleic | C16:1 | 410.2(295.5,546.3) | 206.9(174.4,323.9) | <0.0001 |
| Stearic | C18:0 | 880.8(748.5,1033.5) | 712.68(601.5,789.9) | <0.0001 |
| Oleic | C18:1 | 2762.8(2304.2,3382.2) | 1944.4(1692.4,2481.3) | <0.0001 |
| Linoleic | C18:2 | 4100.4(3050.7,5027.9) | 3449.4(2561.9,4034.8) | 0.0038 |
| α-Linolenic | C18:3 | 78.9(63.7,95.9) | 54.6(41.9,78.1) | <0.0001 |
| Arachidic | C20:0 | 5.37(4.58,7.72) | 5.86(4.39,8.05) | 0.51 |
| Gondioc | C20:1 | 14.4(11.7,19.2) | 11.2(8.5,14.6) | 0.0003 |
| Eicosadienioc | C20:2 | 22.8(17.0,26.2) | 16.5(13.8,19.4) | 0.0002 |
| Dihomogamma-linolenic | C20:3 | 220.9(185.9,279.7) | 140.0(114.5,180.2) | <0.0001 |
| Arachidonic | C20:4 | 850.44(685.7,1098.3) | 615.0(528.1,871.1) | <0.0001 |
| Docosahexaenioc | C20:6 | 181.8(140.4,243.5) | 142.4(111.1,177.8) | 0.0016 |
GLP-1: N=52 for obese, N=61 for lean.
Free IGF-1: N=90 for obese, N=65 for lean.
Octanioc: N=72 for obese, N=66 for lean.
Metabolite has > 10% zero cells.
Nd, not detectable
Consistent with numerous prior reports (Bjorbaek et al., 2004; Trujillo et al., 2006), the adipocyte-derived hormones leptin and adiponectin were reciprocally altered by obesity, such that leptin levels were 3.8-fold higher in obese subjects (p < 0.0001), whereas adiponectin levels were 37% lower (p < 0.0001). Among hormones that have been implicated in control of food intake, the gut-derived orexigenic peptide ghrelin was 30% lower in serum of obese subjects (p < 0.0001), whereas NPY (orexigenic) and PYY (presumed anti-orexigenic) were 15% and 37% higher, respectively (p ≤ 0.0006). Human growth hormone (hGH) trended lower (p = 0.039), and total IGF-1, IGFBP-1, and IGFBP-2 were all dramatically lower in serum of obese subjects (p < 0.0001) (Table 2). We also measured free IGF-1 and found this analyte to be markedly lower in the obese subjects (p = 0.01; Table 2).
Among the pro- and anti-inflammatory cytokines measured, neither interleukin (IL)-6 nor tumor necrosis factor (TNF)-α differed between obese and lean subjects, whereas IL-10 trended lower in the obese subjects (p = 0.025). In contrast, C-reactive protein (CRP) levels were 5.3-fold higher in obese subjects (p < 0.0001) (Table 2). Both IL-6 and TNF-α have been previously implicated in the development of insulin resistance (Wellen et al., 2005; Shoelson et al., 2006), and CRP has been strongly linked to cardiovascular disease (Ridker et al., 1998), as well as to components of the “metabolic syndrome”, including insulin resistance, although a recent study suggests that obesity is the major determinant of the CRP/insulin resistance relationship (Kahn et al., 2006).
Conventional metabolites
Median fasting blood glucose levels were modestly elevated in obese compared to lean subjects (105.0 mg/dl versus 100.5 mg/dl, p = 0.005). Obese subjects had significantly higher levels of non-esterified free fatty acids (p = 0.004) and triglycerides (p < 0.0001), and substantially lower HDL cholesterol (p < 0.0001). Levels of ketones (total and β-hydroxybuyrate), byproducts of lipid catabolism, were not different between the groups, whereas levels of pyruvate (p < 0.0001) and lactate (p = 0.0006), products of carbohydrate catabolism, were higher in serum of obese subjects (Table 2). These findings appear consistent with the increase in RQ observed in the obese subjects (Table 1).
Metabolites measured by mass spectrometry
Consistent with the general picture of perturbation of lipid homeostasis gained from analysis of conventional metabolites, clear differences in individual free and total fatty acids were observed in obese compared to lean subjects (Table 2). Among free fatty acids, 5 of the 8 species measured were significantly higher in obese compared to lean subjects (C14:0, C16:0, C16:1, C18:1, C20:4; p values ranging from 0.0004 to 0.01). The rise in C16:1 (palmitoleate) in insulin resistant and obese subjects is of interest in light of a recent study suggesting that C16:1 promotes insulin sensitivity in skeletal muscle (Cao, et al, 2008). Total fatty acid species were more dramatically increased in the obese subjects, with 13 of the 17 analytes clearly increased in serum of obese subjects, with p < 0.0001 for 9 of these, including C16:1 (Table 2).
We also measured amino acids and acylcarnitines in the serum by MS/MS, and organic acids in the urine by GC/MS. Among a total of 16 amino acids measured in the serum, 7 were not different in obese versus lean subjects (citrulline, histidine, methionine, ornithine, proline, serine, tryptophan). Eight other amino acids were dramatically elevated in obese versus lean participants (alanine, valine, leucine/isoleucine, phenylalanine, tyrosine, glutamate/glutamine, aspartate/asparagine, arginine; p ≤ 0.0007), whereas glycine levels were lower in obese subjects (p = 0.0007) (Table 2). Among 37 acylcarnitine species measured, only 4 were significantly higher in obese subjects (C3, C5, C6, C8:1; p ≤ 0.009) (Table 2). Finally, among 20 organic acids measured in urine by GC/MS, 15 were not different in obese versus lean subjects (methylmalonate, succinate, methylsuccinate, fumarate, glutarate, butyrylglycine, malate, adipate, hexanoyl glycine, suberate, orotate, homovanillic, citrate, pyruvate, lactate). Levels of ethylmalonate were significantly higher, and levels of isobutyryl glycine, isovaleryl glycine, and α-ketoglutarate were lower in urine samples from obese compared to lean subjects (Table 2).
A branched-chain amino acid-related metabolic “signature” in obese subjects
We used principal components analysis (PCA) to consolidate the metabolites referenced above into 18 components that were analyzed for differences between obese and lean subjects. Remarkably, the component that showed the strongest differences between obese and lean groups was comprised of a combination of BCAA (leucine/isoleucine and valine), methionine, Glx (glutamate/glutamine), the aromatic amino acids phenylalanine and tyrosine, and C3 and C5 acylcarnitines (p<0.0001). Furthermore, evaluation of the association between this BCAA-related metabolite component and HOMA reveals a significant linear relationship (Figure 1; r = 0.58, p<0.0001), even after adjusting for obese vs. lean status using a partial Spearman correlation coefficient (r=0.33, p<0.0001). To further ensure the independent relationship of the BCCA-related metabolite component with HOMA, we performed correlation analyses stratified by obese/lean status and adjusted for age, race and sex, which revealed the same magnitude of correlation in obese subjects as in the total subject population (unadjusted Spearman r=0.36, p=0.002; partial Spearman adjusted for age, race, sex r=0.33, p=0.007), with no significant relationship in lean subjects (unadjusted Spearman r=0.17, p=0.18; partial Spearman adjusted for age, race, sex r=0.13, p=0.30), as might be anticipated due to the limited variability of insulin sensitivity within the normal range in this group.
Figure 1. A banched-chain amino acid-related metabolic “signature” correlates with insulin sensitivity.

The figure shows the relationship between insulin sensitivity (HOMA) and a principal component comprised of BCAA-related metabolites including the branched-chain amino acids valine, leucine, and isoleucine, Glx (glutamate + glutamine), the aromatic amino acids phenylalanine and tyrosine, and C3 and C5 acylcarnitines (See Methods and Results for further description). Diamonds, lean subjects (N=67); Squares, obese subjects (N=74).
We note the presence of an outlier among the obese subjects with both severe insulin resistance and a very high score for the BCAA factor (Figure 1). Importantly, when this outlier was removed, we observed the same magnitude of relationship between the BCAA factor and HOMA even when adjusted for obese/lean status, age, race and sex. Thus, this outlier is not a sole driver of the relationship.
Effects of BCAA supplementation in animal feeding studies
To test the idea that BCAA can make a direct contribution to obesity-related comorbidities such as insulin resistance and glucose intolerance, we fed Wistar rats on one of three diets, high-fat (HF), high-fat with supplemented BCAA (HF/BCAA), and standard chow (SC) (Supplemental Table 1). After 13 weeks of feeding, HF-fed rats had gained 170% relative to starting body weight, whereas HF/BCAA and SC-fed rats had gained 151% and 149%, respectively (Figure 2A; p <0.039 for HF versus HF/BCAA or SC groups). The difference in body weight between the HF and HF/BCAA groups was due to different rates of food intake, as HF and SC-fed rats consumed an average of 746 and 782 kcal/rat/week over the 13 week feeding period, whereas HF/BCAA-fed rats consumed 666 kcal/rat/week (Figure 2B); the identical weight gain in the HF/BCAA and SC groups despite different rates of food intake is likely due to the higher rate of energy expenditure in the SC-fed animals (see below).
Figure 2. Effects of BCAA supplementation on body weight, food intake, and insulin signaling.
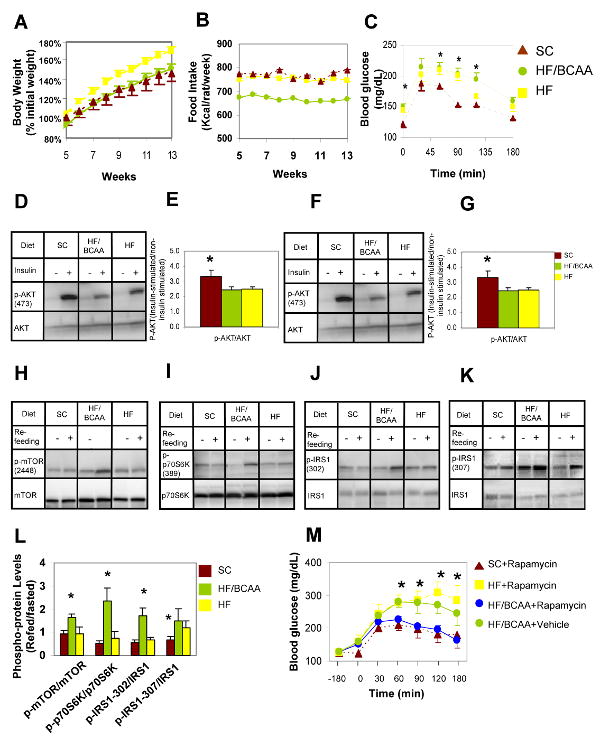
Rats were fed on standard chow (SC), high fat (HF) or HF supplemented with BCAA (HF/BCAA) diets for 12-16 weeks. A) Body weight expressed as % of initial weight; B) Food intake expressed as Kcal/rat/week. Data for panels A and B represent 5-9 animals per group. In panel A, HF was different from both the SC and HF/BCAA groups at all time points, with p < 0.036. In panel B, HF/BCAA was different from both the SC and HF groups at all time points, with p < 0.006. C) Intraperitoneal glucose tolerance test; n = 5-9 rats/group. (*) the SC group was different from the other two groups, with p < 0.036; D) Representative immunoblot of p-AKT levels in skeletal muscle 30 minutes after an acute insulin bolus in overnight fasted rats; E) Quantitative summary of muscle p-AKT studies, expressed as ratio of insulin stimulated: non-insulin stimulated conditions and normalized to total AKT protein; F) Representative immunoblot of p-AKT levels in liver, 30 minutes after an acute insulin bolus in overnight fasted rats; G) Quantitative summary of liver p-AKT studies, expressed as ratio of insulin stimulated: non insulin-stimulated conditions and normalized to total AKT protein levels. For panels E and G, (*) indicates that SC-fed animals had higher levels of p-AKT than the other two groups, with p < 0.033; H) p-mTOR2448, representative immunoblot; I) p-P70S6K1389, representative immunoblot; J) p-IRS-1ser302, representative immunoblot; K) p-IRS1ser307, representative immunoblot; L) Quantitative summary of phosphoproteins normalized to total levels of mTOR, p70S6K1, or IRS-1, respectively. Data in H-L are for skeletal muscle samples from 3-5 animals/group fed on the three diets, fasted for 48 h and then refed on the same diets for 4 h. (*) in panel L indicates higher levels of phosphoproteins in the HF/BCAA group compared to the other two groups, with p < 0.05; M) Rapamycin treatment reverses insulin resistance in HF/BCAA-fed but not HF-fed rats. Data shown are for 5-7 animals per group. (*) p < 0.05, blood glucose significantly different between the HF-fed, rapamycin treated or HF/BCAA-fed, non treated groups versus the SC, rapamycin treated and HF/BCAA, rapamycin treated groups.
Analysis of BCAA and acylcarnitine levels in the blood revealed 80-150% increases in Leu/Ile and 48-109% increases in Val in HF/BCAA-fed rats relative to the other two groups at 3, 6, and 9 weeks of feeding (Supplemental Figures 1A, 1B). Interestingly, over the same time periods, HF fed rats had lower levels of plasma C3 (39-51%, p <0.01) and C5 (29-36%, p <0.08) acylcarnitines compared to SC animals, consistent with a large decrease in RER/RQ that indicates an increased reliance on lipid oxidation for energy production (see below). In contrast, C3 and C5 acylcarnitines were raised to the levels of SC rats in plasma animals fed on the HF/BCAA diet, despite unchanged RER/RQ relative to HF rats (Supplemental Figures 1C, 1D). Further, complete analysis of acylcarnitine species by MS/MS analysis reveals a clear increase in C3 and C5 acylcarnitines in HF/BCAA-fed animals relative to all other groups in skeletal muscle (see below). These results coupled with the known BCAA catabolic pathways suggest that C3 and C5 acylcarnitines are direct products of BCAA catabolism that accumulate with modest BCAA overfeeding.
Despite a rate of body weight gain equivalent to the SC group, HF/BCAA-fed rats were equally insulin resistant as HF-fed rats after 15 weeks of diet feeding based on glucose (IPGTT) (Figure 2C, Supplemental Figure 2A) and insulin (data not shown) tolerance tests. Also, both HF and HF/BCAA-fed rats exhibited impaired phosphorylation (activation) of Akt/protein kinase B in skeletal muscle (Figures 2D, 2E) and liver (Figures 2F, 2G) in response to an acute insulin injection. These experiments demonstrate a contribution of BCAA to development of insulin resistance that is independent of body weight. To exclude the possibility that insulin resistance in the HF/BCAA-fed group was due to the HF diet rather than the supplemented BCAA, we performed an independent feeding study involving ad-lib feeding of SC, HF, or HF/BCAA diets plus a fourth group of animals pair-fed the HF diet (HF-PF) in amounts that matched the lower rate of food intake of the HF/BCAA group. Consistent with the data of Figure 2, rats fed ad-lib on the HF/BCAA diet gained less weight and ate less food than those fed ad-lib on the HF diet; as expected, HF-PF rats had body weights identical to the HF/BCAA group and not significantly different from the SC-fed group (Supplemental Figures 3A, 3B). IPGTT studies performed on all four groups of animals demonstrated clear insulin resistance in the HF and HF/BCAA-fed animals, but not in the HF-PF or SC groups (Supplemental Figures 3C and 3D), proving that moderate fat intake is not sufficient to induce insulin resistance, but requires the presence of an additional factor such as intake of BCAA.
A third feeding study was carried out in which rats were allowed to feed ad-lib either on SC diet or SC supplemented with BCAA (SC/BCAA, Supplemental Table 1) for 12-16 weeks. These studies revealed no differences in body weight, insulin sensitivity assessed by IPGTT, or insulin tolerance tests between the two groups, despite clear increases in BCAA and C3 and C5 acylcarnitine levels in the SC/BCAA compared to the SC group (Supplemental Figure 4). These results demonstrate that the effects of BCAA on feeding behavior and insulin action occur only in the context of a diet that is also high in fat.
Effects of BCAA supplementation on insulin signaling mechanisms
Because BCAA have been reported to interfere with insulin signaling via stimulation of mTOR and S6K1 and phosphorylation of IRS-1 on serine residues (Um et al., 2006; Tremblay, et al, 2007; Krebs, et al., 2007), we investigated the contribution of this pathway to development of insulin resistance in rats fed on the HF, HF/BCAA, or SC diets. In animals fed on the various diets, fasted for 48 h, and then refed on the original diet for 4 h, we found clear increases in phospho-mTOR(ser2448), phospho-S6K1 (thr389), and phospho-IRS1(ser302) in skeletal muscle of HF/BCAA-fed rats compared to either the HF or SC groups (Figures 2H, 2I, 2J, 2K, and 2L). Phospho-IRS1(ser307) levels were raised in both the HF and HF/BCAA groups relative to SC in the fasted/refed state and these levels were also specifically elevated in muscles of fasted HF/BCAA fed rats (Figures 2K, 2L, Supplemental Figure 2B). Fasted and refed HF and HF/BCAA rats had equally increased levels of insulin compared to SC animals (p < 0.04; data not shown), further proving insulin resistance. We also examined phophorylation of IRS-1 serine 636/639 and serine 1101, which have been implicated in diet-induced insulin resistance (Tremblay, et al., 2007; Krebs, et al., 2007), but found no differences in the levels of these modified proteins among the various dietary groups (data not shown).
To eliminate the possibility that the increase in ser/thr phosphorylation of mTOR, S6K1 and IRS-1 was simply due to acute feeding of BCAA, we also performed a “cross-feeding” study, in which HF/BCAA-fed rats were fasted and refed on HF diet, whereas HF and SC-fed rats were fasted and refed on the HF/BCAA diet. Following these manipulations, mTOR, S6K1, and IRS-1(ser302) levels were all significantly elevated in the HF/BCAA-fed group refed on HF diet compared to the other two groups refed on HF/BCAA diet (Supplemental Figure 2C), demonstrating sustained alterations in insulin signaling in the HF/BCAA fed animals that was not due to acute ingestion of excess BCAA in the refeeding period. We measured food intake during both the same-diet refeeding and cross-feeding studies, and found no differences in food intake among the experimental groups (data not shown), thereby eliminating this as a contributing factor to the data shown in Supplemental Figure 2C. Finally, we performed studies on a cohort of rats fed the HF diet with 26% supplementation of 18 amino acids (HF/AA, Supplemental Table 1), such that the diet was isonitrogenous with the HF/BCAA diet. In this set of experiments, the HF and HF/AA groups gained weight at the same rate (187% and 176% weight increases over 16 weeks, respectively), whereas the HF/BCAA group again gained weight at a slow rate similar to the SC fed animals (146% and 131%, respectively). These results show a specific effect of BCAA supplementation that is not a general consequence of increased protein feeding.
To further investigate the role of enhanced mTOR/S6K1/IRS-1 signaling in HF/BCAA-mediated insulin resistance we performed IPGTT experiments in animals treated with the mTOR inhibitor rapamycin. Rapamycin injection clearly reversed the glucose intolerance of HF/BCAA-fed rats, but had no normalizing effect on glucose clearance in the HF rats (Figure 2M), providing a direct demonstration of the importance of altered mTOR/S6K1 signaling in mediating HF/BCAA-induced insulin resistance.
Physiologic measurements in animal feeding studies
Two recent studies have reported changes in energy expenditure in response to transgenic (She et al., 2007) or nutritional (Zhang et al., 2007) manipulation of BCAA or leucine levels, respectively. To investigate this in our own models, we measured total energy expenditure (VO2) and RER (RQ) in rats fed on the HF/BCAA, HF, and SC diets. Feeding of either the HF or HF/BCAA diets lowered VO2 in the dark cycle only, and RER (RQ) in both the dark and light cycles, to similar extents relative to the SC diet (Figure 3). However, no differences were observed in these variables in HF/BCAA versus HF-fed rats.
Figure 3. Indirect calorimetry analysis.

A) RER (RQ); C) VO2 measured continuously over a 34 h period that includes a typical dark/light cycle. Panels B, D represent the average RER (RQ) and VO2, respectively. (*) p<0.05, SC group compared to the other two groups.
Mechanisms underlying BCAA effects in HF diet
Finally, we addressed the question of why BCAA supplementation of HF, but not SC diet contributes to insulin resistance. Recent studies have demonstrated that high fat feeding causes accumulation of a broad range of acylcarnitine species in skeletal muscle, indicative of incomplete oxidation of fatty acids in the mitochondria (Koves et al., 2008; Muoio and Newgard, 2008; An et al., 2004). Accumulation of these incompletely oxidized lipid species in muscle mitochondria may contribute to insulin resistance, since two manuevers that cause their clearance, exercise and transgenic knockout of malonyl CoA decarboxylase, lead to enhanced insulin action (Koves et al., 2008; Muoio and Newgard, 2008). Addition of BCAA to SC diet resulted in no change in a broad array of acylcarnitine species in skeletal muscle relative to animals fed on SC diet alone (Figure 4A). Consistent with our prior studies, HF feeding caused significant accumulation of multiple acylcarnitines in muscle relative to the SC or SC/BCAA groups (Figure 4A). Surprisingly, the HF/BCAA diet also caused accumulation of the same array of muscle acylcarnitines despite the fact that the HF/BCAA animals weighed less and consumed less food than the HF-fed animals. HF/BCAA feeding also caused increased skeletal muscle C3 and C5 acylcarnitine levels relative to all other groups.
Figure 4. BCAA supplementation of HF diet causes accumulation of acylcarnitines in skeletal muscle and chronic activation of mTOR and JNK.
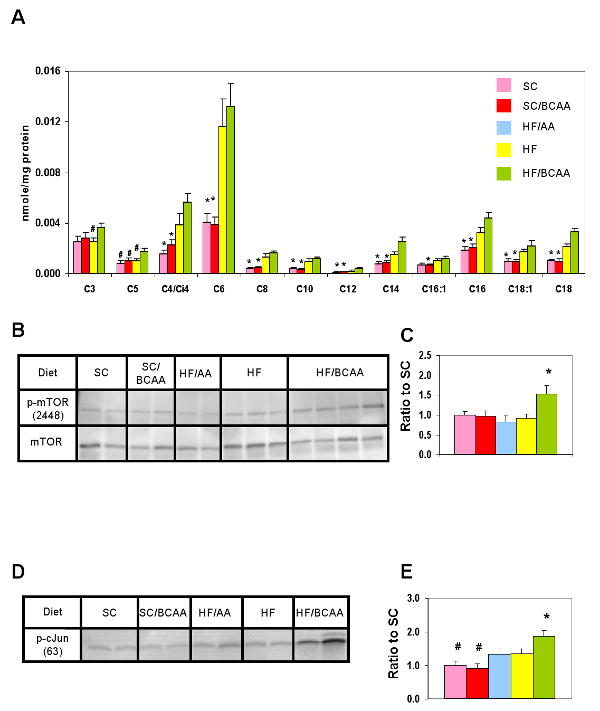
A) Skeletal muscle samples were collected from animals fed on the indicated diets and used for acylcarnitine analysis by MS/MS. Data represent the mean ± SEM for 6 animals per group. (*) p < 0.05 for comparison of SC and SC/BCAA to HF and HF/BCAA groups. B) Representative p-mTOR2448, immunoblot in muscle from overnight fasted SC, SC/BCAA, HF, HF/AA, and HF/BCAA-fed rats; C) Quantitative summary of p-mTOR2448 analyses; D) Representative p-cJUNser63 immunoblot in muscle from the same sets of animals studied in panel B; E) Quantitative summary of p-cJUN analyses. (*) p < 0.05 for comparison of HF/BCAA to the other groups. (#) p<0.05 for comparison of SC and SC/BCAA to the other groups. In panels C and E, n = 6-9 animals/group.
We also compared the effect of BCAA supplementation of the SC and HF diets at the level of mTOR activation in skeletal muscle from fasted rats. Addition of BCAA to the SC diet had no effect on mTOR activity relative to SC feeding alone, whereas BCAA addition to the HF diet increased mTOR activity relative to feeding of HF alone (Figures 4B and 4C). For these studies, the additional control of an isonitrogenous, pan-amino acid supplemented diet (HF/AA) was included, and muscle samples from these animals also had lower levels of phospho-mTOR than the HF/BCAA group, further demonstrating a specific effect of BCAA as opposed to overall nitrogen load.
Finally, the literature contains several examples of parallel regulation of JNK and mTOR and is suggestive of cross-talk between these proteins (Hiratani, et al. 2005; Mussig, et al., 2005). To investigate a possible involvement of JNK in HF/BCAA-mediated impairment of insulin action, muscle samples from all of the foregoing dietary groups were analyzed for JNK activity (Figures 4D and 4E). Relative to SC fed rats, SC/BCAA animals exhibited no increase in JNK activity. HF or HF/AA feeding caused a modest but significant increase in JNK activity relative to the SC groups, whereas HF/BCAA feeding caused a larger increase that was significant compared to all other groups.
Discussion
In the present study, we have applied a comprehensive set of analytical tools to gain a deeper understanding of biochemical, endocrine, inflammatory marker, and physiologic differences between obese and lean humans. A number of the obesity-related changes described herein confirm prior studies, including the higher levels of leptin and the lower levels of adiponectin and ghrelin, the increase in the inflammatory/cardiovascular risk marker, CRP, and the presence of insulin resistance in the obese subjects (Bjorbaek et al., 2004; Trujillo et al., 2006; Reaven et al., 2006; Ridker et al., 1998; Hosoda et al., 2006). However, other findings were not clearly anticipated, either due to inconsistency in the literature, or because novel patterns emerged from our analysis of multiple markers in a single study.
Whereas prior studies have generally indicated that hGH levels are reduced in obesity (Scacchi et al., 1999; Wittchen et al., 2005), IGF-1 levels have been variously reported to be reduced (Marin et al., 1993; Maccario et al., 1999; Schneider et al., 2006) or unchanged (Rasmussen et al., 1994; Maccario et al., 2001). In the current study, we found that obesity was associated with decreases in hGH, IGF-1, IGFBP-1 and IGFBP-2. We believe that these data in aggregate indicate a reduction in bioavailable IGF-1, supported by our novel finding of lower levels of free IGF-1 in obese compared to lean subjects (Table 1).
Hyperlipidemia has been ascribed a role in the pathogenesis of insulin resistance (Muoio and Newgard, 2008; Morino et al., 2006), and in the current study, several perturbations of lipid homeostasis were noted in obese subjects. Among individual free fatty acids, significant but modest increases were noted in endogenously synthesized (“non-essential”) long-chain species (C16:0, C16:1, C18:1) in obese subjects, but not in polyunsaturated (“essential”) species such as C18:2 (linoleic) or C18:3 (linolenic). In contrast, all of these fatty acid species and several more were dramatically elevated in the total fatty acid pool in obese subjects; this pool largely represents fatty acids that circulate in triglycerides or other esterified species, consistent with the strong increase in TG levels in the obese subjects. Accumulation of fatty acids in esterified products may have been driven by the increased propensity of the obese subjects to oxidize glucose and/or amino acids relative to lipids, consistent with their increased RQ. This would lead to increased tissue malonyl CoA levels and suppression of fatty acid oxidation in liver (McGarry, 2002), partitioning lipids towards esterification pathways. Sequestration of a large and diverse pool of fatty acids in esterified species would allow an enhanced rate of their delivery to peripheral tissues, possibly contributing to accumulation of lipid-derived metabolites such as diacylglyerols, ceramides, ketones, prostaglandins, and incompletely oxidized lipids that can impact insulin signaling (Muoio and Newgard, 2008; Morino et al., 2006; Summers, 2006).
We also identified a cluster of obesity-associated changes in specific amino acid, acylcarnitine, and organic acid metabolites in obese compared to lean subjects that was associated with insulin resistance. We hypothesize that these changes may reflect an overload of branched-chain amino acid (BCAA) catabolism in obese subjects. Subjects that become obese on a typical American diet, as in the current study, consume high levels of fat and protein, and dietary protein is comprised of > 20% BCAA (Layman et al., 2006). We propose that when coupled with overnutrition, the relative IGF-1 deficiency that we document in the obese subjects forces the now expanded circulating BCAA pool to be diverted away from protein synthesis and into catabolic pathways.
Consistent with this hypothesis, we observed that levels of the BCAAs valine and leucine/Isoleucine were 20% and 14% higher respectively, in obese compared to lean subjects (p < 0.0001 in both cases). As summarized in Figure 5, other changes also fit the model: 1) The decreased levels of α-ketoglutarate (α-KG) and the increased levels of glutamate. α-KG will be consumed and glutamate synthesized in the first step of BCAA catabolism, transamination; 2) The significant and specific increases in C3 and C5 acylcarnitine levels. C5 acylcarnitines are comprised of α-methylbutyryl and isovalerylcarnitine species; α-methylbutyryl CoA and isovaleryl CoA are intermediates in mitochondrial isoleucine and leucine catabolism, respectively, and these intermediates equilibrate with their cognate acylcarnitine esters. C3 acylcarnitine reflects the propionyl CoA pool; propionyl CoA is a byproduct of both isoleucine and valine catabolism; 3) A high rate of flux through BCAA catabolic pathways and accumulation of glutamate may increase transamination of pyruvate to alanine. Increases in alanine, a highly gluconeogenic amino acid, could contribute to development of glucose intolerance in obesity. Indeed, circulating pyruvate and alanine levels are clearly elevated in the obese subjects; 4) The aromatic amino acids, phenylalanine and tyrosine are elevated in obese compared to lean subjects. This may be explained by the fact that the “large neutral amino acids” (Trp, Phe, Tyr, Leu, Ile, Val), encompassing both BCAA and aromatic amino acids, compete for transport into mammalian cells by the large neutral amino acid transporter (LAT1) (Fernstrom et al., 2005). Assuming that chronic elevations in BCAA impair transport of aromatic amino acids into cells and tissues, this could contribute to reduced production of neurotransmitters such as serotonin (derived from tryptophan) and catecholamines (derived from phenylalanine and tyrosine) in the central nervous system. Indeed, imbalances in the serum molar ratio of tryptophan (Trp) to its large neutral amino acid competitors (sum of Phe + Leu + Ile + Val + Tyr), the so-called “tryptophan ratio” have been related to depression (Chevalier et al., 2005; Capuron et al., 2002) and obesity (Ashley et al., 1985; Breum et al., 2003). In the present study, the tryptophan ratio was depressed in obese versus lean subjects (0.138 versus 0.169, respectively, p<0.0001), driven by elevations in Phe, Tyr, Leu, Ile, and Val (Table 2), coupled with unchanged median tryptophan levels (obese 83.6 μM, lean 88.2 μM); 5) Finally, the exaggerated insulin secretion response to glucose (AIRG) in obese subjects may be due to potentiation of glucose-stimulated insulin secretion by BCAA (Newgard and Matschinsky, 2001).
Figure 5. Schematic Summary of BCAA overload hypothesis.

In the physiological context of overnutrition and low IGF-1 levels, as found in our obese subjects, circulating branched-chain amino acids (BCAA) rise, leading to increased flux of these amino acids through their catabolic pathways. We detected changes in several of the intermediary metabolites of the BCAA catabolic pathway in obese subjects, as indicated by the symbol *. A consequence of increased BCAA levels is the activation of the mTOR/S6K1 kinase pathway and phosphorylation of IRS-1 on multiple serines, contributing to insulin resistance. In addition, increased BCAA catabolic flux may contribute to increased gluconeogenesis and glucose intolerance via glutamate transamination to alanine.
It has been known for some time that obesity is accompanied by an increase in circulating levels of multiple amino acids, including BCAA (Felig et al., 1974; Chevalier, et al, 2005; Um, et al., 2006; Tremblay, et al, 2007; Krebs, et al., 2007). The effects of BCAA, particularly leucine, to activate mTOR and S6K1 are also well known (Um et al., 2006; Tremblay, et al, 2007; Krebs, et al., 2007). It is assumed that similar mechanisms are operative in vivo, based on studies involving infusion of complex amino acid mixtures at rates causing acute 2-7-fold elevations in the levels of 18 amino acids in rodents or humans, and demonstration that such maneuvers impair skeletal muscle glucose uptake and insulin action while increasing hepatic gluconeogenesis (Krebs et al., 2002; Tremblay et al., 2005; Krebs et al., 2003). However, because the experimental design of these foregoing studies resulted in non-physiological increases in a broad array of circulating amino acids, the specific impact of more modest changes in BCAA as observed in obese compared to lean humans could not be predicted.
Therefore, a novel feature of the current work is that that we have tested the effect of supplementation of HF diet with BCAA in amounts that induce a modest rise in BCAA in rodents similar to that observed in human obesity, leading to the demonstration that such changes are sufficient to cause a reduction in food intake, while simultaneously exerting a deleterious effect on insulin sensitivity that is independent of body weight. Moreover, our feeding study in rats was comprised of six arms (SC, SC/BCAA, HF, HF/BCAA, HF/PF, HF/AA) that allowed us to uniquely dissect the interactions between caloric intake and diet composition as it relates to insulin sensitivity. Interestingly, moderate consumption of a HF diet is required but not sufficient to induce impairment in insulin action, as shown by the HF/PF and SC/BCAA supplementation studies summarized in Supplemental Figures 2 and 3. We show that BCAA addition to the HF diet results in the same overaccumulation of even-chained acylcarnitine species in skeletal muscle as caused by HF diet alone, as well as specific accumulation of C3 and C5 acylcarnitine species, even though much less of the HF/BCAA diet was consumed. In contrast, BCAA supplementation of SC diet causes no accumulation of acylcarnitine species. Thus, supplementation of BCAA in a HF diet background may saturate capacity for mitochondrial fuel oxidation in muscle in the same fashion as occurs when HF diet is fed ad-libitum (Koves et al, 2008; Muoio and Newgard, 2008), leading to accumulation of incompletely oxidized lipid-derived metabolites.
If both the HF and HF/BCAA diets cause mitochondrial stress leading to insulin resistance, why does rapamycin reverse the HF/BCAA but not the HF-induced impairment in insulin action? We show that phosphorylation of mTOR, S6K1, and IRS1 Ser302 are all clearly and uniquely enhanced in fasted and refed HF/BCAA-fed animals compared to the other groups (Figure 2). Phosphorylation of IRS1 Ser307 is increased in both the HF and HF/BCAA groups in the fasted/refed state, but is specifically and chronically elevated only in the HF/BCAA group in the fasted state (Figure 2). Consistent with this, mTOR is also uniquely and chronically activated by HF/BCAA feeding relative to the other dietary groups in skeletal muscle from fasted animals, including the HF/AA group with isonitrogenous addition of all the amino acids (Figure 4). Finally, JNK is activated to a greater extent by HF/BCAA feeding than by the HF or HF/AA diets (Figure 4). Taken together, our data show that a common feature of HF and HF/BCAA feeding, the accumulation of mitochondrial acylcarnitines, lead to a common endpoint of insulin resistance, but mediated primarily by chronic activation of mTOR in the case of HF/BCAA feeding, possibly related to the stronger constitutive activation of JNK. This explains why the mTOR inhibitor rapamycin reverses HF/BCAA- but not HF-induced insulin resistance. We note that others have reported that high fat diets alone can lead to activation of mTOR, and that the effects of HF to impair insulin sensitivity are partially reversed by knock-out of S6K1 in mice (Tremblay, et al. 2007). However, S6K1 knockout also affects pancreatic islet function, and the potential additive effect of BCAA supplementation of HF diet, such as occurs in human overnutrition, was not modeled in those prior studies.
Two limitations of our study are acknowledged: 1) Our animal studies were performed in young rats on a rapid growth curve, whereas the human subjects were of middle age. This may help to explain why IGF-1 levels were lower in obese humans than in lean controls, but not in the HF or HF/BCAA rats versus the SC group. However, it can be argued that this makes our finding of BCAA-driven insulin resistance in the rodent model all the more impressive, since this occurred even in a background of normal IGF-1 levels; 2) Humans with type 2 diabetes are not only insulin resistant with regard to glucose metabolism, but are also refractile to insulin regulation of protein metabolism, a condition that is especially apparent in males (Pereira et al., 2008; Gougeon et al., 2008). The studies described here were performed in male rats; additional studies will be required to determine if our findings are also relevant to females.
Out findings should be discussed in the context of three recent studies from other laboratories. In the first, intracerebroventricular (ICV) infusion of leucine caused reduced food intake in mice (Cota et al., 2006), although the effect of dietary BCAA supplementation was not described. Other laboratories have reported that elevations of long-chain acyl CoA levels in the hypothalamus, mediated by increased levels of malonyl CoA, may also constitute an important satiety signal (He et al., 2006; Pocai et al., 2006). Interestingly, in the current study, food intake was reduced only by the HF/BCAA diet and not the HF/AA or SC/BCAA diets, consistent with the idea that lipid and BCAA-derived satiety signals cooperate to control feeding behavior.
A second study reported that mice with knock-out of the mitochondrial branched chain amino-transferase 2 (BCAT2) gene exhibit severe elevations of BCAA coupled with an increase in food consumption and resistance to diet-induced obesity, insulin resistance, and glucose intolerance (She et al., 2007). In addition to the wide disparity in increases in BCAA levels in the BCAT2 knockout mice relative to our BCAA-supplemented rats (14-37-fold versus 0.5-1.5-fold, respectively), a likely explanation for the differences in the two models is that the BCAT knock-out mouse have a complete block in BCAA catabolism, whereas BCAA catabolism is brisk in our HF/BCAA-fed rats. Leucine catabolism is required for this amino acid to inhibit proteolysis, but not protein synthesis (Tischler et al., 1982). The absence of leucine-mediated suppression of proteolysis in BCAT knock-out mice contributes to high rates of protein futile cycling, which consumes large amounts of energy. Thus, the increased food intake and resistance to diet-induced obesity in BCAT knock out mice is likely due to a high rate of glucose and fatty acid oxidation required to maintain ATP stores at a level sufficient to support protein futile cycling. In contrast, HF/BCAA-fed rats actually have lower rates of total energy expenditure (VO2) than SC-fed rats (Figure 4), allowing BCAA signaling pathways that lead to reduced food intake and insulin resistance to emerge. Moreover, lean humans trend towards higher energy expenditure when normalized for lean body mass relative to obese subjects (Table 1), the opposite of what was found in the BCAT2 knock-out mice. For these reasons, we believe that our feeding model is a more accurate reflection of the metabolic effects of BCAA overfeeding in human obesity.
Finally, a third study by Zhang, et al. reported that supplementation of leucine in the drinking water of mice had no effects in the context of SC diet, consistent with the current findings, but contrary to our results, caused an improvement in glucose tolerance when given to HF animals (Zhang et al., 2007). There are multiple differences between the two studies that should be noted: 1) We fed all 3 branched-chain amino acids as a supplement to the food, whereas Zhang, et al. gave leucine alone in the drinking water; 2) Leucine has no effect on consumption of HF diet in the Zhang, et al. study, whereas BCAA supplementation reduced consumption of the HF diet in our study. Conversely, Zhang et al report that leucine supplementation of HF diet increases energy expenditure (VO2) and slightly lowers RQ, whereas we find that HF/BCAA feeding does not affect VO2 or RQ relative to HF feeding; 3) Zhang et al. did not analyze insulin signaling pathways, so we cannot determine if mTOR, S6K, IRS-1 serine phosphorylation or other variables were altered by leucine supplementation. We again suggest that our study more faithfully models human obesity, in which all three BCAA are elevated and energy expenditure is not increased.
Our intent in applying a multidimensional analytical platform to the study of human obesity was to provide a deeper understanding of metabolism-related variables that differ between lean and obese individuals. The “BCAA overload” hypothesis just described, derived from human data and corroborated in an animal study, suggests that in the context of a poor dietary pattern that includes high fat consumption, BCAA may make an independent contribution to development of insulin resistance and diabetes.
Experimental Procedures
Subjects
We report baseline results from 74 obese individuals and 67 lean controls. Obese individuals enrolled from several local weight loss programs, including the Weight Loss Maintenance Study (WLM) (n=45), an NHLBI-sponsored multi-center clinical trial, the Duke University bariatric surgery program (n = 13), the Duke Diet and Fitness Center (n = 9), the low-carbohydrate, ketogenic diet program (n = 4), or Structure House (n = 3). These programs are described in full detail elsewhere (Haqq et al., 2005). Lean control subjects were recruited from the surrounding community and were selected for similar age, gender and race distribution compared to the 45 WLM enrollees. All study measurements were obtained before 10 am, and after an overnight fast.
Both obese and lean individuals were excluded from participation if they had significant cardiopulmonary, renal or liver disease, active malignancy, or were taking diabetes medication, systemic corticosteroids, or weight loss medication. The studies were approved by the Duke Institutional Review Board, and all participants provided written informed consent.
Anthropometric measurements
Weight, height and waist circumference were measured by standard methods.
Measures of behavior
Dietary intake was measured by self-administered Block Food Frequency Questionnaire (Harlan and Block, 1990). Usual physical activity was measured by the self-administered International Physical Activity Questionnaire (IPAQ) (Craig et al., 2003).
Physiologic measurements
Methods used for measurement of body composition by dual energy X-ray absorptiometry (DEXA) and abdominal computerized tomography (CT), and resting energy expenditure (REE) by indirect calorimetry have been described previously (Haqq, et al., 2005).
Insulin sensitivity was measured by Homeostasis Model Assessment (HOMA), using the formula: HOMA = (fasting insulin in uU/mL * fasting glucose in mM)/22.5 (Matthews et al., 1985). Insulin sensitivity (Si) and acute insulin response to glucose (AIRg) were measured by intravenous glucose tolerance test (IV-GTT) in a subset of 26 obese and 26 lean subjects, using the insulin-modified frequently sampled IVGTT (Bergman et al., 1981; Houmard et al., 2002).
Biochemical Measurements
Blood and urine samples were obtained after an overnight fast to measure the analytes described below. A detailed description of blood and urine sample preparation and coefficients of variation for these assays has been published (Haqq et al., 2005).
Hormone analysis
RIAs were performed for leptin, insulin, C-peptide, glucagon, total ghrelin, total adiponectin, and peptide YY (PYY) (Linco, St. Charles, MO), and Neuropeptide Y (NPY) (Alpco, Salem, NH) in plasma samples, and pancreatic polypeptide (PP) (Alpco), human growth hormone (hGH), total insulin-like growth factor-I (IGF-I) and 3 IGF-1 binding proteins (IGFBP1, IGFBP2, IGFBP3) (DSL, Webster, TX) in serum samples. ELISAs were used to meaure amylin and total gastric inhibitory polypeptide (GIP) (Linco), resistin (Biovendor, Candler, NC), and free IGF-1 (Diagnostic Systems Laboratories, Webster, TX) in plasma samples. Total GLP-1 was measured in plasma, using an assay from MesoScale Devices (Gaithersburg, MD),
Conventional metabolite analysis
Plasma glucose, lactate, total cholesterol, HDL- and LDL-cholesterol, and triglycerides were measured with kits from Roche Diagnostics (Indianapolis, IN), and free fatty acids (total) and ketones (total and 3-hydroxybutyrate) with kits from Wako (Richmond, VA). Plasma pyruvate was measured as described (Hansen and Freier, 1978).
Cytokine analysis
ELISA-based assays (Biosource, Camarillo, CA) were used to measure IL-6, IL-10, and TNF-α in serum. C-reactive protein (CRP) was measured in plasma using a kit from Roche.
Serum acylcarnitines and amino acids
Acylcarnitines and amino acids were analyzed by tandem mass spectrometry (MS/MS) as described previously (An et al., 2004; Millington et al., Wu et al., 2004, Chace, et al., 1995, Ferrara, et al., 2008).
Total fatty acids, serum free fatty acids, and organic acids
For determination of total fatty acids (free + esterified) in plasma samples, fatty acid residues were aggressively transesterified to their methyl esters in a solution of 4 % v/v acetyl chloride in methanol (Lepage and Roy, 1986). To measure non-esterified free fatty acids, separate serum samples were gently methylated using iodomethane and purified by solid-phase extraction (Patterson, et al, 1999). For analysis of urinary organic acids, analytes were extracted in ethyl acetate, dried, and then converted to trimethyl silyl esters by N,O-bis (trimethylsilyl) trifluoroacetamide, with protection of alpha-keto groups by oximation with ethoxyamine hydrochloride. Derivatized fatty and organic acids were analyzed by capillary gas chromatography/mass spectrometry (GC/MS) using a Trace DSQ instrument (Thermo Electron Corporation, Austin, TX).
All MS analyses employed stable-isotope-dilution with internal standards from Isotec (St. Louis, MO), Cambridge Isotope Laboratories (Andover, MA), and CDN Isotopes (Pointe-Claire, Quebec, CN). A list of all internal standards utilized in these studies has been published (Ferrara, et al., 2008).
Animals and dietary regimens
All procedures described in this article involving animals were approved by the Duke University Institutional Animal Care and Use Committee. Male Wistar rats (150-175 g, Charles River) were housed in pairs with free access to water and feeding of the various diets shown in Supplemental Table 1 for 12-16 weeks prior to biochemical and physiological analyses. We also performed a “refeeding and crossfeeding” study, in which we fed HF/BCAA, HF, or SC diets, fasted animals for 48 h, and then either refed the same diets as consumed prior to fasting, or crossfed, such that HF/BCAA-fed animals received HF diet during refeeding, and HF and SC-fed animals received HF/BCAA diet. All refeeding was performed for a period of 4 h. We also fed rats on HF/BCAA, HF or SC diet, fasted for 24 h, and injected with 0.5 mg/kg rapamycin dissolved in ethanol, or vehicle (ethanol) intraperitoneally 3 h prior to IPGTT. Food intake and body weights were monitored weekly.
Glucose and insulin tolerance tests in rats
For glucose tolerance tests, rats were fasted for twenty-four hours with free access to water and given an intraperitoneal injection of glucose (1 g/kg of body weight; 45% glucose). Blood was obtained from the tail vein immediately prior to glucose injection and every 30 minutes thereafter for a period of 180 minutes, and glucose levels were measured with BD Logic® glucose meter. For insulin tolerance test experiments, insulin (0.75 IU/kg body weight; Humulin regular, Eli Lilly) was injected intraperitoneally to ad lib-fed animals. Blood samples were obtained from the tail vein immediately prior to and every 30 minutes after the insulin injection for a period of 180 minutes.
Indirect Calorimetry
Indirect calorimetry was performed using an 8 chamber Oxymas system (Columbus Instruments, Columbus, OH, USA). Rats fed on the various diets described above were acclimatized to the system for 24 h prior to measurement.
Immunoblot analysis of insulin signaling proteins
Approximately 100 mg of gastrocnemius muscle or liver samples were homogenized and prepared for electrophoresis as described (An, et al., 2004). Blots were incubated overnight at 4 °C with anti-AKT, anti-phospho (Ser 473)-AKT, anti-IRS1, anti-phospho (Ser 302)-IRS1, anti-phospho (Ser307)-IRS1, anti-mTOR, anti-phospho (Ser 2448)-mTOR, anti-S6K1, or anti-phospho (Thr 389)-S6K1 antibodies (New England Biolabs). Signals were detected using HRP-conjugated secondary antibody and the ECL™ Western Blotting Analysis System (Amersham Biosciences, Piscataway, NJ) and quantified with the VersaDoc Imaging system (Bio-Rad). JNK assay was performed using a nonradioactive JNK kinase assay kit (New England Biolabs).
Statistical analysis
For the human metabolic profiling data, results are presented as medians and interquartile ranges, and Wilcoxon rank sum tests were used to compare all individual analytes in lean versus obese subjects. We also performed unsupervised multivariate analyses (principal components analysis, PCA) using software procedures in SAS version 9.1 (SAS Institute, Inc., Cary, NC). The level of each of the PCA-derived metabolite components was then compared in lean versus obese subjects using Wilcoxon rank sum testing. In addition, correlation between PCA-derived components and insulin resistance (as measured by HOMA) was assessed. Details of the PCA and correlation analyses are described in Supplementary Materials. In analysis of the human profiling data, as a reasonable compromise between type-I (false positive) and type-II (false negative) error, a nominal p-value of 0.01 was used for declaring a statistically significant result, with no adjustment for multiple comparisons.
For the animal feeding studies, data are presented as averages ± standard errors, and student T-test was used for statistical analysis.
Supplementary Material
Acknowledgments
This work was supported by a sponsored research agreement from Glaxo SmithKline, and we would like to thank Drs. Derek Nunez, Jeff Cobb, William Wilkison, Nandu Gattu, Terry Walker, Nick Livingston, and Kenneth Batchelor for their support and advice. The work was also supported by NIH grant 1K23-RR-021979 (to AMH). Some participants in this study enrolled in the Weight Loss Maintenance Study (WLM) at Duke, an NHLBI-sponsored multi-center clinical trial, supported by U01 HL068734-02 (to LPS). The authors also gratefully acknowledge Laveina Dash, Tonya Milligan, Lori Aiken, Chelle Yin, Janice Lee, Lorraine Elliott, Lauren C Naliboff, Erin L Chu, Dr William Kraus, and Dr Jarol Boan for their advice and technical assistance during this study. We thank Drs Larry Gene Moss, Debbie Muoio, Jens Juul Holst, and Thomas Coffman for critical review of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- An J, Muoio DM, Shiota M, Fujimoto Y, Cline GW, Shulman GI, Koves TR, Stevens R, Millington D, Newgard CB. Hepatic expression of malonyl-CoA decarboxylase reverses muscle, liver and whole-animal insulin resistance. Nat Med. 2004;10:268–74. doi: 10.1038/nm995. [DOI] [PubMed] [Google Scholar]
- Ashley DVM, Fleury MO, Golay A, Maeder E, Leathwood PD. Evidence for diminished brain 5-hydroxytryptamine biosynthesis in obese diabetic and non-diabetic humans. Am J Clin Nutr. 1985;42:1240–1245. doi: 10.1093/ajcn/42.6.1240. [DOI] [PubMed] [Google Scholar]
- Bergman RN, Phillips LS, Cobelli C. Physiologic evaluation of factors controlling glucose tolerance in man: Measurement of insulin sensitivity and beta-cell glucose sensitivity from the response to intravenous glucose. J Clin Invest. 1981;68:1456–67. doi: 10.1172/JCI110398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorbaek C, Kahn BB. Leptin signaling in the central nervous system and periphery. Recent Prog Horm Res. 2004;59:305–331. doi: 10.1210/rp.59.1.305. [DOI] [PubMed] [Google Scholar]
- Breum L, Rasmussen MH, Hilsted J, Fernstrom JD. Twenty-four hour plasma tryptophan concentrations and ratios are below normal in obese subjects and are not normalized by substantial weight reduction. Am J Clin Nutr. 2003;77:1112–1118. doi: 10.1093/ajcn/77.5.1112. [DOI] [PubMed] [Google Scholar]
- Cao H, Gerhold K, Mayers JR, Wiest MM, Watkins SM, Hotamisligil GS. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell. 2008;134:933–944. doi: 10.1016/j.cell.2008.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuron L, Ravaud A, Neveu PJ, Miller AH, Maes M, Dantzer R. Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol Psych. 2002;7:468–473. doi: 10.1038/sj.mp.4000995. [DOI] [PubMed] [Google Scholar]
- Chace DH, Hillman SL, Millington DS, Kahler SG, Roe CR, Naylor EW. Rapid diagnosis of maple syrup urine disease in blood spots from newborns by tandem mass spectrometry. Clin Chem. 1995;41:62–68. [PubMed] [Google Scholar]
- Chevalier S, Marliss EB, Morais JA, Lamarche M, Gougeon R. Whole-body protein anabolic response is resistant to the action of insulin in obese women. Am J Clin Nutr. 2005;82:355–365. doi: 10.1093/ajcn.82.2.355. [DOI] [PubMed] [Google Scholar]
- Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312:927–930. doi: 10.1126/science.1124147. [DOI] [PubMed] [Google Scholar]
- Craig CL, Marshall AL, Sjostrom M, Bauman AE, Booth ML, Ainsworth BE, Pratt M, Ekelund U, Yngve A, Sallis JF, Oja P. International Physical Activity Questionnaire: 12-country reliability and validity. MedSci Sports Exerc. 2003;35:1381–95. doi: 10.1249/01.MSS.0000078924.61453.FB. [DOI] [PubMed] [Google Scholar]
- Felig P, Wahren J, Hendler R, Brundin T. Splanchnic glucose and amino acid metabolism in obesity. J Clin Invest. 1974;53:582–590. doi: 10.1172/JCI107593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernstrom JD. Branched-chain amino acids and brain function. J Nutr. 2005;135:1539S–1546S. doi: 10.1093/jn/135.6.1539S. [DOI] [PubMed] [Google Scholar]
- Ferrara CT, Wang P, Neto EC, Stevens RD, Bain JR, Wenner BR, Ilkayeva OR, Keller MP, Blasiole DA, Kendziorski C, Yandell BS, Newgard CB, Attie AD. Genetic network of liver metabolism revealed by integration of metabolic and transcriptional profiling. PLoS Genetics. 2008;14:e1000034. doi: 10.1371/journal.pgen.1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gougeon R, Morais JA, Chevalier S, Pereira S, Lamarche M, Marliss EB. Determinants of whole-body protein metabolism in subjects with and without type 2 diabetes. Diabetes Care. 2008;231:128–133. doi: 10.2337/dc07-1268. [DOI] [PubMed] [Google Scholar]
- Harlan LC, Block G. Use of adjustment factors with a brief food frequency questionnaire to obtain nutrient values. Epidemiology. 1990;1:224–31. doi: 10.1097/00001648-199005000-00008. [DOI] [PubMed] [Google Scholar]
- Haqq AM, Lien LF, Boan J, Arlotto M, Slentz CA, Muehlbauer MJ, Rochon J, Gallup D, McMahon RL, Bain JR, Stevens R, Millington D, Butler MD, Newgard CB, Svetkey LP. The Study of the Effects of Diet on Metabolism and Nutrition (STEDMAN) weight loss project: rationale and design. Contemp Clin Trials. 2005;26:616–25. doi: 10.1016/j.cct.2005.09.003. [DOI] [PubMed] [Google Scholar]
- He W, Lam TK, Obici S, Rossetti L. Molecular disruption of hypothalamic nutrient sensing induces obesity. Nature Neuro. 2006;9:227–233. doi: 10.1038/nn1626. [DOI] [PubMed] [Google Scholar]
- Hedley AA, Ogden CL, Johnson CL, Carroll MD, Curtin LR, Flegal KM. Prevalence of overweight and obesity among US children, adolescents, and adults, 1999-2002. JAMA. 2004;291:2847–50. doi: 10.1001/jama.291.23.2847. [DOI] [PubMed] [Google Scholar]
- Hiratani K, Haruta T, Tani A, Kawahara J, Usui I, Kobayashi M. Role of mTOR and JNK in serine phosphorylation, translocation and degradation of IRS-1. Biochem Biophys Res Commun. 2005;30:836–842. doi: 10.1016/j.bbrc.2005.07.152. [DOI] [PubMed] [Google Scholar]
- Hosoda H, Kojima M, Kangawa K. Biological, physiological, and pharmacological aspects of ghrelin. J Pharmacol Sci. 2006;100:398–410. doi: 10.1254/jphs.crj06002x. [DOI] [PubMed] [Google Scholar]
- Houmard JA, Tanner CJ, Yu C, Cunningham PG, Poires WJ, MacDonald KG, Shulman GI. Effect of weight loss on insulin sensitivity and intramuscular long-chain fatty acyl-CoAs in morbidly obese subjects. Diabetes. 2002;51:2959–2963. doi: 10.2337/diabetes.51.10.2959. [DOI] [PubMed] [Google Scholar]
- Kahn SE, Zinman B, Haffner SM, O'Neill MC, Kravitz BG, Yu D, Freed MI, Herman WH, Holman RR, Jones NP, Lachin JM, Viberti GC, ADOPT Study Group Obesity is a major determinant of the association of C-reactive protein levels and the metabolic syndrome in type 2 diabetes. Diabetes. 2006;55:2357–64. doi: 10.2337/db06-0116. [DOI] [PubMed] [Google Scholar]
- Kaiser HF. The application of electronic computers to factor analysis. Educational and Psychological Measurement. 1960;20:141–151. [Google Scholar]
- Krebs M, Krssak M, Bernroider E, Anderwald C, Brehm A, Meyerspeer M, Nowotny P, Roth E, Waldhausl W, Roden M. Mechanism of amino acid-induced skeletal muscle insulin resistance in humans. Diabetes. 2002;51:599–605. doi: 10.2337/diabetes.51.3.599. [DOI] [PubMed] [Google Scholar]
- Krebs M, Brehm A, Krssak M, Anderwald C, Bernroider E, Nowotny P, Roth E, Chandramouli V, Landau BR, Waldhausl W, Roden M. Direct and indirect effects of amino acids on hepatic glucose metabolism in humans. Diabetologia. 2003;46:917–925. doi: 10.1007/s00125-003-1129-1. [DOI] [PubMed] [Google Scholar]
- Krebs M, Brunmair B, Brehm A, Artwohl M, Szendroedi J, Nowotny P, Roth E, Furnsinn C, Promintzer M, Anderwald C, Bischof M, Roden M. The mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes. 2007;56:1600–1607. doi: 10.2337/db06-1016. [DOI] [PubMed] [Google Scholar]
- Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Lopaschuk GD, Muoio DM. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal musle insulin resistance. Cell Metabolism. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Layman DK, Walker DA. Potential importance of leucine in treatment of obesity and the metabolic syndrome. J Nutr. 2006;136:319S–323S. doi: 10.1093/jn/136.1.319S. [DOI] [PubMed] [Google Scholar]
- Lepage G, Roy CC. Direct transesterification of all classes of lipids in a one-step reaction. J Lipid Res. 1986;27:114–20. [PubMed] [Google Scholar]
- Maccario M, Ramunni J, Oleandri SE, Procopio M, Grottoli S, Rossetto R, Savio P, Aimaretti G, Camanni F, Ghigo E. Relationship bewteen IGF-1 and age, gender, body mass, fat distribution, metabolic and hormonal variables in obese patients. Int J Obes Relat Metab Disord. 1999;23:612–618. doi: 10.1038/sj.ijo.0800889. [DOI] [PubMed] [Google Scholar]
- Maccario M, Tassone F, Gauna C, Oleandri SE, Aimaretti G, Procopio M, Grottoli S, Pflaum CD, Stasburger CJ, Ghigo E. Effects of short-term administration of low-dose rhGH on IGF-1 levels in obesity and Cushing's syndrome: indirect evaluation of sensitivity to GH. Eur J Endocrinology. 2001;144:251–256. doi: 10.1530/eje.0.1440251. [DOI] [PubMed] [Google Scholar]
- Marin P, Kvist H, Lindstedt G, Sjostrom L, Bjorntorp P. Low concentrations of insulin-like growth factor-1 in abdominal obesity. Int J Obes Relat Metab Disord. 1993;17:83–89. [PubMed] [Google Scholar]
- McGarry JD. Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes. 2002;51:607–614. doi: 10.2337/diabetes.51.1.7. [DOI] [PubMed] [Google Scholar]
- Millington DS, Kodo N, Norwood DL, Roe CR. Tandem mass spectrometry: a new method for acylcarnitine profiling with potential for neonatal screening for inborn errors of metabolism. J Inherit Metab Dis. 1990;13:321–4. doi: 10.1007/BF01799385. [DOI] [PubMed] [Google Scholar]
- Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. 2006;(Suppl):S9–S15. doi: 10.2337/db06-S002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muoio DM, Newgard CB. Molecular and metabolic mechanisms of insulin resistance and β-cell failure in type 2 diabetes. Nature Rev Mol Cell Biol. 2008;9:193–205. doi: 10.1038/nrm2327. [DOI] [PubMed] [Google Scholar]
- Mussig K, Fiedler H, Staiger H, Weigert C, Lehmann R, Schleicher ED, Haring HU. Insulin-induced stimulation of JNK and the PI 3-kinase/mTOR pathway leads to phosphorylation of serine 318 or IRS-1 in C2C12 myocytes. Biochem Biophys Res Comm. 2005;335:819–825. doi: 10.1016/j.bbrc.2005.07.154. [DOI] [PubMed] [Google Scholar]
- Newgard CB, Matschinsky FM. Substrate control in insulin release. In: Jefferson J, Cherrington A, editors. Handbook of Physiology. Oxford University Press; 2001. pp. 125–152. [Google Scholar]
- Patterson BW, Zhao G, Elias N, Hachey DL, Klein S. Validation of a new procedure to determine plasma fatty acid concentration and isotopic enrichment. J Lipid Res. 1999;40:2118–2124. [PubMed] [Google Scholar]
- Pereira S, Marliss EB, Morais JA, Chevalier S, Gougeon R. Insulin resistance of protein metabolism in type 2 diabetes. Diabetes. 2008;57:56–63. doi: 10.2337/db07-0887. [DOI] [PubMed] [Google Scholar]
- Pocai A, Lam TK, Obici S, Gutierrez-Juarez R, Muse ED, Arduini A, Rossetti L. Restoration of hypothalamic lipid sensing normalizes energy and glucose homeostasis in overfed rats. J Clin Invest. 2006;116:1081–1091. doi: 10.1172/JCI26640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen MH, Frystyk J, Andersen T, Breum L, Christiansen JS, Hilsted J. The impact of obesity, fat distribution, and energy restriction on insulin-like growth factor-1, IGF binding protein-3, insulin, and growth hormone. Metabolism. 1994;43:315–319. doi: 10.1016/0026-0495(94)90099-x. [DOI] [PubMed] [Google Scholar]
- Reaven GM. The metabolic syndrome: is this diagnosis necessary? Am J Clin Nutr. 2006;83:1237–47. doi: 10.1093/ajcn/83.6.1237. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Buring JE, Shih J, Matias M, Hennekens CH. Prospective study of C-reactive protein and the risk of future cardiovascular events among apparently healthy women. Circulation. 1998;98:731–3. doi: 10.1161/01.cir.98.8.731. [DOI] [PubMed] [Google Scholar]
- Scacchi M, Pincelli A, Cavagnini F. Growth hormone in obesity. Int J Obesity. 1999;23:260–271. doi: 10.1038/sj.ijo.0800807. [DOI] [PubMed] [Google Scholar]
- Schneider HJ, Saller B, Kotsche J, Marz W, Erwa W, Wittchen HU, Stalla GK. Opposite associations of age-dependent insulin-like growth factor-I standard deviation scores with nutritional state in normal weight and obese subjects. Eur J Endocrinology. 2006;154:699–706. doi: 10.1530/eje.1.02131. [DOI] [PubMed] [Google Scholar]
- She P, Reid TM, Bronson SK, Vary TC, Hajnal A, Lynch CJ, Hutson SM. Disruption of BCATm in mice leads to increased energy expenditure associated with activation of a futile protein turnover cycle. Cell Metabolism. 2007;6:181–194. doi: 10.1016/j.cmet.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowers JR. Obesity as a cardiovascular risk factor. Am J Med. 2003;115(8A):37S–41S. doi: 10.1016/j.amjmed.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 2006;45:42–72. doi: 10.1016/j.plipres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Tischler ME, Desautels M, Goldberg AL. Does leucine, leucyl-tRNA, or some metabolite of leucine regulate protein synthesis and degradation in skeletal and cardiac muscle? J Biol Chem. 1982;257:1613–1621. [PubMed] [Google Scholar]
- Tremblay F, Krebs M, Dombrowski L, Brehm A, Bernroider E, Roth E, Nowotny P, Waldhausl W, Marette A, Roden M. Overactivation of S6 kinase 1 as a cause of human insulin resistance during increased amino acid availability. Diabetes. 2005;54:2674–2684. doi: 10.2337/diabetes.54.9.2674. [DOI] [PubMed] [Google Scholar]
- Tremblay F, Brule S, Um SH, Li Y, Masuda K, Roden M, Sun XJ, Krebs M, Polakiewicz RD, Thomas G, Marette A. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc Natl Acad Sci USA. 2007;104:14056–14061. doi: 10.1073/pnas.0706517104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay F, Lavigne C, Jacques H, Marette A. Role of dietary proteins and amino acids in the pathogenesis of insulin resistance. Ann Rev Nutr. 2007;27:293–310. doi: 10.1146/annurev.nutr.25.050304.092545. [DOI] [PubMed] [Google Scholar]
- Trujillo ME, Scherer PE. Adipose tissue-derived factors: impact on health and disease. Endocrine Reviews. 2006 Dec 7; doi: 10.1210/er.2006-0033. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Um SH, D'Alessio D, Thomas G. Nutrient overload, insulin resistance and ribosomal protein S6 kinase 1, D6K1. Cell Metab. 2006;3:393–402. doi: 10.1016/j.cmet.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittchen HU, Glaesmer H, Marz W, Stalla GK, Lennert H, Zeiher AM, Silber S, Koch U, Boher S, Pittrow D, Ruf G, DETECT study group Cardiovascular risk factors in primary care patients: methods and baseline prevalence results from the DETECT program. Current Medical Research and Opinion. 2005;12:619–629. doi: 10.1185/030079905X38187. [DOI] [PubMed] [Google Scholar]
- Wu JY, Kao HJ, Li SC, Stevens R, Hillman S, Millington D, Chen YT. ENU mutagenesis identifies mice with mitochondrial branched-chain aminotransferase deficiency resembling human maple syrup urine disease. J Clin Invest. 2004;113:434–40. doi: 10.1172/JCI19574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Guo K, LeBlanc RE, Loh D, Schwartz GJ, Yu YH. Increasing dietary leucine intake reduces diet-induced obesity and improves glucose and cholesterol metabolism in mice via multimechanisms. Diabetes. 2007;56:1647–1654. doi: 10.2337/db07-0123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.