

Abstract
We report the mode of action of a proteomimetic compound that binds to the exterior surface of gp120 and blocks HIV-1 entry into cells. Using a one cycle time-of-addition study and antibody competition binding studies, we have determined that the compound blocks HIV-1 entry through modulation of key protein-protein interactions mediated by gp120. The compound exhibits anti-HIV-1 replication activities against several pseudotype viruses derived from primary isolates and the resistant strains isolated from existing drug candidates with equal potency. Together, these data provide evidence that the proteomimetic compound represents a novel class of HIV-1 viral entry inhibitor that functions through protein surface recognition in analogy to an antibody.
Keywords: calix[4]arene scaffold, HIV-entry inhibitor, proteomimetic inhibitor, protein surface recognition
The development of HIV-1 therapies remains a formidable challenge and is of paramount importance in the search for maximal efficacy and minimal resistance. The majority of the well-established HIV-1 drugs target three main viral enzymes: reverse transcriptase, integrase and protease 1. However, the emergence of multi-drug resistant HIV-1 strains has propelled the development of new drug candidates with novel mechanisms of inhibition, such as HIV-entry and fusion inhibitors 2. Nearly 20% of newly diagnosed HIV-1 patients show resistance to the existing drug classes 3. Despite the elucidation of the molecular machinery involved both in HIV-entry and-fusion steps 4, enfuvirtide5 and maraviroc6 are the only entry- inhibitors approved by the FDA. We report herein a novel proteomimetic inhibitor of HIV-1 viral entry and the characterization of its antiviral activities against a panel of HIV-1 Envs pseudoviruses from primary isolates, its inhibitory mechanism and its retained potency against strains that are insensitive to a known gp120/CD4 inhibitor.
HIV viral entry into a host cell requires three main stages: binding of the gp120 protein on the viral envelope to CD4 cell receptor, a conformational change in gp120 that permits binding to other receptors on the cell, and a conformational change in gp41 that leads to fusion of the envelope and host cell membrane 4,7,8. A broad diversity of neutralizing antibodies have been generated from memory B cells in response to HIV in HIV-infected patients. Many of these high affinity neutralizing antibodies are targeted to the gp120 variable loops, the CD4-binding site, and the co-receptor-binding site 9. These studies validate that host response to HIV-entry and-fusion comprises multiple antibody responses with neutralizing activities against several epitopes on gp120. The interaction of viral gp120 with the CD4 receptor on the cell surface thus provides a viable target 10. A soluble form of CD4 was first designed to interrupt this process 11, however, its low activity and rapid clearance impeded further development of the approach. An immunoglobulin molecule (PRO-542) 12,13 containing the gp120-binding motif was developed and resulted in a longer plasma life. Concurrent efforts have focused on designing effective, potent and selective small molecules that inhibit HIV entry. FDA approved HIV-fusion inhibitor, enfuvirtride, binds to an intermediate in the fusion process and prevents it from proceeding thus inhibiting replication of HIV. Meanwhile, BMS-378806 was shown to potently inhibit both laboratory-adapted HIV-1 strains and retain high activity against primary isolates 14, 15. The resistant viral strain derived from BMS-378806 has also been profiled and used to confirm a mechanism that involves the targeting of viral entry by inhibition of the binding of HIV envelope gp120 protein to CD4 receptors via a specific and competitive mechanism with a 1:1 stoichiometry, similar to that of the soluble CD416. The other two compounds originally claimed as HIV integrase inhibitors, Zintevir 17, 18 and L-chicoric acid19, were later shown to act as inhibitors of gp120-CD4 binding, with efficacy in the low micromoler range. In surveying the spectrum of HIV inhibitors, the development of new molecular scaffolds that target viral entry with broad activities would be highly desirable.
Herein, we describe a novel strategy based on a proteomimetic approach that targets key protein-protein interactions in HIV-entry and results in the inhibition of HIV replication. The discontinuous character of critical residues over a large area of the protein-protein interface makes their replacement by peptides or peptidomimetic derivatives challenging. To prepare effective synthetic scaffolds, we adopted an antibody mimetic approach involving the generation of macrocycles with controlled molecular dimensions to complement multiple groups on the surface of gp120.20-22 This biomolecule mimetic strategy to therapeutic design has recently been comprehensively reviewed23. By mimicking interactions within the gp120 domain using synthetic molecules, we expected to modulate the HIV-entry process. We describe the development and evaluation of a potent macrocyclic inhibitor that blocks HIV-entry, results in inhibition of HIV replication in primary isolates and maintains potency against strains resistant to existing drug candidates.
We have recently described compound 1 (Figure 1A) as a new sub-micromolar inhibitor of HIV infection24. Compound 1, based on a tetrabutoxy-calix[4]arene scaffold, adopts a cone conformation and the projected aromatic isophthalate spacers at the upper rim play an essential role in its anti-HIV activity24. Moreover, compound 1 also retains potency against laboratory HIV strains in different cell lines while maintaining low cytotoxicity. In the present study, we have investigated the stage in the infection process on which compound 1 exerts its antiviral activity. HIV replication requires many steps, including entry of the virus, reverse transcription of RNA, and integration of DNA in succession25. Since the anionic periphery at the upper rim of 1 likely reduces its cell-permeability, we hypothesized that it targets extracellular complexes. To address the point of action, we performed cell-based time of addition assays over one HIV replication cycle (Figure 1B). Compound 1 at two different concentrations was added one hour before, simultaneously with, and three hours after HIV-1 IIIB infection and one viral replication cycle was monitored in TZM-b1 cells. We observed dose-dependent responses in the inhibition of viral replication by 1 at both 1μM and 0.33μM concentrations. There was a correlation in the potency of 1 depending whether it was added one-hour before, simultaneously with or three-hours after viral infection, confirming that it was acting at the viral entry stage.
Figure 1.

Results for the time-of-addition inhibition of HIV-replication by compound 1. (A) Molecular structures of compound 1 and compound 2. (B) Time-dependent inhibition of one-cycle HIV-replication at two different concentrations, 1μM (gray) and 0.33μM (black), of compound 1.
A key first step in the viral entry process involves the protein-protein interaction between the CD4 receptor on the cell surface and gp120 on the HIV envelope. The possibility that compound 1 functions by blocking CD4 and gp120 binding was investigated by using an ELISA assay (Figure 2) where the inhibition of binding between pre-coated soluble CD4 and HIV-1 IIIB gp120 is expressed as percent of control binding in the absence of inhibitors. Compound 1 demonstrated a dose-dependent inhibition of CD4 and GP120 association with an EC50 in the submicromolar range (Figure 2). The importance of eight negative charges on the upper rim of 1 was shown by the weak activity of tetraacid tetraester 2 (Figure 1A) which had an EC50 of >70 uM (Figure 2), consistent with the reported in vitro SAR studies 24. These results suggest that the observed modulation of HIV replication by compound 1 is due to its blocking the binding of soluble CD4 to HIV IIIB gp120.
Figure 2.

Results for the inhibition of CD4-GP120 binding. Dose-dependent inhibition of CD4-GP120 binding by compound 1 (blue) and compound 2 (magenta). Each data point in the figure represents mean +/− SD of 5 independent experiments.
To investigate the possible molecular target and binding site for compound 1 at the viral entry stage, we employed gp120-antibody competition experiments and docking studies. From the crystal structure it appears that the third variable region (V3) of HIV-1 gp120 is essential for coreceptor binding 8. Binding of gp120 to cell-surface CD4 positions the V3 loop such that its coreceptor-binding tip protrudes 30 Å from the core toward the target cell membrane. Monoclonal antibody 447-52D is a well-defined HIV-1 V3 antibody whose antigenic site is located at the tip of V3 loop (GPGR) of gp120. Compound 1 is able to block the binding of antibody 447-52D to the loop of gp120 in a dose-dependent manner (Figure 3). Negative control 2 shows no effect confirming that the binding of 1 to gp120 is dependent on the overall peripheral charges on its upper rim. Furthermore, another gp120 glycan binding monoclonal antibody 2G12 was used to address the specificity of binding between 1 and gp120. Increasing concentrations of 1 did not affect the interaction between antibody 2G12 and the glycoprotein regions of gp120. Similar inhibition potency for 1 was observed with two other V3 loop-binding antibodies, 39F and PK70, suggesting that the principal target for 1 is a site near or within the V3 loop of gp120. We show the specificity as compound 1 cannot inhibit the binding between gp120 and glycan antibody 2G12. Rather, our experimental data suggests that it can inhibit the binding between V3 loop and V3 antibodies. Since the interaction between V3 and V3 antibodies is quite specific, we reason that V3 loop can be a potential target site for compound 1. Collectively, these data suggest that the compound can interact with V3 loop.
Figure 3.
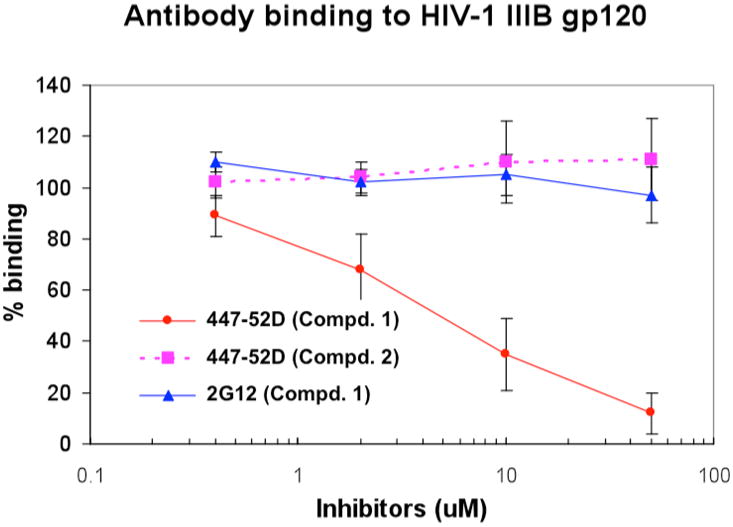
Inhibition of antibodies (447-52D and 2G12) binding to gp120 by compound 1 and compound 2. Each-data point in the figure represents Mean +/− SD of 5 independent experiments.
Further insight into the possible interaction site of 1 with gp120 was gained through computational docking studies. Compound 1 is polyanionic and it is reasonable to predict that it can interact with other structural motifs on gp120. Using the crystal structure reported by Kwong et al. 4 and the MOE molecular modeling program, 1 was allowed to dock with and sample the whole surface of gp120. In the highest scoring structure, 1 adopts a cone conformation with the polar periphery pointed towards the solvent-exposed surface of gp120 and making contact with the region where the neutralizing antibody 17b binds (Figure 4A). Superimposing antibody 17b on the docked structure shows an overlap on the surface of gp120 of the putative binding domain for 1 and that of the antibody. The binding interface between the antibody 17b and gp120 involves a complementary central hydrophobic core with an array of peripheral electrostatic interactions. The antibody contact surface is very acidic, containing three Asp and three Glu residues, although hydrophobic contacts predominate at the centre (Figure 4B). The corresponding gp120 surface that contacts the antibody contains of a hydrophobic centre surrounded by a highly basic periphery (3 Lys, 1 Arg). These docking studies suggest that compound 1, with anionic peripheral functionalities, has the potential to bind to the CD4 induced site on gp120.
Figure 4.

Docking study of compound 1 binding to gp120. (A) CD4 is shown in ribbon (yellow). gp120 is shown in solid surfaces. The antibody 17b is shown in stick representation (grey and magenta). Docking of compound 1 onto the crystal structure. (B) Close up of the region where compound 1 docks onto gp120. Shown in ribbons: gp120 (blue), CD4 (yellow). The negatively charged polar residues of the antibody 17b (grey and magenta ribbons) contacting gp120 are shown as red sticks.
Using the CD4/gp120 ELISA binding assay, antibody competition experiments and docking studies, we have identified possible recognition sites for compound 1 on gp120. To gain insight into the binding region, we have attempted to isolate HIV resistant strains cultured in the presence of compound 1 but have not been successful. Our data suggest that multiple interactions are needed at the potential binding site, which may contribute to the slow development of the resistant strains. A future goal will be to determine the exact region of gp120 that receives the most selective pressure for mutation and then to use site directed mutagenesis and co-crystalization to identify exact site of gp120 binding. Such an effort should lead to the development of new generations of inhibitors with asymmetric functionalities to increase potentcy. To establish if compound 1 could provide a broad coverage against a panel of clinical isolates with inherent heterogeneity of the HIV-1 glycoprotein, we evaluated the antiviral potencies of compound 1 towards two different clades of clinical isolates. We then demonstrated that the anti-HIV activities of compound 1 against laboratory HIV strains could be further translated to the primary isolates strain from human patients (Table 1). Compound 1 exhibits potent antiviral activities against pseudotype viruses with genetically distinct Envs derived from subtypes B and C with a wide spectrum of activity.
Table 1.
Results of compound 1 against pseudotype viruses with Env from primary HIV-1 isolates.
| Clade B | Compound 1(uM) |
|---|---|
| NL4-3 | 0.65 +/− 0.07 |
| 5768.4 | 4.1 +/−0.71 |
| 7165.18 | 4.5 +/− 0.62 |
| SS1196.1 | 2.8 +/− 0.86 |
| RHPA4259.7 | 3.1 +/−0.82 |
| RE J 04541.67 | 1.9+/−0.42 |
| WIT04160.33 | 1.8+/−0.37 |
| Clade C | |
| Du123.6 | 1.4+/−0.18 |
| Du151.2 | 3.1 +/−0.56 |
| Du422.1 | 1.8+/−0.31 |
| ZM197M.PB7 | 2.7 +/− 0.49 |
| ZM109F.PB4 | 3.9 +/− 0.36 |
BMS-378806 is a recently discovered small molecule that targets the binding of host-cell CD4 with viral gp120 protein and inhibits the first steps of HIV-1 infection 15,17. However, resistant strains have also been isolated against this compound. We envisioned that multi-site binding on gp120 could lead to compound 1 being less sensitive to the mutations found in these HIV strains. To test this, we compared the ability of 1 to inhibit viral fusion of a BMS-378806 resistant strain in a cell-based assay (Figure 5). YU-2 is a HIV-1 molecular clone and YU-2 (T/P) is a BMS-378806 (Figure 5B) resistant HIV-1 strain from primary isolates. Even though it exhibited less antiviral activity against YU-2 strains than BMS-378806, compound 1 was equally potent against both BMS-378806 resistant and sensitive viruses. From these results, we have shown that BMS-378806 resistant virus remained sensitive to compound 1. Moreover, a second BMS-378806 sensitive/resistant virus pair (DH012 and DH012-T/P) was also investigated against compound 1 and showed similar results (data not shown). These studies establish the feasibility of targeting HIV entry processes with a proteomimetic compound and demonstrate that compound 1 is effective against viral strains that are resistant to another HIV-1 entry inhibitor.
Figure 5.

Results for the inhibition of gp120 mediated membrane fusion by compound 1 and BMS-378806. Dose-dependent inhibition of gp120 mediated membrane fusion and comparison of sensitivity of mutated strain (YU-2(T/P)) of gp120 towards compound 1 and BMS 378806.
In conclusion, we have shown that a proteomimetic inhibitor 1 can bind to the HIV-1 envelope protein gp120 and modulate viral entry process. Compound 1 is a surface binder of gp120, which its mode of inhibition is advantageous since the inhibitor remained active against a broad range of primary isolates from patients. When challenged against clinically isolated resistant strains developed from a current drug candidate, compound 1 maintained its potency suggesting a potential approach to tackling the emergence of resistant strains. This molecular scaffold should provide a new strategy for designing antiviral agents aimed at HIV-1 viral entry. Further work will include identification of the exact binding site on gp120 and detail mechanistic studies to design further improved compounds in both specificity and potency.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (GM35208 to A.D.H.; AI-38204 to Y-C.C. and AI- 065310 to C-H. C.) for financial support of this work.
Footnotes
Supporting Information: Methods and materials used for cell-based fusion assay, ELISA protocol for gp120/CD4 binding experiments, competition of antibody binding by an (ELISA) assay, and pseudotype virus infection assay for time-of-addition studies are available. Synthetic experimental procedures, analytical and spectral characterization data of compounds 1 and 2.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Antiretroviral drugs used in the treatment of HIV infection [Google Scholar]
- 2.Marcelin AG, Ceccherini-Silberstein F, Perno CF, Calvez V. Curr Opin HIV AIDS. 2009;4:531. doi: 10.1097/COH.0b013e328331d4b1. [DOI] [PubMed] [Google Scholar]
- 3.Little SJ, Holte S, Routy JP, Daar ES, Markowitz M, Collier AC, Koup RA, Mellors JW, Connick E, Conway B, Kilby M, Wang L, Whitcomb JM, Hellmann NS, Richman DD. N Engl J Med. 2002;347:385. doi: 10.1056/NEJMoa013552. [DOI] [PubMed] [Google Scholar]
- 4.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Nature. 1998;393:648. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burton A. Lancet Infect Dis. 2003;3:260. doi: 10.1016/s1473-3099(03)00620-0. [DOI] [PubMed] [Google Scholar]
- 6.Sax PE. AIDS Clin Care. 2007;19:75. [PubMed] [Google Scholar]
- 7.Weiss CD. AIDS Rev. 2003;5:214. [PubMed] [Google Scholar]
- 8.Huang CC, Tang M, Zhang MY, Majeed S, Montabana E, Stanfield RL, Dimitrov DS, Korber B, Sodroski J, Wilson IA, Wyatt R, Kwong PD. Science. 2005;310:1025. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheid JF, Mouquet H, Feldhahn N, Seaman MS, Velinzon K, Pietzsch J, Ott RG, Anthony RM, Zebroski H, Hurley A, Phogat A, Chakrabarti B, Li Y, Connors M, Pereyra F, Walker BD, Wardemann H, Ho D, Wyatt RT, Mascola JR, Ravetch JV, Nussenzweig MC. Nature. 2009;458:636. doi: 10.1038/nature07930. [DOI] [PubMed] [Google Scholar]
- 10.Franke R, Hirsch T, Overwin H, Eichler J. Angew Chem Int Ed Engl. 2007;46:1253. doi: 10.1002/anie.200603274. [DOI] [PubMed] [Google Scholar]
- 11.Fisher RA, Bertonis JM, Meier W, Johnson VA, Costopoulos DS, Liu T, Tizard R, Walker BD, Hirsch MS, Schooley RT, et al. Nature. 1988;331:76. doi: 10.1038/331076a0. [DOI] [PubMed] [Google Scholar]
- 12.Allaway GP, Davis-Bruno KL, Beaudry GA, Garcia EB, Wong EL, Ryder AM, Hasel KW, Gauduin MC, Koup RA, McDougal JS, et al. AIDS Res Hum Retroviruses. 1995;11:533. doi: 10.1089/aid.1995.11.533. [DOI] [PubMed] [Google Scholar]
- 13.Jacobson JM, Lowy I, Fletcher CV, O'Neill TJ, Tran DN, Ketas TJ, Trkola A, Klotman ME, Maddon PJ, Olson WC, Israel RJ. J Infect Dis. 2000;182:326. doi: 10.1086/315698. [DOI] [PubMed] [Google Scholar]
- 14.Wang T, Zhang Z, Wallace OB, Deshpande M, Fang H, Yang Z, Zadjura LM, Tweedie DL, Huang S, Zhao F, Ranadive S, Robinson BS, Gong YF, Ricarrdi K, Spicer TP, Deminie C, Rose R, Wang HG, Blair WS, Shi PY, Lin PF, Colonno RJ, Meanwell NA. J Med Chem. 2003;46:4236. doi: 10.1021/jm034082o. [DOI] [PubMed] [Google Scholar]
- 15.Lin PF, Blair W, Wang T, Spicer T, Guo Q, Zhou N, Gong YF, Wang HG, Rose R, Yamanaka G, Robinson B, Li CB, Fridell R, Deminie C, Demers G, Yang Z, Zadjura L, Meanwell N, Colonno R. Proc Natl Acad Sci USA. 2003;100:11013. doi: 10.1073/pnas.1832214100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang HG, Williams RE, Lin PF. Curr Pharm Des. 2004;10:1785. doi: 10.2174/1381612043384565. [DOI] [PubMed] [Google Scholar]
- 17.Rusconi S, Scozzafava A, Mastrolorenzo A, Supuran CT. Curr Top Med Chem. 2007;7:1273. doi: 10.2174/156802607781212239. [DOI] [PubMed] [Google Scholar]
- 18.Jing N, Hogan ME. J Biol Chem. 1998;273:34992. doi: 10.1074/jbc.273.52.34992. [DOI] [PubMed] [Google Scholar]
- 19.Robinson WE., Jr Antiviral Res. 1998;39:101. doi: 10.1016/s0166-3542(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 20.Sun J, Wang DA, Jain RK, Carie A, Paquette S, Ennis E, Blaskovich MA, Baldini L, Coppola D, Hamilton AD, Sebti SM. Oncogene. 2005;24:4701. doi: 10.1038/sj.onc.1208391. [DOI] [PubMed] [Google Scholar]
- 21.Blaskovich MA, Lin Q, Delarue FL, Sun J, Park HS, Coppola D, Hamilton AD, Sebti SM. Nat Biotechnol. 2000;18:1065. doi: 10.1038/80257. [DOI] [PubMed] [Google Scholar]
- 22.Sun J, Blaskovich MA, Jain RK, Delarue F, Paris D, Brem S, Wotoczek-Obadia M, Lin Q, Coppola D, Choi K, Mullan M, Hamilton AD, Sebti SM. Cancer Res. 2004;64:3586. doi: 10.1158/0008-5472.CAN-03-2673. [DOI] [PubMed] [Google Scholar]
- 23.Yin H, Hamilton AD. Angew Chem Int Ed Engl. 2005;44:4130. doi: 10.1002/anie.200461786. [DOI] [PubMed] [Google Scholar]
- 24.Tsou LK, Dutschman GE, Gullen EA, Telpoukhovskaia M, Cheng YC, Hamilton AD. Bioorg Med Chem Lett. 2010;20:2137. doi: 10.1016/j.bmcl.2010.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lever AM, Jeang KT. Int J Hematol. 2006;84:23. doi: 10.1532/IJH97.06112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.