

Abstract
Toll-like receptors (TLRs) are essential receptors of the innate immune system and are first responders for protection against bacterial and viral pathogens. Recently, several TLRs have also been implicated in regulating cell death and survival in non-pathogen injuries such as stroke and oxidative stress. Investigating the role of TLRs during central nervous system damage is an important focus of research that may reveal new mechanisms underlying the cellular response to injury and survival. Retinal pigmented epithelium (RPE) cells form an epithelial layer underneath the neural retina that maintains the function of photoreceptors and are the primary cell type affected in the retinal disease age-related macular degeneration (AMD). Predicted loss of function polymorphisms in the TLR3 gene are associated with protection from AMD but the role of TLR3 in regulating RPE survival during AMD-like injury, such as high oxidative stress, is not known. Therefore the purpose of this study is to evaluate the effect of TLR3 signaling on RPE viability during oxidative stress. We demonstrated that TLR3 activation in the presence of oxidative stress injury significantly increased RPE cell viability, in contrast to TLR3 reducing cell viability in the absence of cellular injury. Furthermore, we show signal transducer and activator of transcription 3 (STAT3) signaling as an essential mediator of TLR3-regulated protection of RPE cells. STAT3 signaling was increased by TLR3 activation and knockdown of STAT3 transcripts using siRNA abolished the protective effect of TLR3 during oxidative stress. Together, these results demonstrate a novel pro-survival role for TLR3 signaling within the RPE during injury. These findings support the concept that dysregulation of TLR3 activity may contribute to the development of AMD, suggesting that precise regulation of the TLR3 pathway during AMD-associated injury could be of therapeutic interest.
Keywords: Toll-like receptor 3, Retinal pigmented epithelium, Oxidative stress, STAT3
1. Introduction
The toll-like receptor (TLR) family of innate immune system receptors respond to multiple pathogen-associated molecular patterns, resulting in activation of nuclear factor kappa B (NF-κB) signaling and release of cytokines that trigger inflammatory responses (Chen et al., 2008b; Takeda and Akira, 2004). TLRs also regulate both cellular degeneration and survival during non-pathogen injury. The expression of TLRs such as TLR2 and TLR4 are increased in neurodegenerative brains (Walter et al., 2007) and chronic release of inflammatory cytokines induces neurodegeneration (Campbell et al., 1993; Drouin-Ouellet and Cicchetti, 2012). Furthermore, a pathoregulatory role of TLRs in neurodegeneration is supported by findings of increased neuronal survival in the retina and brain in TLR4 null mice (Caso et al., 2007; Dvoriantchikova et al., 2010; Kilic et al., 2008). Interestingly, an opposite role of TLR signaling during injury has also been reported, most likely due to the variability of injury and activation sites as well as the kinetics of TLR signaling (Bsibsi et al., 2006, 2010; van Noort and Bsibsi, 2009). TLR3 activation within astrocytes may play a neuroprotective role rather than a pro-inflammatory response (Bsibsi et al., 2006, 2010). TLR3 induction in astrocytes lead to enhanced neuronal survival and attenuation of astrocytic gliosis in human organotypic cortical brain slices (Bsibsi et al., 2006, 2010). TLR3 also protected cells within the arterial wall during vascular disease (Cole et al., 2011). Therefore, the precise activity of TLR3 in regulating cell death and survival pathways in various cell types and in different injury conditions remains to be understood.
TLRs are expressed throughout in the retina, with TLR3 having the highest expression within the retinal pigmented epithelium (RPE) (Kumar et al., 2004). The RPE is a monolayer of tightly packed, interconnected epithelial cells between the photoreceptors of the retina and the choroidal blood supply. The RPE provides essential cellular support and maintenance of photoreceptor functions including phagocytosis of photoreceptor outer segments, nutrient supply to the retina, maintaining the visual cycle, and removal of reactive oxygen species (Strauss, 1995). As a result of this close interaction, RPE dystrophy leads to photoreceptor death and contributes to several degenerative diseases of the retina such as age-related macular degeneration (AMD) (Kinnunen et al., 2012; Marmorstein et al., 1998). Several cellular signaling pathways, including oxidative stress and dysregulated immune system activation, are known to contribute to RPE atrophy and subsequent photoreceptor loss in AMD. Furthermore, predicted loss of function polymorphisms of the TLR3 gene were implicated in reduced AMD progression (Edwards et al., 2008; Yang et al., 2008). However, the effect of TLR3 on RPE survival during AMD-like injury needs to be directly investigated at the cellular and molecular levels.
In this study, we examined the role of TLR3 during oxidative stress injury in RPE cells. Our experimental data demonstrate that TLR3 activation protects RPE cells from oxidative stress induced death, in contrast to a pro-death effect of TLR3 when activated in the absence of oxidative stress. Our results also implicate STAT3 as a downstream mediator of TLR3-induced protection during oxidative stress. Therefore, the effect of TLR3 activation on cellular viability depends on the presence of injury, suggesting that TLR3 is an important factor in cell survival and death during retinal disease.
2. Methods
2.1. Cell culture
All procedures involving mice were performed according to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Animal Care and Use Committee at the University of Miami. Primary mouse RPE cultures were generated from wild type and TLR3 knockout mice obtained from Jackson Laboratory (Bar Harbor, Maine) using a protocol modified from Chen et al. (2008a). The wild type control mice are strain B6;129SF2/J and the TLR3 knockout mice are strain B6;129S1-Tlr3tm1Flv/J which have a targeted mutation resulting in a truncated non-functional TLR3 (Alexopoulou et al., 2001). Mice were genotyped by PCR using primers specific for the TLR3 wild type allele (341 bp) and mutant allele (208 bp) (Table 1). Briefly, eyes were enucleated at postnatal day 21 and anterior chamber, lens, and retina were removed (Chen et al., 2008a). The remaining eyecup was treated with 0.25% trypsin for 1 h and the RPE was removed with gentle shaking. Cells were plated at a density of 15,000 cells/ml in 96-well plates and maintained in DMEM/F12 media supplemented with 1× essential amino acids, 10% fetal bovine serum (FBS), 100 U/ml of penicillin and 100 μg/ml of streptomycin, and 1× fungizone. The cultures were passaged once before use and assayed at 80% density. The purity of the cultures was verified by morphology and QPCR analysis of RPE, neuronal and glial cell marker genes (Table 1).
Table 1.
Primer sequences used in this study.
| Gene | Sequence | |
|---|---|---|
| ARP | Forward | 5′-ATCTGCTGCATCTGCTTG-3′ |
| Reverse | 5′-CGACCTGGAAGTCCAACTAC-3′ | |
| Rig-1 | Forward | 5′-ACCAGAGCACTTGTGGACGCTT-3′ |
| Reverse | 5′-ACTTCTGTGCCGGGAGGGTCA-3′ | |
| STAT3 | Forward | 5′-ACAGATTGCCTGCATTG-3′ |
| Reverse | 5′-CTGCTAATGACGTTATCCAGT-3′ | |
| TLR3 | Mutant | 5′-GCCAGAGGCCACTTGTGTAG-3′ |
| Wildtype | 5′-GCAACCCTTTCAAAAACCAG-3′ | |
| Common | 5′-AATTCATCAGTGCCATGAGTTT-3′ | |
| Glutamate synthetase | Forward | 5′-CTTGGCTCTTAGGGGAACTG-3′ |
| Reverse | 5′-GAGTCATCGTGGCAAGAGAA-3′ | |
| RPE65 | Forward | 5′-TGGATCTCTGTTGCTGGAAAGGGT-3′ |
| Reverse | 5′-AGGCTGAGGAGCCTTCATAGCATT-3′ | |
| Tyrosinase | Forward | 5′-ATGAAGCACCAGGGTTTCTG-3′ |
| Reverse | 5′-TCAGGTGTTCCATCGCATAA-3′ | |
| Rhodopsin | Forward | 5′-GTCAGCCACCACACAGAAGG-3′ |
| Reverse | 5′-CTGGCTCGTCTCCGTCTTG-3′ | |
| TLR3 siRNA | Forward | 5′-GGAUAGGUGCCUUUCGA-3′ |
| Reverse | 5′-UGACGAAAGGCACCUAUGC-3′ | |
| STAT3 | Forward | 5′-GAGUUGAAUUAUCAGCUUA-3′ |
| siRNA | Reverse | 5′-UAAGCUGAUAAUUCAACUC-3′ |
The ARPE-19 cell line is a non-transformed cell line derived from adult human RPE cells that shares many properties with RPE in vivo, including polarization, tight junction formation, phagocytosis of rod outer segments and immunologic responses (Dunn et al., 1996). The cells were purchased from ATCC (Manassas, VA) and experiments were conducted using early passages at sub-confluent densities. The cell cultures were maintained in 5% CO2 at 37 °C in Dulbecco’s modified Eagle’s medium/F12 (DMEM/F12) (Invitrogen Carlsbad, CA) and supplemented with 10% fetal bovine serum (FBS), 100 U/ml of penicillin and 100 μg/ml of streptomycin, and passaged every 3 days.
2.2. Viability assays
Primary RPE cultures and ARPE-19 cells were plated at a density of 10,000–15,000 cells/ml into 96-well plates. Cells at approximately 90% density were incubated with normal growth media only (“untreated”), or growth media supplemented with 100 μg/ml poly(I:C) (to induce TLR3 signaling) (Invivogen, San Diego, CA), 0.8–1.6 mM paraquat (to induce oxidative stress), or a combination of poly(I:C) and paraquat. These concentrations are within the range used by other studies (Fragoso et al., 2012; Kumar et al., 2004; Shiose et al., 2011). Cell viability was measured after 24 h of treatment using the Cell Titer Blue assay (Promega, Madison, WI) by adding the Cell Titer Blue reagent to the wells for 2 h followed by quantification using an ELISA plate reader (excitation 530 nm, emission 590 nm). Cell viability was compared among treatments and normalized to untreated cells.
2.3. Immunohistochemistry
ARPE-19 cells were grown on glass coverslips coated with 2% gelatin or in a precoated multiwell chamber slide. The cells were incubated in normal growth media with or without 100 μg/ml poly(I:C) for 24 h to induce TLR3 signaling. The cells were then fixed in 4% paraformaldehyde in PBS for 20 min at room temperature and permeabilized in 50% methanol/50% acetone solution at −20 °C. Slides were incubated in rabbit anti-phosphorylated STAT3 antibody and mouse anti-p65 antibody (Cell Signaling Technology, Beverly, MA) overnight at 4 °C. The slides were washed three times in PBS and labeled with goat anti-rabbit HRP-conjugated and goat anti-mouse Alexa 488 secondary antibodies, respectively, followed by incubation with the Tyramide signal amplification kit (Invitrogen, Carlsbad, CA). The slides were viewed using a Zeiss Axiovert 200 fluorescent microscope and the percent of cells with active STAT3 signaling was calculated by counting the number of nuclear phospho-STAT3 positive cells and dividing by the total number of DAPI-positive cells (Fragoso et al., 2012). Microscopic settings, including exposure times, were kept constant for comparisons between antibody and control staining. Negative controls included secondary antibodies only and isotype control antibody.
2.4. siRNA knockdown
TLR3, RIG-1, and STAT3 transcripts were knocked-down in ARPE-19 cells using the Silencer siRNA Kit AM1640 (Ambion, Carlsbad, CA). A scrambled siRNA was used as a control. Sequences for TLR3 and STAT3 siRNA are listed in Table 1. RIG-1 was specifically knocked down using ON-TARGETplus SMARTpool siRNA (Dharmacon, Lafayette, CO). Cells were plated in a 96-well plate at 10,000 cells/ml and transfected with siRNA using siPORT (Ambion Carlsbad, CA) following the manufacturer’s protocol as described in Silva et al. (2010). Knock-down efficiency was assessed by QPCR and Western blotting from lysates of siRNA transfected cells 24 h post transfection.
2.5. Quantitative PCR analysis
Total RNA was isolated from siRNA transfected APRE-19 cell cultures using TRIzol (Invitrogen, Carlsbad, CA), according to the manufacturer’s instructions, and cDNA was synthesized from 1 μg of total RNA using Thermoscript (Invitrogen, Carlsbad, CA). Quantitative PCR analysis was performed using SYBR green (Bio-Rad, Hercules, CA) with the Mastercycler Real-time PCR system (Eppendorf, Hauppauge, NY). Relative transcript levels of each gene were calculated using the delta-delta Ct method (Nolan et al., 2006). The housekeeping gene acidic ribosomal phospho-protein (ARP) was used as a normalization control (Hackam et al., 2004).
2.6. Western blot
Cell lysates were prepared in buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% NP40, 0.05% SDS) containing proteinase and phosphatase inhibitors. Twenty microliters of lysate were resolved in 4–12% SDS–PAGE gels using Tris–glycine buffer and the proteins were then transferred onto polyvinylidene fluoride membranes and probed using the antibodies that detect total TLR3 (Abcam, Cambridge, MA), phosphorylated STAT3 (Cell Signaling Technology, Beverly, MA), total STAT3 (Cell Signaling Technology, Beverly, MA), and β-actin (Sigma, St. Louis, MO), as described by Nakamura and Hackam (2010). Bands were quantified using NIH Image J and the values were normalized to β-actin to correct for loading differences.
2.7. Statistical analysis
Statistical analysis of the data was conducted by Student’s t-test and ANOVA. A p-value less than 0.05 was considered to be significant. Mean ± standard deviation are shown.
3. Results
3.1. TLR3 activation protects primary RPE cell cultures from oxidative stress
TLR3 signaling has different consequences on cell survival in the presence or absence of injury in various cell types (Bsibsi et al., 2006, 2010; Cole et al., 2011; Estornes et al., 2012; Shiose et al., 2011). To determine the effects of TLR3 activation on RPE cell viability during oxidative stress, we employed a reductionist approach using mouse primary RPE cultures. TLR3 signaling was induced in RPE cells using the prototypic ligand polyinosinic:polycytidylic acid (poly(I:C), 100 μg/ml) a synthetic double stranded RNA that is commonly used as a TLR3 activator (Kumar et al., 2004; Shiose et al., 2011). Oxidative stress was induced using paraquat, which indirectly generates oxygen radicals through metabolization in the mitochondria and is often used to model oxidative stress injury in the retina (Cingolani et al., 2006).
Primary mouse RPE cultures were established from wild type mice and the purity of the cultures was confirmed by morphology (Fig. 1A) and PCR amplification of RPE-specific genes (Fig. 1B). As shown in Fig. 1C, treatment of the RPE cultures with 1.6 mM paraquat to induce oxidative stress led to a 40% reduction in viability (n = 3, p < 0.01) compared with untreated cells. In contrast, activation of TLR3 signaling by poly(I:C) in the presence of paraquat significantly increased cellular viability (n = 3, p < 0.01). Notably, poly(I:C) treatment of RPE obtained from TLR3 knock-out (KO) mice did not rescue cellular viability in the presence of oxidative stress (Fig. 1D), indicating the requirement of TLR3 signaling for cellular protection under these conditions.
Fig. 1.
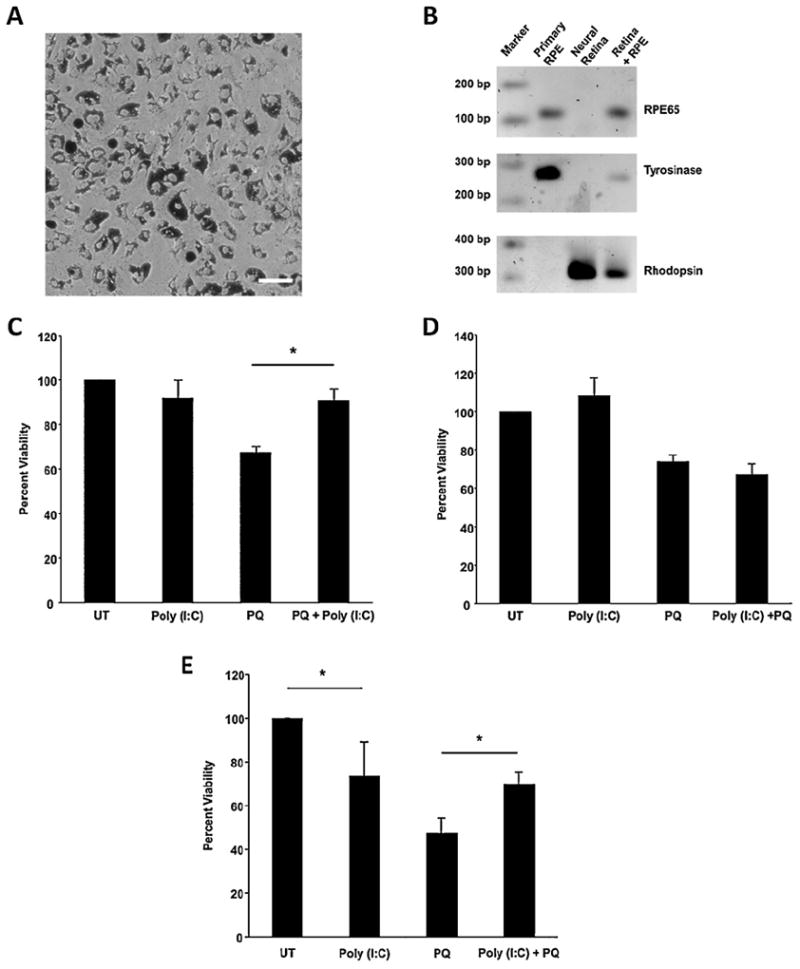
TLR3 activation protects primary mouse RPE cultures and ARPE-19 cells from oxidative stress. (A) Representative image of wild type mouse RPE primary cells after 5 days in culture showing pigmented cells with typical RPE preconfluent morphology (20× magnification, scale bar represents 100 μm). (B) PCR amplification of RPE cell markers RPE65 (112 bp) and Tyrosinase (276 bp) and the photoreceptor marker rhodopsin (315 bp). RPE primary cultures are enriched for RPE markers and do not express other retinal cell markers. (C) Poly(I:C) significantly increased cell survival of primary RPE cultures obtained from wild type mice in the presence of paraquat compared with oxidative stress alone or poly(I:C) treatment alone (n = 3, *p < 0.05). (D) Poly(I:C) did not increase cell survival of primary mouse RPE cultures obtained from TLR3 KO mice in the presence of paraquat compared with oxidative stress alone or poly(I:C) treatment alone (n = 3, *p < 0.05). (E) Poly(I:C) significantly increased survival of ARPE-19 cells in the presence of paraquat compared with oxidative stress alone or poly(I:C) treatment alone. Poly(I:C) alone decreased cell viability compared with untreated cells. Viability was measured using Cell Titer Blue assay. UT, untreated (growth media only); PQ, paraquat.
3.2. TLR3 activation rescues ARPE-19 cells from oxidative stress
In order to better understand the mechanisms by which TLR3 signaling regulates cellular viability in the presence of injury, we moved to an RPE cell line model. The ARPE-19 cell line shares many properties with RPE cells in vivo, and offers the advantages of lower variability and ease of transfection compared with primary cultures. Similar to the primary RPE cultures above, we observed that TLR3 activation in the presence of oxidative stress injury rescued ARPE-19 cells by 50% (n = 5, p < 0.05) (Fig. 1E). Interestingly, TLR3 activation in the absence of injury lead to approximately 25% reduction in cell viability (n = 5, p < 0.05), which is consistent with reported findings (Shiose et al., 2011). Because of the similarities between primary mouse RPE cultures and the ARPE-19 cells in response to poly(I:C) and oxidative stress, we continued to use the ARPE-19 to further identify the molecular basis for the protective effects of TLR3 activation during oxidative stress.
3.3. TLR3 signaling is required for RPE cell rescue during oxidative stress
Poly(I:C) is known to activate receptors other than TLR3, such as RIG-1 (Kleinman et al., 2012; Slater et al., 2010). Therefore, to confirm that poly(I:C)-dependent survival in ARPE-19 cells occurred though the TLR3 signaling pathway, we knocked down TLR3 signaling using TLR3 specific siRNA. TLR3 siRNA reduced TLR3 transcript levels by 84%, as measured by QPCR (n = 3, p < 0.01) (Fig. 2A), and protein levels by 46%, as measured by Western blotting (n = 3, p < 0.05) (Fig. 2B and C), compared with cells transfected with scrambled siRNA control 24 h after transfection. Furthermore, poly(I:C) treatment did not increase the viability of TLR3 siRNA transfected cells exposed to oxidative stress, in contrast to poly(I:C) treatment of control siRNA transfected cells (Fig. 2D). These results confirm the requirement for TLR3 in cell rescue. Additionally, to test the specificity of the TLR3 siRNA, we examined the expression of TLR4 in TLR3 siRNA transfected cells. TLR4, which can also protect cells from oxidative stress, was unchanged by TLR3 siRNA transfection, as measured by QPCR (Fig. 2E) (Komori et al., 2012; Yi et al., 2012).
Fig. 2.
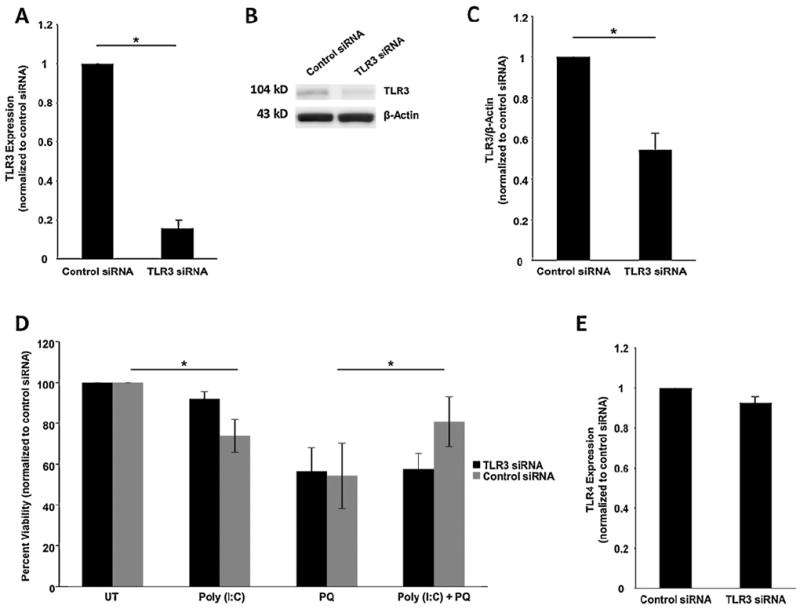
Poly(I:C) protection of APRE-19 cells during oxidative stress is TLR3-dependent. (A) TLR3 specific siRNA decreased TLR3 RNA expression by 84%, measured by QPCR at 24 h post-transfection (n = 3, *p < 0.05). (B and C) Protein expression of TLR3 was reduced by 46%, measured by Western blotting using an anti-TLR3 antibody. Detection of β-actin was used as a loading control for normalization. (D) Poly(I:C) did not rescue cells from paraquat treatment when TLR3 was knocked down by siRNA. Control siRNA transfection still resulted in rescue of cell viability when treated with poly(I:C) and oxidative stress, similar to untransfected cells in Fig. 2 (*p < 0.05, n = 5). (E) To examine specificity of TLR3 siRNA, TLR4 expression was measured in cells transfected with TLR3 siRNA. TLR4 expression was not reduced by TLR3 siRNA compared with control siRNA as measured by QPCR (n = 3).
To identify whether the RIG-1 pathway contributes to poly(I:C)-dependent protection of RPE, we knocked down RIG-1 transcripts using specific siRNA. RIG-1 siRNA reduced RIG-1 expression by 80% (n = 3, p < 0.01) compared with cells transfected with scrambled siRNA, measured by QPCR 24 h post transfection (Fig. 3A). RIG-1 was not detectable by Western blotting. As shown in Fig. 3B, RIG-1 siRNA transfected cells showed approximately 20% cell death when exposed to poly(I:C) (Fig. 3B). In the presence of oxidative stress and poly(I:C), RIG-1 siRNA transfected cells increased cell survival by 50% compared with oxidative stress only treated cells (Fig. 3B). These results confirm that RIG-1 signaling is not responsible for protecting RPE cells from oxidative stress.
Fig. 3.
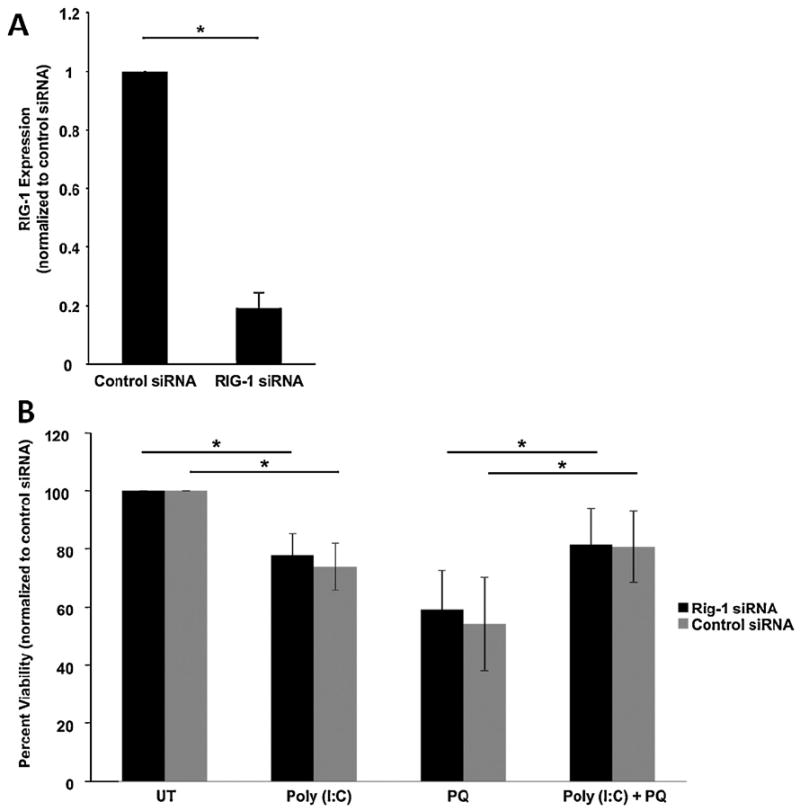
Poly(I:C) induced protection of ARPE-19 cells is independent of the RIG-1 pathway. (A) RIG-1 specific siRNA decreased RIG-1 RNA expression measured by QPCR compared with control siRNA (n = 3). (B) RIG-1 siRNA transfected cells show no difference in cell viability compared with control siRNA transfected cells in each treatment. Knockdown of RIG-1 results in a modest increase in cell death from poly(I:C) by 20% compared to untreated cells, similar to control transfected cells. Both control siRNA and RIG-1 transfections resulted in rescue of cell viability when treated with poly(I:C) and oxidative stress similar to untransfected cells in Fig. 2 (*p < 0.05, n = 5) indicating that RIG-1 does not play a role in poly(I:C) induced protection. ARPE-19 cell cultures were treated with poly(I:C) and/or 0.8 mM paraquat for 24 h and viability was measured using Cell Titer Blue assay. UT, untreated (growth media only); PQ, paraquat.
3.4. TLR3 signaling activates the STAT3 pathway
STAT3 is a transcription factor that is activated by multiple cytokines and serves various functions in the cell, including regulating proliferation and survival (Aaronson and Horvath, 2002; Fragoso et al., 2012; Zhang et al., 2003). Because STAT3 activation regulates several anti-apoptotic pathways (Liu et al., 2010; Luo et al., 2011; Sharma et al., 2011) and has been associated with protection of retinal cells during injury, including oxidative stress (Fragoso et al., 2012; Zhang et al., 2003), it is a compelling candidate regulator of TLR3-induced protection. To investigate whether the STAT3 pathway is a potential mechanism of TLR3-induced protection during oxidative stress, we first tested whether TLR3 activates STAT3 signaling in RPE cells. When the STAT3 signaling pathway is activated, STAT3 is phosphorylated and is translocated into the nucleus. A commonly used marker of STAT3 activation is detection of nuclear phospho-STAT3. Immunocytochemistry was used to examine STAT3 activation in ARPE-19 cells incubated with poly(I:C), using antibodies against phospho-STAT3. Co-detection of nuclear p65 was used as a marker for NF-κB signaling and TLR3 activation.
As shown in Fig. 4, 78% of cells in the poly(I:C)-treated cultures had nuclear phospho-STAT3 staining, indicating that TLR3 activation increased STAT3 activation 24 h after treatment (Fig. 4A and B, n = 3, p < 0.05). Further, phospho-STAT3 and total STAT3 levels were increased in cells treated with poly(I:C) compared with untreated cells by 41% and 31% respectively, measured by Western blotting (Fig. 4C–F, n = 3, p < 0.05), which confirms the IHC findings. Interestingly, cells treated with both poly(I:C) and paraquat showed an increase of total STAT3 levels by approximately 2.5-fold compared with paraquat only treated cells (Fig. 4D, n = 3, p < 0.05), suggesting that the total amount of STAT3 that is available for activation is important for promoting survival during injury.
Fig. 4.
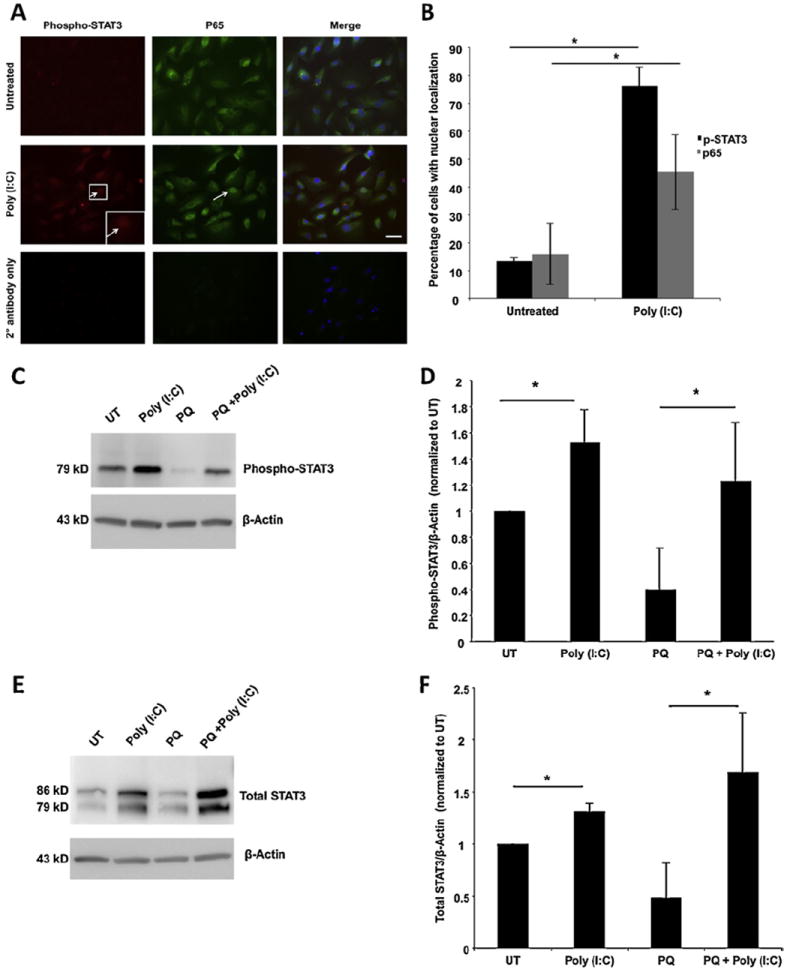
TLR3 activation increases STAT3 expression and signaling. (A and B) Activation of STAT3 in ARPE-19 cells was performed by immunodetection using anti phospho-STAT3 antibody. TLR3 activation was confirmed using an anti-p65 antibody. Poly(I:C) induces STAT3 phosphorylation in approximately 70% of cells and p65 in approximately 50% of cells after 24 h of treatment indicating that TLR3 activation leads to increased STAT3 signaling. Arrows show nuclear localization; inset shows higher magnification; 20× magnification, scale bar represents 50 μm. (C–F) Poly(I:C) treatment increased both total and phospho-specific STAT3 compared with untreated cells after 24 h. Paraquat treatment decreased both total and phospho-STAT3 levels compared with untreated cells. Treatment of cells with both poly(I:C) and paraquat increased both total STAT3 and phospho-STAT3 expression compared with untreated cells. Detection of β-actin was used as a loading control for normalization (n = 3, *p < 0.05).
A time-course analysis was conducted to determine the dynamics of TLR3-dependent regulation of STAT3. TLR3 activation resulted in a time dependent increase in total STAT3 between 0 and 8 h of treatment (Fig. 5A and C), similar to previous work in other cell types (Dai et al., 2006). The increase in phosphorylated STAT3 by TLR3 was not apparent in the immediate time-points but reached significance by 5 and 8 h and then dropped slightly at 24 h (Figs. 4 and 5). These results indicate that while TLR3 signaling increases STAT3 transcription and translation, phosphorylation of STAT3 may be an indirect effect of TLR3 activation, for example, through production of cytokines and activation of the JAK/STAT pathway.
Fig. 5.
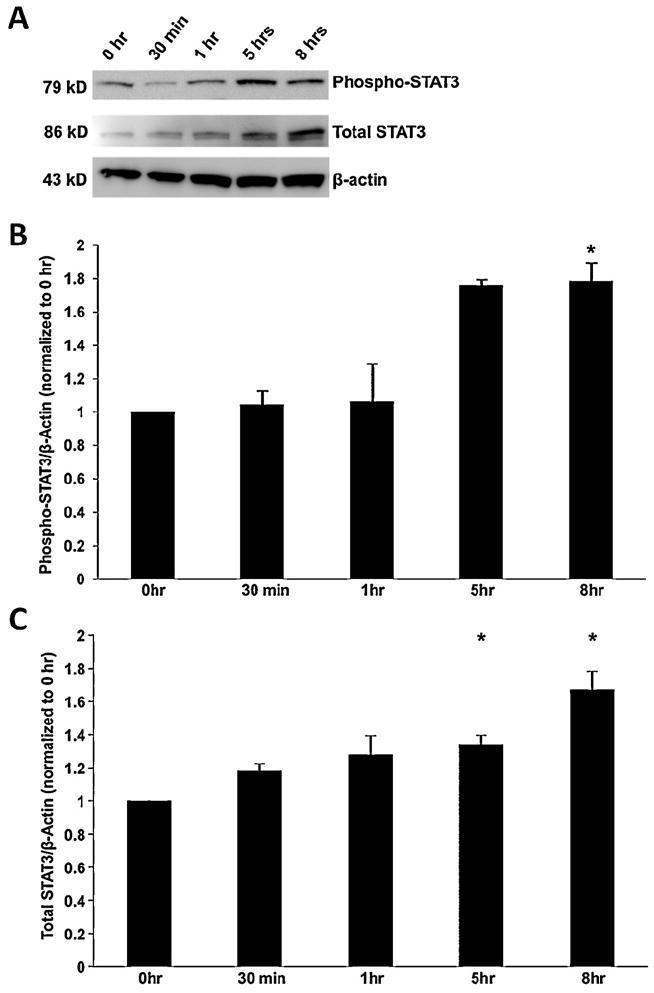
Total and phosphorylated STAT3 increases with poly(I:C) stimulation in a time dependent manner. (A–C) Protein expression of phosphorylated and total STAT3 levels 0–8 h after TLR3 activation by poly(I:C), measured by Western blotting. Both phosphorylated and total STAT3 were significantly increased at 5 h and 8 h post stimulation compared to 0 h treatment (n = 3, *p < 0.05). β-Actin levels were measured as a loading control.
3.5. STAT3 is required for TLR3 rescue of RPE cells from oxidative stress
To identify whether STAT3 mediates TLR3 dependent protection of RPE cells during oxidative stress, we knocked down STAT3 expression in the presence of TLR3 activation and measured RPE cell viability. The siRNA approach was used instead of chemical inhibitors because STAT3 siRNA is more specific to the STAT3 molecule than most available inhibitor compounds. STAT3 siRNA decreased STAT3 expression by 79% measured by QPCR (n = 3) (Fig. 6A) and by 34% (n = 3) measured by Western blotting when compared with cells transfected with scrambled siRNA (Fig. 6B and C) 24 h after transfection. Furthermore, poly(I:C) did not induce protection of STAT3 siRNA transfected ARPE-19 cells exposed to oxidative stress, in contrast to control siRNA transfected cells (Fig. 6D, n = 3, p < 0.05). This finding is equivalent to the results with TLR3 siRNA (Fig. 2), and indicates that STAT3 is required for TLR3 induced protection. Additionally, transfection of STAT3 siRNA only resulted in a decrease in the amount total STAT3 present in cells, and did not cause a significant reduction in the proportion of phosphorylated STAT3, compared with control siRNA (Fig. 7), indicating that downregulation of STAT3 by siRNA does not affect its phosphorylation.
Fig. 6.
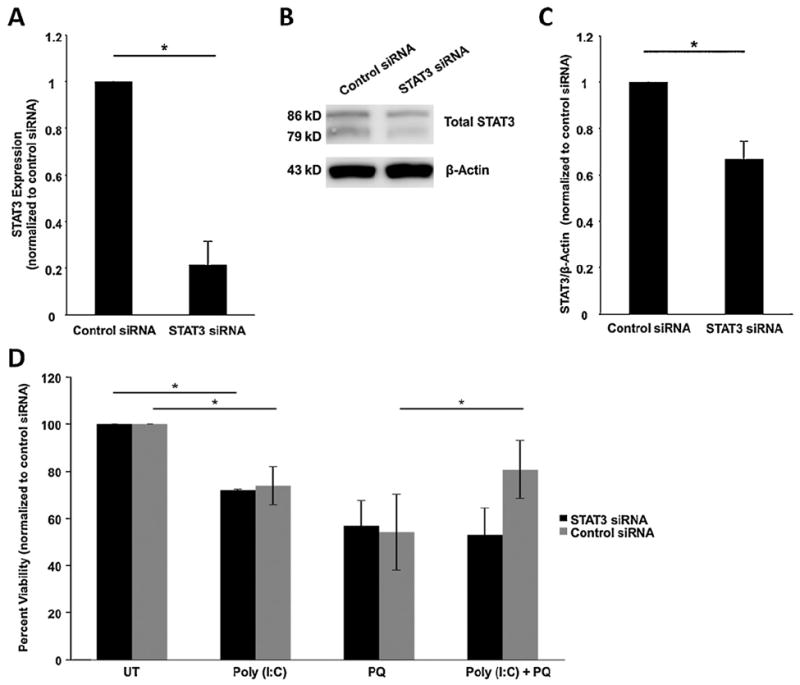
TLR3 induced protection of ARPE-19 cells during oxidative stress is STAT3 dependent. (A) STAT3 specific siRNA decreased STAT3 RNA expression by 79% measured by QPCR compared with control siRNA transfected cells 24 h after transfection (n = 3, *p < 0.05). (B and C) Protein expression of STAT3 was reduced measured by Western blotting. β-Actin levels were measured as a loading control. (D) Knocking down STAT3 using siRNA resulted in no rescue in poly(I:C) and oxidative stress conditions compared with oxidative stress only treated cells. Control siRNA resulted in 50% rescue of cell when treated with poly(I:C) and oxidative stress (n = 5, *p < 0.05). ARPE-19 cell cultures were treated with poly(I:C) and/or 0.8 mM paraquat for 24 h and viability was measured using Cell Titer Blue assay. UT, untreated (growth media only); PQ, paraquat.
Fig. 7.
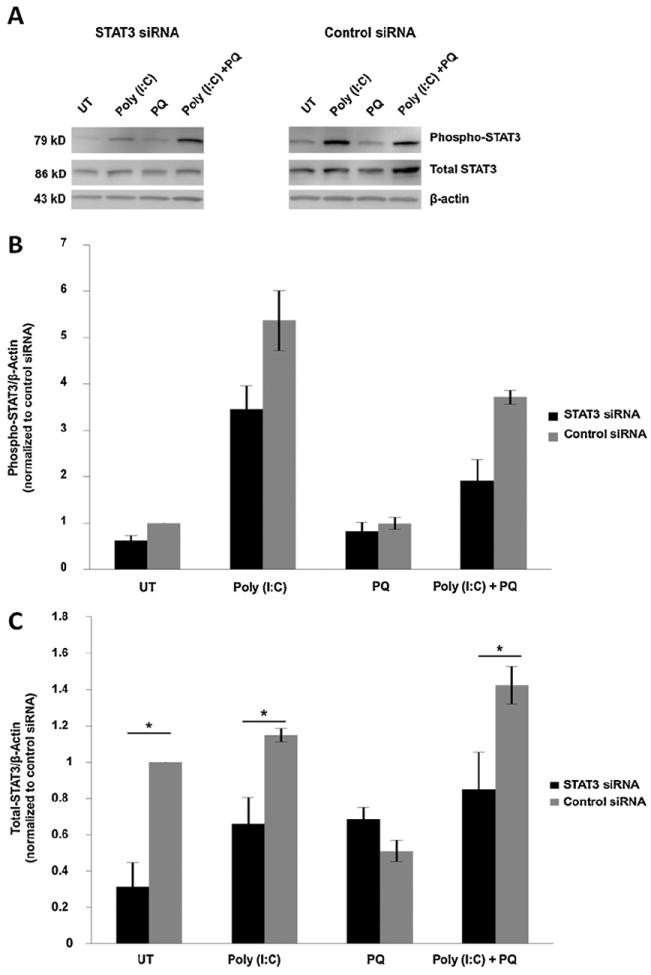
Effect of STAT3 knock-down on STAT3 activation. STAT3 siRNA did not significantly alter phosphorylation of STAT3. (A and B) Protein expression of phosphorylated STAT3 in STAT3 siRNA treated cells did not significantly decrease compared with control siRNA treated cells, as measured by Western blotting. (C) Total STAT3 levels in STAT3 siRNA treated cells were significantly decreased compared with control siRNA treated cells (n = 3, *p < 0.05). β-Actin levels were measured as a loading control.
4. Discussion
Under pathogen and infectious conditions, TLR3 serves as an innate immunity sensor that induces inflammation and promotes cell death. In this study, we examined the role of TLR3 on RPE cell survival during non-pathogen-mediated oxidative stress injury. We demonstrated that TLR3 serves as a mediator of cellular protection in an oxidative stress injury model, and that it requires STAT3 signaling. Therefore, this study suggests a novel protective role for TLR3 signaling during disease-like injury in the RPE.
Several studies have demonstrated that TLR3 signaling induced cellular apoptosis and retinal inflammation and degeneration. Kleinman et al. (2012) showed that intravitreal injections of dsRNA in wild type mice lead to disruption of RPE structure and retinal cell loss through activation of TLR3 signaling, and that surface TLR3 activation on RPE triggered caspase-3 mediated cell death under non-injury conditions. Shiose et al. (2011) showed that TLR3 ablation protected mice from cone-rod dystrophies and activation of TLR3 lead to retinal degeneration in wild type mice. Our study showed a similar result in that ARPE-19 cells treated with poly(I:C) without paraquat had increased death (Fig. 1E). However, when poly(I:C) and paraquat treatments were combined, there was a significant increase in cell viability compared with cells in oxidative stress conditions alone (Fig. 1C and E). This finding indicates that TLR3 signaling may have protective properties in the presence of an injury paradigm such as oxidative stress, contrary to its role as an initiator of cell death under non-injury conditions.
Our findings in RPE are supported by other studies that also showed a dual role for TLR3 as a pathogenic or protective pathway in other cell types. Jin et al. (2011) showed contrasting activities of TLR3 as a pathogenic or protective pathway in response to Theiler’s virus-induced demyelinating disease. TLR3-mediating signaling during viral infection protected axons of the brain and spinal cord against demyelinating disease, whereas TLR3 signaling prior to viral infection increased pathogenesis (Jin et al., 2011). Additionally, Bsibsi et al. (2006) showed that activation of TLR3 signaling in astrocytes triggers secretion of neuroprotective mediators leading to neuronal protection of the brain. Other TLRs show similar properties: TLR4 activation can lead to both photoreceptor protection from oxidative stress or increased cell death during oxidative injury depending on the timing of TLR4 signaling (Yi et al., 2012). TLR9 has also been shown to have opposing roles in the development of lupus in that it can both activate autoimmune responses as well as induce tolerance (Ehlers and Ravetch, 2007).
The TLR family has the ability to induce tolerance to subsequent insults and injury after initial exposure to a ligand, in a phenomenon known as preconditioning. TLR preconditioning has been shown to promote neuroprotection of the brain after ischemic injury through TLR3 or TLR7 activation prior to injury (Leung et al., 2012; Stevens et al., 2011). In the retina, preconditioning of Muller glia-photoreceptor cultures with TLR4 activation protected against oxidative stress damage (Yi et al., 2012). In this study, we used paraquat, which induces oxidative stress over time through free radicals produced during processing in the mitochondria (Bus and Gibson, 1984). It is possible that a preconditioning paradigm occurred in this study due to the rapid activation of TLR3 by poly(I:C) and the slower initiation of oxidative stress. TLR3 activation may have promoted cellular tolerance to oxidative stress, leading to cellular protection. Future studies will identify if a preconditioning effect occurs in this model.
In this study, we identified a link between STAT3 and TLR3 mediated protection in the RPE. We demonstrated that TLR3 activation induced STAT3 signaling and that knockdown of STAT3 using siRNA blocked the protective effects of TLR3 during oxidative stress. These findings further support STAT3 as a cellular survival pathway in the RPE (Fragoso et al., 2012; Zhang et al., 2003). Studies conducted by Barry et al. (2009), showed that STAT3 activation protected primary neonatal rat ventricular myocytes from oxidative stress damage and Sarafian et al. (2010) showed that disruption of STAT3 signaling leads to decreased protection from oxidative stress in astrocytes. Additionally, we demonstrated that oxidative stress decreased STAT3 levels in the RPE, whereas TLR3 activation was able to increase STAT3 levels during oxidative stress, possibly resulting in enough activation to promote resistance to injury. Although this study identified STAT3 as an important element in TLR3-dependent protection from oxidative stress, the mechanism by which TLR3 interacts with the STAT3 pathway is not yet known. From the dynamics of the time course in Fig. 5, it is possible that TLR3 activation directly upregulates STAT3 expression, while STAT3 phosphorylation may be indirectly increased from cytokine secretion and activation of the Jak/STAT pathway following TLR3 stimulation. Future experiments will focus on the mechanism of TLR3/STAT3 interaction, and how TLR3/STAT3 promotes RPE cell survival in this context.
A leading cause of visual impairment in the age group of 65 and older in the western world is AMD. AMD results from RPE atrophy, leading to death of photoreceptors in the macula region of eye and subsequent progressive loss of central vision (Cai et al., 2000). The initial pathogenesis involves degeneration of the RPE (Green et al., 1985). AMD is a disease that has multiple genetic and environmental factors (Chen et al., 2010). Oxidative stress and, more recently, innate immunity, have been proposed as major contributors to AMD (Cingolani et al., 2006; Lu et al., 2006). Both of these factors have been studied independently (Detrick and Hooks, 2010; Usui et al., 2009); to our knowledge, our work is the first to examine them simultaneously within the RPE. In this study, we showed that activation of the innate immunity receptor TLR3 in the presence of oxidative stress leads to RPE cell protection in culture. In contrast, TLR3 activation alone leads to RPE cell loss.
Although this study is limited to in vitro experiments, it suggests that TLR3 may be an important target for therapeutic intervention for patients with AMD and other diseases in which high oxidative stress is a key factor. It is important to note that TLR3 may have a differential response in vivo in the presence or absence of the many environmental and genetic factors that contribute to disease, which would lead to either further progression or attenuation of AMD. Indeed, a pathogenic role for TLR3 is supported by evidence showing accumulation of Alu-repeat derived dsRNA, which is a ligand for TLR3, in RPE from AMD eyes (Kaneko et al., 2011; Kleinman et al., 2012). It is possible that the timing (acute vs chronic) of TLR3 activation, presence of injury, or type of activator (endogenous or viral ligands) influences the effect on the RPE. It is currently unclear how TLR3 could be activated in vivo: Potential sources of TLR3 activation are dsRNA released from dying necrotic cells (Bernard et al., 2012; Cavassani et al., 2008) following injury or from non-injury conditions such as build-up of endogenous dsRNA (Kaneko et al., 2011), mRNA (Kariko et al., 2004) or other endogenous activators, such as stathmin (Bsibsi et al., 2010). TLR3 is also activated by UV light (Bernard et al., 2012), which is an injury relevant to the RPE. Future experiments will examine the role of TLR3 activation during oxidative stress using animal models of AMD.
Acknowledgments
This study was supported by a Research to Prevent Blindness Ernest & Elizabeth Althouse Special Scholar Award, the Karl Kirchgessner Foundation, NIH grant RO1 EY017837, and a Fight for Sight Student Fellowship. Institutional support to BPEI was from a Research to Prevent Blindness Unrestricted Grant and an NEI Center Core Grant P30EY014801.
Abbreviations
- TLR3
toll-like receptor 3
- RPE
retinal pigmented epithelium
- Poly(I:C)
polyinosinic:polycytidylic acid
- AMD
age-related macular degeneration
- STAT3
signal transducer and activator of transcription 3
References
- Aaronson DS, Horvath CM. A road map for those who don’t know JAK-STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Barry SP, Townsend PA, McCormick J, Knight RA, Scarabelli TM, Latchman DS, Stephanou A. STAT3 deletion sensitizes cells to oxidative stress. Biochemical and Biophysical Research Communications. 2009;385:324–329. doi: 10.1016/j.bbrc.2009.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard JJ, Cowing-Zitron C, Nakatsuji T, Muehleisen B, Muto J, Borkowski AW, Martinez L, Greidinger EL, Yu BD, Gallo RL. Ultraviolet radiation damages self noncoding RNA and is detected by TLR3. Nature Medicine. 2012 doi: 10.1038/nm.2861. http://dx.doi.org/10.1038/nm.2861 [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- Bsibsi M, Bajramovic JJ, Vogt MH, van Duijvenvoorden E, Baghat A, Persoon-Deen C, Tielen F, Verbeek R, Huitinga I, Ryffel B, Kros A, Gerritsen WH, Amor S, van Noort JM. The microtubule regulator stathmin is an endogenous protein agonist for TLR3. Journal of Immunology. 2010;184:6929–6937. doi: 10.4049/jimmunol.0902419. [DOI] [PubMed] [Google Scholar]
- Bsibsi M, Persoon-Deen C, Verwer RW, Meeuwsen S, Ravid R, Van Noort JM. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia. 2006;53:688–695. doi: 10.1002/glia.20328. [DOI] [PubMed] [Google Scholar]
- Bus JS, Gibson JE. Paraquat: model for oxidant-initiated toxicity. Environmental Health Perspectives. 1984;55:37–46. doi: 10.1289/ehp.845537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Nelson KC, Wu M, Sternberg P, Jr, Jones DP. Oxidative damage and protection of the RPE. Progress in Retinal and Eye Research. 2000;19:205–221. doi: 10.1016/s1350-9462(99)00009-9. [DOI] [PubMed] [Google Scholar]
- Campbell IL, Abraham CR, Masliah E, Kemper P, Inglis JD, Oldstone MB, Mucke L. Neurologic disease induced in transgenic mice by cerebral over-expression of interleukin 6. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:10061–10065. doi: 10.1073/pnas.90.21.10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–1608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, Hogaboam CM, Kunkel SL. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. Journal of Experimental Medicine. 2008;205:2609–2621. doi: 10.1084/jem.20081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Muckersie E, Robertson M, Fraczek M, Forrester JV, Xu H. Characterization of a spontaneous mouse retinal pigment epithelial cell line B6-RPE07. Investigative Ophthalmology and Visual Science. 2008a;49:3699–3706. doi: 10.1167/iovs.07-1522. [DOI] [PubMed] [Google Scholar]
- Chen R, Alvero AB, Silasi DA, Steffensen KD, Mor G. Cancers take their toll—the function and regulation of toll-like receptors in cancer cells. Oncogene. 2008b;27:225–233. doi: 10.1038/sj.onc.1210907. [DOI] [PubMed] [Google Scholar]
- Chen Y, Bedell M, Zhang K. Age-related macular degeneration: genetic and environmental factors of disease. Molecular Interventions. 2010;10:271–281. doi: 10.1124/mi.10.5.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani C, Rogers B, Lu L, Kachi S, Shen J, Campochiaro PA. Retinal degeneration from oxidative damage. Free Radical Biology and Medicine. 2006;40:660–669. doi: 10.1016/j.freeradbiomed.2005.09.032. [DOI] [PubMed] [Google Scholar]
- Cole JE, Navin TJ, Cross AJ, Goddard ME, Alexopoulou L, Mitra AT, Davies AH, Flavell RA, Feldmann M, Monaco C. Unexpected protective role for toll-like receptor 3 in the arterial wall. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:2372–2377. doi: 10.1073/pnas.1018515108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Sayama K, Yamasaki K, Tohyama M, Shirakata Y, Hanakawa Y, Tokumaru S, Yahata Y, Yang L, Yoshimura A, Hashimoto K. SOCS1-negative feedback of STAT1 activation is a key pathway in the dsRNA-induced innate immune response of human keratinocytes. Journal of Investigative Dermatology. 2006;126:1574–1581. doi: 10.1038/sj.jid.5700294. [DOI] [PubMed] [Google Scholar]
- Detrick B, Hooks JJ. Immune regulation in the retina. Immunologic Research. 2010;47:153–161. doi: 10.1007/s12026-009-8146-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin-Ouellet J, Cicchetti F. Inflammation and neurodegeneration: the story ‘retolled’. Trends in Pharmacological Sciences. 2012;33:542–552. doi: 10.1016/j.tips.2012.07.002. [DOI] [PubMed] [Google Scholar]
- Dunn KC, Aotaki-Keen AE, Putkey FR, Hjelmeland LM. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Experimental Eye Research. 1996;62:155–169. doi: 10.1006/exer.1996.0020. [DOI] [PubMed] [Google Scholar]
- Dvoriantchikova G, Barakat DJ, Hernandez E, Shestopalov VI, Ivanov D. Toll-like receptor 4 contributes to retinal ischemia/reperfusion injury. Molecular Vision. 2010;16:1907–1912. [PMC free article] [PubMed] [Google Scholar]
- Edwards AO, Chen D, Fridley BL, James KM, Wu Y, Abecasis G, Swaroop A, Othman M, Branham K, Iyengar SK, Sivakumaran TA, Klein R, Klein BE, Tosakulwong N. Toll-like receptor polymorphisms and age-related macular degeneration. Investigative Ophthalmology and Visual Science. 2008;49:1652–1659. doi: 10.1167/iovs.07-1378. [DOI] [PubMed] [Google Scholar]
- Ehlers M, Ravetch JV. Opposing effects of toll-like receptor stimulation induce autoimmunity or tolerance. Trends in Immunology. 2007;28:74–79. doi: 10.1016/j.it.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Estornes Y, Toscano F, Virard F, Jacquemin G, Pierrot A, Vanbervliet B, Bonnin M, Lalaoui N, Mercier-Gouy P, Pacheco Y, Salaun B, Renno T, Micheau O, Lebecque S. dsRNA induces apoptosis through an atypical death complex associating TLR3 to caspase-8. Cell Death and Differentiation. 2012;19:1482–1494. doi: 10.1038/cdd.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragoso MA, Patel AK, Nakamura RE, Yi H, Surapaneni K, Hackam AS. The Wnt/beta-catenin pathway cross-talks with STAT3 signaling to regulate survival of retinal pigment epithelium cells. PLoS ONE. 2012;7:e46892. doi: 10.1371/journal.pone.0046892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green WR, McDonnell PJ, Yeo JH. Pathologic features of senile macular degeneration. Ophthalmology. 1985;92:615–627. [PubMed] [Google Scholar]
- Hackam AS, Strom R, Liu D, Qian J, Wang C, Otteson D, Gunatilaka T, Farkas RH, Chowers I, Kageyama M, Leveillard T, Sahel JA, Campochiaro PA, Parmigiani G, Zack DJ. Identification of gene expression changes associated with the progression of retinal degeneration in the rd1 mouse. Investigative Ophthalmology and Visual Science. 2004;45:2929–2942. doi: 10.1167/iovs.03-1184. [DOI] [PubMed] [Google Scholar]
- Jin YH, Kaneyama T, Kang MH, Kang HS, Koh CS, Kim BS. TLR3 signaling is either protective or pathogenic for the development of Theiler’s virus-induced demyelinating disease depending on the time of viral infection. Journal of Neuroinflammation. 2011;8:178. doi: 10.1186/1742-2094-8-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, Cho WG, Kleinman ME, Ponicsan SL, Hauswirth WW, Chiodo VA, Kariko K, Yoo JW, Lee DK, Hadziahmetovic M, Song Y, Misra S, Chaudhuri G, Buaas FW, Braun RE, Hinton DR, Zhang Q, Grossniklaus HE, Provis JM, Madigan MC, Milam AH, Justice NL, Albuquerque RJ, Blandford AD, Bogdanovich S, Hirano Y, Witta J, Fuchs E, Littman DR, Ambati BK, Rudin CM, Chong MM, Provost P, Kugel JF, Goodrich JA, Dunaief JL, Baffi JZ, Ambati J. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011;471:325–330. doi: 10.1038/nature09830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for toll-like receptor 3. Journal of Biological Chemistry. 2004;279:12542–12550. doi: 10.1074/jbc.M310175200. [DOI] [PubMed] [Google Scholar]
- Kilic U, Kilic E, Matter CM, Bassetti CL, Hermann DM. TLR-4 deficiency protects against focal cerebral ischemia and axotomy-induced neurodegeneration. Neurobiology of Disease. 2008;31:33–40. doi: 10.1016/j.nbd.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Kinnunen K, Petrovski G, Moe MC, Berta A, Kaarniranta K. Molecular mechanisms of retinal pigment epithelium damage and development of age-related macular degeneration. Acta Ophthalmologica. 2012;90:299–309. doi: 10.1111/j.1755-3768.2011.02179.x. [DOI] [PubMed] [Google Scholar]
- Kleinman ME, Kaneko H, Cho WG, Dridi S, Fowler BJ, Blandford AD, Albuquerque RJ, Hirano Y, Terasaki H, Kondo M, Fujita T, Ambati BK, Tarallo V, Gelfand BD, Bogdanovich S, Baffi JZ, Ambati J. Short-interfering RNAs induce retinal degeneration via TLR3 and IRF3. Molecular Therapy. 2012;20:101–108. doi: 10.1038/mt.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori H, Watanabe H, Shuto T, Kodama A, Maeda H, Watanabe K, Kai H, Otagiri M, Maruyama T. alpha1-acid glycoprotein up-regulates CD163 via TLR4/CD14 protein pathway: possible protection against hemolysis-induced oxidative stress. Journal of Biological Chemistry. 2012;287:30688–30700. doi: 10.1074/jbc.M112.353771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar MV, Nagineni CN, Chin MS, Hooks JJ, Detrick B. Innate immunity in the retina: toll-like receptor (TLR) signaling in human retinal pigment epithelial cells. Journal of Neuroimmunology. 2004;153:7–15. doi: 10.1016/j.jneuroim.2004.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung PY, Stevens SL, Packard AE, Lessov NS, Yang T, Conrad VK, van den Dungen NN, Simon RP, Stenzel-Poore MP. Toll-like receptor 7 preconditioning induces robust neuroprotection against stroke by a novel type I interferon-mediated mechanism. Stroke. 2012;43:1383–1389. doi: 10.1161/STROKEAHA.111.641522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Li PK, Li C, Lin J. Inhibition of STAT3 signaling blocks the anti-apoptotic activity of IL-6 in human liver cancer cells. Journal of Biological Chemistry. 2010;285:27429–27439. doi: 10.1074/jbc.M110.142752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Hackett SF, Mincey A, Lai H, Campochiaro PA. Effects of different types of oxidative stress in RPE cells. Journal of Cellular Physiology. 2006;206:119–125. doi: 10.1002/jcp.20439. [DOI] [PubMed] [Google Scholar]
- Luo W, Bodary PF, Shen Y, Wickenheiser KJ, Ohman MK, Guo C, Bahrou KL, Myers MG, Jr, Eitzman DT. Leptin receptor-induced STAT3-independent signaling pathways are protective against atherosclerosis in a murine model of obesity and hyperlipidemia. Atherosclerosis. 2011;214:81–85. doi: 10.1016/j.atherosclerosis.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein AD, Finnemann SC, Bonilha VL, Rodriguez-Boulan E. Morphogenesis of the retinal pigment epithelium: toward understanding retinal degenerative diseases. Annals of the New York Academy of Sciences. 1998;857:1–12. doi: 10.1111/j.1749-6632.1998.tb10102.x. [DOI] [PubMed] [Google Scholar]
- Nakamura RE, Hackam AS. Analysis of Dickkopf3 interactions with Wnt signaling receptors. Growth Factors. 2010;28:232–242. doi: 10.3109/08977191003738832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan T, Hands RE, Bustin SA. Quantification of mRNA using real-time RT-PCR. Nature Protocols. 2006;1:1559–1582. doi: 10.1038/nprot.2006.236. [DOI] [PubMed] [Google Scholar]
- Sarafian TA, Montes C, Imura T, Qi J, Coppola G, Geschwind DH, Sofroniew MV. Disruption of astrocyte STAT3 signaling decreases mitochondrial function and increases oxidative stress in vitro. PLoS ONE. 2010;5:e9532. doi: 10.1371/journal.pone.0009532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Yang B, Xi X, Grotta JC, Aronowski J, Savitz SI. IL-10 directly protects cortical neurons by activating PI-3 kinase and STAT-3 pathways. Brain Research. 2011;1373:189–194. doi: 10.1016/j.brainres.2010.11.096. [DOI] [PubMed] [Google Scholar]
- Shiose S, Chen Y, Okano K, Roy S, Kohno H, Tang J, Pearlman E, Maeda T, Palczewski K, Maeda A. Toll-like receptor 3 is required for development of retinopathy caused by impaired all-trans-retinal clearance in mice. Journal of Biological Chemistry. 2011;286:15543–15555. doi: 10.1074/jbc.M111.228551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AK, Yi H, Hayes SH, Seigel GM, Hackam AS. Lithium chloride regulates the proliferation of stem-like cells in retinoblastoma cell lines: a potential role for the canonical Wnt signaling pathway. Molecular Vision. 2010;16:36–45. [PMC free article] [PubMed] [Google Scholar]
- Slater L, Bartlett NW, Haas JJ, Zhu J, Message SD, Walton RP, Sykes A, Dahdaleh S, Clarke DL, Belvisi MG, Kon OM, Fujita T, Jeffery PK, Johnston SL, Edwards MR. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathogens. 2010;6:e1001178. doi: 10.1371/journal.ppat.1001178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens SL, Leung PY, Vartanian KB, Gopalan B, Yang T, Simon RP, Stenzel-Poore MP. Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury. Journal of Neuroscience. 2011;31:8456–8463. doi: 10.1523/JNEUROSCI.0821-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss O. The retinal pigment epithelium. In: Kolb H, Fernandez E, Nelson R, editors. SourceWebvision: The Organization of the Retina and Visual System [Internet] University of Utah Health Sciences Center; Salt Lake City (UT): 1995. [Google Scholar]
- Takeda K, Akira S. TLR signaling pathways. Seminars in Immunology. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Usui S, Komeima K, Lee SY, Jo YJ, Ueno S, Rogers BS, Wu Z, Shen J, Lu L, Oveson BC, Rabinovitch PS, Campochiaro PA. Increased expression of catalase and superoxide dismutase 2 reduces cone cell death in retinitis pigmentosa. Molecular Therapy. 2009;17:778–786. doi: 10.1038/mt.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Noort JM, Bsibsi M. Toll-like receptors in the CNS: implications for neurodegeneration and repair. Progress in Brain Research. 2009;175:139–148. doi: 10.1016/S0079-6123(09)17509-X. [DOI] [PubMed] [Google Scholar]
- Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, Bode B, Manietta N, Walter J, Schulz-Schuffer W, Fassbender K. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cellular Physiology and Biochemistry. 2007;20:947–956. doi: 10.1159/000110455. [DOI] [PubMed] [Google Scholar]
- Yang Z, Stratton C, Francis PJ, Kleinman ME, Tan PL, Gibbs D, Tong Z, Chen H, Constantine R, Yang X, Chen Y, Zeng J, Davey L, Ma X, Hau VS, Wang C, Harmon J, Buehler J, Pearson E, Patel S, Kaminoh Y, Watkins S, Luo L, Zabriskie NA, Bernstein PS, Cho W, Schwager A, Hinton DR, Klein ML, Hamon SC, Simmons E, Yu B, Campochiaro B, Sunness JS, Campochiaro P, Jorde L, Parmigiani G, Zack DJ, Katsanis N, Ambati J, Zhang K. Toll-like receptor 3 and geographic atrophy in age-related macular degeneration. The New England Journal of Medicine. 2008;359:1456–1463. doi: 10.1056/NEJMoa0802437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi H, Patel AK, Sodhi CP, Hackam DJ, Hackam AS. Novel role for the innate immune receptor toll-like receptor 4 (TLR4) in the regulation of the Wnt signaling pathway and photoreceptor apoptosis. PLoS ONE. 2012;7:e36560. doi: 10.1371/journal.pone.0036560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SS, Wei JY, Li C, Barnstable CJ, Fu XY. Expression and activation of STAT proteins during mouse retina development. Experimental Eye Research. 2003;76:421–431. doi: 10.1016/s0014-4835(03)00002-2. [DOI] [PubMed] [Google Scholar]