

Abstract
Purpose
To discover drugs lowering PrPSc in prion-infected cultured neuronal cells that achieve high concentrations in brain to test in mouse models of prion disease and then treat people with these fatal diseases.
Methods
We tested 2-AMT analogs for EC50 and PK after a 40 mg/kg single dose and 40–210 mg/kg/day doses for 3 days. We calculated plasma and brain AUC, ratio of AUC/EC50 after dosing. We reasoned that compounds with high AUC/EC50 ratios should be good candidates going forward.
Results
We evaluated 27 2-AMTs in single-dose and 10 in 3-day PK studies, of which IND24 and IND81 were selected for testing in mouse models of prion disease. They had high concentrations in brain after oral dosing. Absolute bioavailability ranged from 27–40%. AUC/EC50 ratios after 3 days were >100 (total) and 48–113 (unbound). Stability in liver microsomes ranged from 30–>60 min. Ring hydroxylated metabolites were observed in microsomes. Neither was a substrate for the MDR1 transporter.
Conclusions
IND24 and IND81 are active in vitro and show high AUC/EC50 ratios (total and unbound) in plasma and brain. These will be evaluated in mouse models of prion disease.
Keywords: antiprion drugs, drug discovery, IND24, IND81, prion disease
INTRODUCTION
Among the neurodegenerative disorders, one group of diseases is caused by an alternatively folded prion protein (PrP) isoform (1–3). PrP is converted from its cellular isoform, PrPC, to an alternative conformer, PrPSc, which accumulates and causes CNS dysfunction. The conversion of PrPC into PrPSc can occur spontaneously, be induced by an autosomal dominant mutation of the PrP gene or result from exposure to exogenous PrPSc (4). The infectious nature of prions results from the ability of PrPSc to self-propagate by stimulating the misfolding of PrPC. Spontaneous cases account for >85% of all human PrP prion disease, the most common form of which is sporadic Creutzfeldt-Jakob disease (CJD). The inherited forms account for 10–15% of all cases and are typified by Gerstmann-Sträussler-Scheinker (GSS) disease (5). The infectious form represents <1% of all cases and is epitomized by kuru that was transmitted among the Fore people of New Guinea by ritualistic cannibalism (6). Currently, there is no effective treatment for any PrP prion diseases.
CJD shares histopathological findings of aggregated mis-folded protein deposits in the brain with other human neurodegenerative diseases and proteinopathies, including Alzheimer’s disease (AD); Parkinson’s disease (PD); tauopathies, such as frontotemporal dementia (FTD); Huntington’s disease (HD); and amyotrophic lateral sclerosis (ALS) (7,8). AD, HD, PD, and tauopathies involve misprocessing of specific cellular proteins into alternate non-native isoforms that produce cellular toxicity; pathogenic proteins then propagate in a prion-like process (9–15). PrPSc forms insoluble fibrils, then aggregates, some form of which is neurotoxic (16,17). Oligomers may be more neurotoxic than aggregates although this remains controversial. Lowering levels of PrPSc in the brain by halting its formation or increasing its clearance should be therapeutically desirable.
Infectious forms of animal prions can be propagated in vitro in prion-infected murine neuroblastoma cell lines (ScN2a) (18). Compounds reported to be active in vitro in lowering PrPSc levels are known drugs approved for other indications, or small chemical sets expected to have bioactivity, based on results from cell-based assays (19–22). Drugs or experimental compounds reported to have antiprion activity include acridines (e.g., quinacrine) (23,24); tricyclic antidepressants; analogs of statins (25); pyrazolones (26); indole-3-glyoxylamides (27); and pyridyl hydrazones (28), including “Compd B.” Except for Compd B, all have failed to substantially extend survival in prion-infected animals. Larger polyanionic or polycationic compounds, or polyamidoamine dendrimers (PAMAM), have antiprion activity in cells (29), but are not practical as drugs, because of drug delivery, safety, or other issues.
Recently, we discovered a set of molecules containing the 2-aminothiazole (2-AMT) moiety using high-throughput screening (HTS) of almost 10,000 diverse chemical compounds (30) and reported medicinal chemistry efforts to identify more potent and drug-like 2-AMT analogs (31). Preliminary studies indicate that the 2-AMTs do not decrease the expression of PrPC or denature PrPSc, suggesting that they likely exert their antiprion activity by inhibiting the formation or enhancing the clearance of PrPSc (30). Twenty-seven 2-AMT analogs were synthesized and tested in vitro for potency and in two rounds of PK screening. In the first round, single-dose PK studies focused on AUC, AUC/EC50 ratios, and Cmax/EC50 in brain and plasma as criteria for further advancement. In the second round, 10 were evaluated in multiple-dose PK studies to determine 3-day concentrations (C3-day) after dosing as part of a liquid diet. This is the least invasive way to dose mice in efficacy studies that could last up to 300 days or more. From C3-day in lasma and brain, we calculated AUC and AUC/EC50 ratios. IND24 and IND81 were selected for subsequent studies in prion-infected mouse models.
Success treating bacterial infection while preventing resistance is correlated with dosing regimens that achieve and maintain high multiples of AUC/MIC, time above the MIC, or both (32,33). We reasoned that if the same principles apply in the treatment of prion diseases, and in vitro EC50 determinations have predictive value, like MIC, then IND24 and IND81 would be suitable candidates to advance, because both showed brain and plasma AUC/EC50 ratios, after 3 days of dosing, greater than 100 based on total concentration, and 113 (IND24) and 48 (IND81) based on unbound brain concentration when dosed at 210 mg/kg/day.
Overall, the in vitro activity and high AUC/EC50 ratios in vivo in brain and plasma for total and unbound drug predict good extension in survival for IND24 and IND81 in RML-infected mouse models of prion disease. Good extension in survival was previously reported for Compd B in an RML-infected mouse model of prion disease (28). It would be important to compare the effects of IND24 and IND81 in the RML mouse model with Compd B serving as a positive control. Therefore, we evaluated its PK at various doses and showed that at a dose of 100 mg/kg/day, Compd B had similar AUC/EC50 ratios in brain and plasma compared with IND24 and IND81 based on total and unbound concentrations. Compd B has a phenylhydrazone group, which can be activated by cytochrome P450 (CYP450) to reactive intermediates, making it potentially unsuitable as a drug for humans or animals (34). IND24 and IND81 lack a phenyl-hydrazone moiety and were well tolerated when dosed at 210 mg/kg/day for two weeks.
MATERIALS AND METHODS
Materials
All twenty-seven 2-AMT analogs were synthesized at small scale (up to 1 g) at the Small Molecule Discovery Center at UCSF. Lead compounds IND24 and IND81 were subsequently synthesized at 100- to 200-g scales at ChemVeda (Hyderabad, India). One hundred mg of Compd B [(E)-5-(4-(2-(pyridin-4-ylmethylene)hydrazinyl)phenyl)oxazole] along with the synthetic scheme was generously provided by Professor Katsumi Dohura (Tohoku University, Sendai, Japan), subsequently synthesized at UCSF, and finally scaled-up to 100 g quantities at ChemPartner (Shanghai, China). Warfarin (positive control for protein binding assay) and chlorowarfarin (internal standard for warfarin) were obtained from Toronto Research Chemicals (Ontario, Canada). Blank sodium heparinized plasma from mouse (CD-1) and human was obtained from Bioreclamation (Hicksville, NY), and Dulbecco’s phosphate-buffered saline (PBS) from Invitrogen (Carlsbad, CA). The rapid equilibrium dialysis (RED) devices and reusable base plate were obtained from Thermo Scientific (Rockford, IL). Mouse, rat, and human liver microsomes were from Xenotech (Lenexa, KS).
FVB mice (bred at the Hunter’s Point animal facility at UCSF or purchased from Charles River, Hollister, CA) were used for all PK studies. Dose formulations for in vivo PK studies contained DMSO (Thermo Fisher Scientific, Rockford, IL), propylene glycol (Sigma-Aldrich, St. Louis, MO), α-tocopheryl polyethylene glycol 1,000 succinate (TPGS; Eastman Chemical Co., Kingsport, TN), absolute ethanol (Fisher Scientific, Pittsburgh, PA), labrosol (Gattefosse, France), and polyethylene glycol 400 (PEG400; Hampton Research, Aliso Viejo, CA). Rodent liquid diet was obtained from Bio-Serv (French-town, NJ). Brain tissue was homogenized using a Precellys 24 (Bertin Technologies, France) tissue homogenizer. LC/MS/MS analysis was performed using an API 4,000 triple quadruple mass spectrometer (Applied Biosystems) with Analyst 1.4.2 software, coupled to a Shimadzu CBM-20A controller, LC20AD pumps, and SIL-5000 auto sampler (Shimadzu Scientific, Columbia, MD). Compounds were separated on either a BetaBasic C18 or a BDS Hypersil C8 column (both 3 μm, 50×2 mm; Thermo Scientific, Rockford, IL) using a gradient between 0.1% formic acid in water and 0.1% formic acid in acetonitrile (ACN). HPLC-grade ACN and water were obtained from VWR Scientific (Radnor, PA).
Chemistry
The general procedure for synthesis of the 2-AMTs and Compd B are depicted in Schemes 1 and 2, respectively. Details of the synthesis of the 27 2-AMTs are given in the Supplementary Material. Compd B was previously described (28) and our implementation of the synthesis, including a route to Compd B, is described in the Supplementary Material.
Scheme 1.
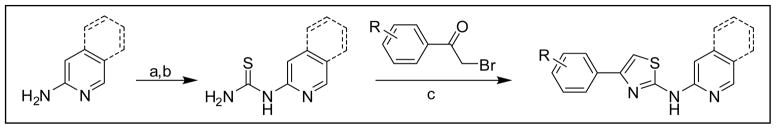
General procedure for synthesis of antiprion 2-AMTs. Conditions: (a) BzNCS, acetone, reflux; (b) NaOH, MeOH, reflux; (c) EtOH, reflux.
Scheme 2.
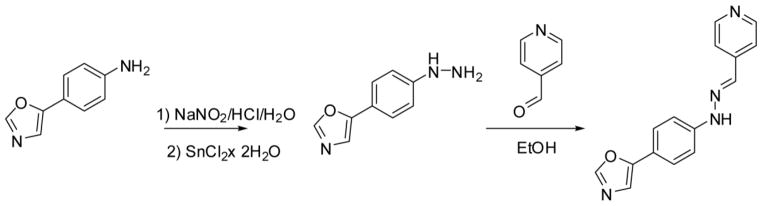
Synthesis of Compd B.
Cell-Based Assays
To identify and confirm hits that lower levels of PrPSc, concentration-effect (EC50) curves were performed by ELISA (Lu et al., in preparation). Mouse N2a neuroblastoma cells (ATCC) transfected with full-length mouse PrP and infected with the RML strain of mouse-adapted scrapie prions (ScN2a-cl3 cells; (35)) were seeded into black-walled, clear-bottomed, tissue culture–treated plates (Greiner) and incubated with the compounds at a final test concentration ranging from 1 nM to 10 μM. After 5 days’ incubation at 37°C in a humidified and 5% CO2-enriched environment, lysates were generated as previously described and transferred to high-binding ELISA plates (Greiner) coated with D18 primary antibody for overnight incubation at 4°C. The next day, the plates were washed with Tris-buffered saline Tween-20 (TBST) before addition of 100 μL of a 1:1,000 dilution of HRP-conjugated D13 antibody in 1% BSA/PBS (1-h incubation at room temperature). After incubation with the D13 antibody, the plates were washed with TBST, 100 μL of 2,2′-azino-bis(3-ethylbenzothiazo-line-6-sulphonic acid (ABTS) was added to each well for 10 min, and absorbance at 405 nm was read using a Spec-traMax M5 plate reader (Molecular Devices, Sunnyvale, CA).
To evaluate cell viability (35), mouse ScN2a-cl3 cells were seeded into 96-well, black polystyrene plates (Greiner) and treated with compound as described above for the ELISA plates. After 5 days, the growth media was aspirated, the plates washed once with PBS (250 μL/well), and the plates aspirated dry. Calcein-AM (100 μL/well, 5 μg/mL solution in calcium-and magnesium-free PBS) was added, and the plates were incubated at 37°C for 45 min. Fluorescent emission intensity was quantified using a Spectramax M5 plate reader, excitation/emission spectra of 485 nm/530 nm.
Calculated Physicochemical Parameters
Calculated physicochemical parameters for the 27 2-AMTs, including tPSA (A), xlogP, and number of hydrogen bond donors and acceptors were determined using SARvision (http://www.chemapps.com) to explore any relationships between physicochemical and PK parameters.
Fasted-State Simulated Intestinal Fluid Solubility
Intestinal solubility was estimated using fasted-state simulated intestinal-fluid (FaSSIF) solubility. Aliquots of the 10-mM DMSO stocks were transferred to pH 2 (HCl) buffer, pH 7.4 (phosphate) buffer with and without 0.05% polysorbate 80 (PS80), or FaSSIF to give a target concentration of 250 μM solute and 2.5% DMSO. After equilibration at room temperature overnight, the solutions were filtered and solute concentration determined by fast gradient HPLC with UV/VIS/MS detection with reference to 1, 5, 10, 50, 100, and 250 μM analytical standards. These analytical standards were prepared from a 500-μM intermediate stock solution made by diluting 10 μl of the 10-mM sample stocks into 190 μl of 50:50 (v/v) ACN/water. Aliquots of 0.4, 2, 4, 20, 40, and 100 μl were transferred in duplicate into a 96-well plate and the volumes were made up to 200 μl per well with ACN/water. The plate was then heat-sealed with a foil sheet. Prior to sample filtration, filters were primed with 600 μl of sample to resolve potential adsorption problems. Both sample replicates were collected through the primed filter. Duplicate determinations were made in all cases.
Media Solubility
To evaluate media solubility, two protocols were followed. In the first, solutions were prepared as described above. After overnight equilibration, samples were centrifuged for 30 min, without filtration, and supernatant analyzed by HPLC/UV/VIS with reference to three standards (5, 100, 250 μM). In the second protocol, turbidimetric analysis, dilutions of the compound of interest were prepared in media with target concentrations of 5, 10, 20, 50, 100, 200, and 300 μM. Media without solute was included for background. These solutions were evaluated using a light scattering technique and a Nepheloskan instrument. A reading that was greater than or equal to 3 times background was considered the limit of solubility.
Hepatic Microsomal Stability
Stability of nine 2-AMT compounds and Compd B were determined in vitro in mouse, rat, and human liver microsomes to compare rates of disappearance across species. Aliquots of DMSO solute stocks were diluted into ACN and then into assay buffer (PBS, pH 7.4,±0.05% PS80). Final experimental solute concentrations were 1 μM (0.6% ACN, 0.01% DMSO). To commercially available mouse, rat, or human hepatic microsomes (~0.5 mg/mL final concentration), NADPH (1 mM) or PBS was added. The resulting mixture was incubated at 37°C, and aliquots removed at 0, 1, 10, 30, and 60 min, then quenched with ACN containing 2 μM carbamazepine (internal standard). After centrifugation at 12,000×g for 10 min, the supernatant was analyzed by LC/MS for remaining starting material. Duplicate incubations were run for each time-point. The percentage of solute remaining at the end of the incubation was used to determine in vitro half-life (t1/2) using the calculations below (36).
where (-k) is the slope of the linear regression line from the plot of log percent remaining versus incubation time.
Identification of Metabolites of IND24 and IND81
Metabolite identification was only performed for IND24 and IND81 because they were the only two that were selected for evaluation in RML-infected mouse models. The goal was to identify metabolites in human microsomes and determine if these would also be observed in mouse, rat, and dog. Metabolites were separated on a Kinetex 100-Å column (2.6 μm, 100×2.1 mm, Phenomenex, Torrance, CA) at ambient temperature. The mobile phase consisted of 0.1% formic acid (Solvent A) and ACN (Solvent B), and was delivered at 0.200 mL/min for 50 min. The initial composition of solvent B was maintained at 1% for 5 min and then increased in a linear manner as follows: 30% at 20 min; 50% at 25 min, maintained at 50% for 3 min; and then increased to 90% at 40 min. Solvent B was maintained at 90% for up to 45 min and then decreased to 1% in the next 2 min. The column was allowed to equilibrate at 1% solvent B for 5 min prior to the next injection. The HPLC effluent going to the mass spectrometer was directed to waste through a divert valve for the initial 5 min after sample injection. Mass spectrometric analyses were performed on a ThermoFinnigan LTQ Orbitrap mass spectrometer (ThermoScientific; Waltham MA), which was interfaced to an Agilent HP-1100 HPLC system (Agilent Technologies, Palo Alto, CA) and equipped with an electro-spray ionization source (ESI). The parameters for the ESI source were: capillary temperature 325°C; source voltage 3.5 kV; source current 100 μA; capillary voltage 33.0 V. The mass spectrometer was operated in a positive-ion mode with data-dependent scanning.
The Orbitrap mass analyzer was calibrated according to the manufacturer’s directions using a mixture of caffeine, Met-Arg-Phe-Ala (MRFA) peptide and Ultramark 1621. The parent compounds and their metabolites were detected by full-scan mass analysis from m/z 150–1000 at a resolving power of 60,000 [at m/z 400, full width at half maximum (FWHM); 1-s acquisition] with data-dependent MS/MS analyses triggered by the most abundant ion. This was followed by MS3 of the most abundant product ion. The resolving power used for multiple-stage mass analysis was the same as the full-scan mass analysis. The CID was conducted with an isolation width of 3 Da, normalized collision energy of 35 for MS/MS and MS3, activation q of 0.25 and an activation time of 30 ms. Default automatic gain control (AGC) target ion values were used for MS, MS/MS, and MS3 analyses. The data obtained were analyzed using Xcalibur v2.1 software (ThermoScientific; Waltham MA). Four-decimal monoisotopic masses of the parent compounds and their oxidative metabolites calculated using ChemBioDraw Ultra software version 11.0 (Cambridge-Soft; Cambridge, MA) were used to interpret further the fragment ions.
Cytochrome P450 Phenotyping
Because IND24 and IND81 were the only compounds selected for in vivo testing in RML-infected mice, they were the only ones subjected to cytochrome P450 phenotyping. IND24 and IND81 were incubated in pooled human liver microsomes (HLMs; 0.5 mg/mL microsomal protein) in the presence or absence of selective inhibitors of P450s, specifically 10 μM furafylline with 15-min preincubation (CYP1A2), 5 μM sulfaphenazole (CYP2C9), 5 μM (+)-N-3-benzylnirvanol (CYP2C19), 1 μM quinidine (CYP2D6), and 1 μM ketoconazole (CYP3A4). IND24 and IND81 concentration was 1 μM and the final organic solvent of the incubation mixture was less than 0.1%. Duplicate incubations were performed and the reaction stopped after 60 min by the addition of 0.1% formic acid in ACN containing internal standard. After vortexing and centrifugation at 12,000×g for 10 min, the supernatant was analyzed by LC/MS/MS.
Involvement in the metabolism of a specific CYP enzyme was estimated based on the following equations:
where TC is the test compound.
CYP450 inhibition or induction studies were not performed at this time to identify potential drug-drug interactions, because CJD is a rapidly fatal disease and dose adjustment could be made for drugs that would need to be coadministered.
Bidirectional MDCK-MDR1 Cell Permeability
Permeability studies were performed to determine which, if any, were substrates for P-glycoprotein (P-gp). MDCK-MDR1 cells were grown to confluence for 5–10 days on 1-μm filters in 24-well plates (37). Aliquots of DMSO stocks were diluted into Hank’s balanced salt solution (HBSS), pH 7.4, containing 25 mM +0.05% PS80 to give 10 μM solute concentration. The solute-containing donor solutions were transferred to either the apical or basolateral chamber of the permeability diffusion apparatus. Receiver solutions consisted of HBSS (pH 7.4) containing 25 mM HEPES +0.05% PS80. Sequential samples of transported solute were taken at 20-min intervals using an automated liquid-handling platform. The concentration of transported solute during each sampling interval was determined by HPLC/UV/MS. Permeability coefficients were calculated for each sampling interval. The average and standard deviation from the intervals are reported. Mass balance in the system was ascertained by comparing the sum of total transported solute and remaining donor solute with the starting mass of solute and is expressed as a percentage of donor solute at time zero. Significant deviations from 100% (generally less than 70%) suggest solute adsorption to the apparatus or monolayer, or chemical or metabolic instability during the course of the experiment. In the event of mass balance less than 70%, the cell monolayers were extracted with ACN and analyzed for the solute of interest. Determinations were conducted in duplicate.
Plasma Protein, Brain Tissue, and Cell-Culture Media Binding
Binding studies were only performed for IND24, IND81, and Compd B because these were the only three selected for in vivo evaluation in RML-infected mice, in order to ultimately evaluate in vitro–in vivo (IVIV) correlations based on total and unbound drug in plasma, brain tissue, and cell-culture media. Stock solutions of 0.1 mM of IND24, IND81, Compd B, and warfarin (positive control) were prepared in DMSO and diluted 100-fold in mouse and human plasma, or in mouse brain homogenate (prepared by diluting brain samples from untreated FVB mice 5-fold with water and homogenizing using a Precellys 24 tissue homogenizer), or in cell-culture media that was supplemented with 10% FBS, to yield a final concentration of 1 μM. The final DMSO concentration in plasma was 1%. In vitro protein binding was determined by using a rapid equilibrium dialysis (RED) device containing a dialysis membrane with a molecular weight cut-off of ~8,000 daltons. The experiments were run in duplicate (warfarin) or triplicate (IND24, IND81, and Compd B). A 200-μl aliquot of the spiked plasma, brain homogenate, or cell-culture media sample was placed in the sample chamber and 350 μl of Dulbecco’s PBS, pH 7.4, was placed in the buffer chamber of the insert. The unit was covered with a sealing tape and incubated at 37°C with shaking at 100 rpm for 4 h. At the end of the incubation period, 50-μl aliquots each from the sample and buffer chambers were pipetted into separate microcentrifuge tubes. The buffer sample was diluted with 50 μl of appropriate blank plasma/brain homogenate/cell-culture media, and an equal volume of PBS, pH 7.4, was added to the plasma/brain homogenate/cell-culture media samples. For analysis, 150 μl of ACN containing internal standard was added to 50-μl aliquots of the samples and centrifuged at ~12,000×g for 10 min. The supernatants were analyzed by LC/MS/MS. Plasma protein binding and binding in cell-culture media was calculated as follows:
The fraction unbound (fu) value in the diluted brain tissue (fumeas) was calculated as above. This was converted to the undiluted fu value (fubrain) using the following equation (38),
for which D is the dilution factor of the brain tissue.
In Vivo Studies
Single-Dose Pharmacokinetic Studies
Twenty-seven 2-AMTs and Compd B synthesized and tested for antiprion potency in dose-titration EC50 studies using an ELISA-based assay were selected for first-round testing in oral PK studies at a single dose of 40 mg/kg. It was important to identify 2-AMT compounds from these studies that showed good exposure, especially in the brain, to advance to 3-day studies.
Protocols for PK studies employing mice were reviewed and approved by the UCSF Institutional Animal Care and Use Committee (IACUC). Female FVB mice, weighing approximately 25 g, were used for all in vivo PK studies. Mice were housed with free access to food and water, and were maintained on 12-h light/dark cycles for 1 week before dosing studies were initiated.
For the single dose of 40 mg/kg, compounds were dissolved in a formulation containing 5% propylene glycol, 35% TPGS and 60% PEG400, and administered by oral gavage. Two animals per time point were used. At specified time points after dosing (0.5, 1, 2, 4, 6, and 24 h), animals were euthanized by CO2, and blood for plasma (by cardiac puncture) and brain samples were collected from each. The heparinized blood samples were centrifuged to obtain plasma. Brain samples were weighed, diluted 10-fold with water, and then homogenized using a Precellys 24 tissue homogenizer. The brain and plasma samples were flash-frozen on dry ice and then stored at −80°C until analysis.
IND24, IND81, and Compd B were also evaluated in single-dose PK studies at 1 mg/kg intravenous (IV) injection and 10 mg/kg oral gavage to characterize several PK parameters, including Vss, CL, t1/2, Cmax, and AUC. For IV dosing through tail injection, IND81 and Compd B were dissolved in either 10% DMSO in PEG400/water (1:1) or 10% DMSO; IND24 was dissolved in 10% ethanol in PEG400/water (1:1). For the single-dose administration at 10 mg/kg, compounds were dissolved in a formulation containing 20% propylene glycol, 5% ethanol, 5% labrosol, and 70% PEG400, and administered by oral gavage. Two animals per time point were used. At specified time points after dosing (5 min, 0.25, 0.5, 1, 2, 4, and 6 h for IV dosing, or 0.25, 0.5, 1, 2, 4, 6, and 24 h for oral dosing), animals were euthanized by CO2. Blood and brain samples were obtained from each. The heparinized blood samples were centrifuged to obtain plasma. Brain samples were weighed, diluted five-fold with water, and then homogenized using a Precellys 24 tissue homogenizer. The brain and plasma samples were flash-frozen on dry ice and then stored at −80°C until analysis.
Multidose Pharmacokinetic Studies
Ten compounds were selected from among the 27 2-AMTs for the second round of screening in 3-day multiple-dose (MD) PK studies at PO doses of 40, 80, 130, and 210 mg/kg, chosen based on AUC and ratios of AUC/EC50 and Cmax/EC50 from the single-dose experiments. We used a liquid diet to facilitate easy drug administration, as well as to serve as the daily source of all water and food. In addition, Compd B at doses of 25, 50, 100, and 150 mg/kg/day was administered for 3 days in liquid diet. This dosing approach was chosen because the mouse bioassay studies that would be used to assess drug effects on survival would be expected to run for 110 to 300 days, or more; gavage dosing for such a long period is not feasible and more stressful to mice. For the MD PK studies with the 2-AMT compounds, 4.5 g of the 2-AMT analogs was added to 15 mL of pure PEG400, vortexed, and sonicated to ensure dissolution, then stored at 4°C until needed. This highly concentrated PEG400 solution was subsequently diluted to final dosing concentrations and added to the rodent liquid diet.
Wild-type FVB mice weigh ~25 g and typically drink 20 mL of liquid diet per day, allowing an estimate of daily drug consumption. A single-dosing cohort consisted of three mice in a shared cage and liquid diet was provided at the start of the study in sufficient volume (~200 mL) to last for the entire 3-day period. Mice typically consume their diet during the dark cycle (12 h), so while a dosing interval is 24 h, consumption of food, water, and drug occurred during a ~12 h period on each of 3 days. Three hours after the end of the final dosing period, animals were euthanized by CO2, followed by collection of plasma (cardiac puncture) and removal of whole brain. Samples were processed and stored as described under single-dose PK studies. IND24 and IND81 were also dosed at 210 mg/kg/day for 14 days to evaluate drug tolerance; methods of administration and sample collection were as described above.
Sample Analysis
Plasma and brain homogenate samples were extracted using a protein-precipitation method and analyzed by specific LC/MS/MS methods developed for each compound dosed in vivo. The analytical method accuracy and precision were monitored by analyzing quality control (QC) samples that were prepared by the same methods as the plasma or brain homogenate samples. The brain and plasma concentration data were used to calculate the maximal concentration (Cmax), area under the concentration-time curve from time 0 to the last time point (AUClast), and absolute bioavailability (%F) by noncompartmental analysis with sparse sampling performed using Phoenix WinNonlin 6.1 software (Pharsight, Mountain View, CA), following single-dose studies. From the multiple-dose PK data, concentrations (C3-day) in brain and plasma were determined from the samples taken 3 h after the end of the dark cycle on the third day. The AUC for plasma and brain after 3 days of oral dosing was estimated (39) as the product of C3-day × τ, where τ is the dosing interval (12 h) because mice eat and drink predominantly during their dark cycle, and this would generate a more conservative estimate for AUC. We know of no other method to estimate AUC after 3 days of dosing using the liquid diet. Other approaches require an assumption of linear PK, which is not observed based on plasma or brain concentrations at the dose of 210 mg/kg/day when compared with the 3-day data at 40 mg/kg or based on single-dose findings after doses of 10 or 40 mg/kg. The ratios of AUC/EC50 were determined from total or unbound drug concentrations in plasma, brain, and cell-culture media. For the unbound ratios, we used unbound concentrations for both AUC and EC50, after appropriate corrections for binding. The C3-day measured and the AUC values estimated are lower than those observed at the end of the dosing interval because the brain and plasma samples were collected 3 h after the end of dosing (the light cycle began at 6:00 AM; brain and plasma samples were collected at 9:00 AM.)
LC/MS Assays
For LC/MS quantification for all 2-AMTs, samples and their respective internal standards were injected into either a BetaBasic C18 or BDS Hypersil C8 column. The solvent system used for separation was composed of water and ACN containing 1% formic acid. For quantification of IND24 and IND81, samples (along with a proprietary internal standard) were injected onto a BetaBasic C18 column maintained at room temperature. The amount of ACN in the gradient was increased from 75% ACN to 95% ACN over 2.5 min, held for 0.5 min, and then re-equilibrated to 75% ACN over 1.4 min. Data acquisition used MRM in the positive-ion mode, and the transitions monitored were m/z 344→226 for IND24; m/z 351→233 for IND81; and m/z 363→245 for internal standard.
For quantification of Compd B, samples (along with a proprietary internal standard) were injected onto a BDS Hypersil C8 column maintained at room temperature. The amount of ACN in the gradient was increased from 25% ACN to 95% ACN over 2.0 min, held for 1.0 min, and then re-equilibrated to 25% ACN over 1.4 min. Data acquisition used MRM in the positive-ion mode, and the transitions monitored were m/z 265→160 for Compd B and m/z 321→253 for internal standard.
RESULTS
Antiprion Potency and Calculated Physicochemical Parameters
Potency was defined as the EC50 value, calculated as the total or unbound drug concentration at which there was a 50% reduction in PrPSc levels. We chose to define potency in terms of EC50 and not IC50 values since IC50 estimates are traditionally used where a specific target is to be inhibited. When we measured changes in PrPSc levels, we did not know the specific target. We defined the concentration producing a 50% reduction in PrPSc as the EC50 value, in a target-agnostic paradigm for which such values were determined irrespective of effects on cell viability (EC50 >10 μM). Figure 1 depicts EC50 curves and extrapolated values for IND24 (1.27 μM) and IND81 (1.95 μM) based on total compound concentration. Corresponding values for unbound concentrations, after correcting for protein binding, are 325 and 452 nM, respectively. Concentration-effect relationships (EC50) were obtained using eight concentrations per curve ranging from 10 nM to 32 μM, increasing by half-log increments. The EC50 values and physicochemical parameters for the twenty-seven 2-AMT compounds and Compd B are shown (Table I). Calculated parameters included total polar surface area (tPSA), xlogP, and number of hydrogen bond donors and acceptors. The calculated values, such as xlogP and PSA, were generally within the range of acceptable values for CNS drugs.
Fig. 1.
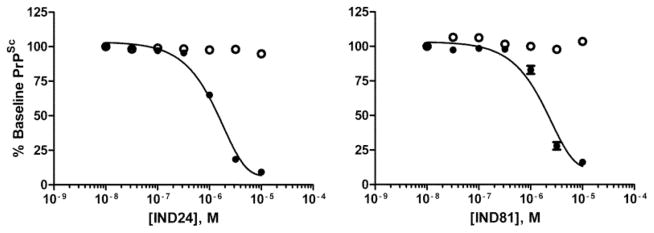
Potency (EC50) curves as measured by ELISA (solid circles) for IND24 (1.27 μM and 325 nM for total and unbound compound, respectively) and IND81 (1.95 μM and 452 nM for total and unbound compound, respectively). Cell viability by calcein (open circles) indicated that LC50 values for both IND24 and IND81 were >10 μM. Solid lines show results based on total drug. All EC50 values are based on n = 3.
Table I.
2-AMT Analogs Synthesized and Tested For Potency With Calculated Parameters
| Compd | Structure | EC50 (μM)a | MW | tPSAb | xlogP | HBA/HBDc |
|---|---|---|---|---|---|---|
| IND2 |
|
1.66 | 297.4 | 47.0 | 4.41 | 4/1 |
| IND7 |
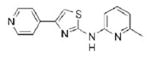
|
1.00 | 268.3 | 50.7 | 3.05 | 4/1 |
| IND22 |
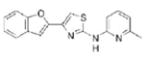
|
1.46 | 307.4 | 51.3 | 5.17 | 4/1 |
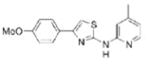
| IND24 |
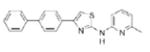
|
1.27 | 343.4 | 37.8 | 6.27 | 3/1 |
| IND26 |
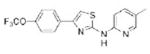
|
>10.0 | 351.4 | 47.0 | 5.81 | 4/1 |
| IND29 |

|
1.57 | 268.3 | 50.7 | 3.13 | 4/1 |
| IND33 |
|
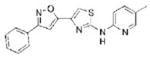
0.94 | 334.4 | 64.1 | 4.29 | 5/1 |
| IND36 |
|
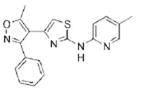
>32.0 | 348.4 | 64.1 | 4.57 | 5/1 |
| IND38 |
|
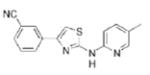
2.29 | 292.4 | 61.6 | 4.16 | 4/1 |
| IND42 |
|
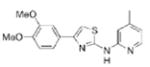
1.00 | 327.4 | 56.3 | 4.37 | 5/1 |
| IND43 |
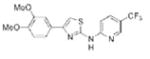
|
2.53 | 381.4 | 56.3 | 5.08 | 5/1 |
| IND44 |
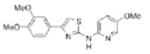
|
0.99 | 343.4 | 65.5 | 4.05 | 6/1 |
| IND46 |
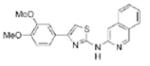
|
0.11 | 363.4 | 56.3 | 5.38 | 5/1 |
| IND47 |
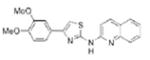
|
0.43 | 363.4 | 56.3 | 5.51 | 5/1 |
| IND48 |
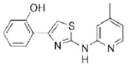
|
>10.0 | 283.4 | 58.0 | 4.07 | 4/2 |
| IND49 |
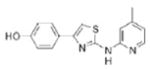
|
>10.0 | 283.4 | 58.0 | 4.07 | 4/2 |
| IND52 |
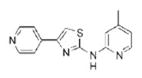
|
4.95 | 268.3 | 50.7 | 3.13 | 4/1 |
| IND57 |
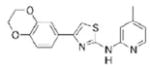
|
>10 | 325.4 | 56.3 | 3.95 | 5/1 |
| IND64 |
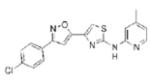
|
0.85 | 368.8 | 64.1 | 4.91 | 5/1 |
| IND74 |
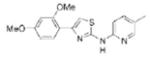
|
3.13 | 327.4 | 56.3 | 4.37 | 5/1 |
| IND76 |
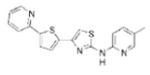
|
1.23 | 350.5 | 50.7 | 4.83 | 4/1 |
| IND78 |
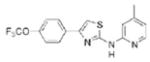
|
>32.0 | 351.4 | 47.0 | 5.81 | 4/1 |
| IND81 |
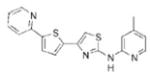
|
1.95 | 350.5 | 50.7 | 4.83 | 4/1 |
| IND82 |
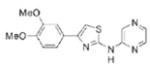
|
15.6 | 314.4 | 69.2 | 2.78 | 6/1 |
| IND85 |
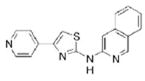
|
0.31 | 304.4 | 50.7 | 4.14 | 4/1 |
| IND86 |
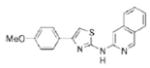
|
0.34 | 333.4 | 47.0 | 5.41 | 4/1 |
| IND91 |
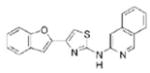
|
0.92 | 343.4 | 51.3 | 6.25 | 4/1 |
| Compd B |
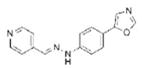
|
0.25 | 264.3 | 63.6 | 2.62 | 5/1 |
EC50 values usually based on n = 3 or more
tPSA = total polar surface area relating to N and O atoms (TPSA_NO) as calculated in Vortex v2011, Dotmatics Limited
HBA = H-bond acceptor; HBD = H-bond donor
Solubility
Determinations of solubility at pH 2.0 and pH 7.4 in FaS-SIF, and in cell-culture media supplemented with 10% FBS were performed for nine selected 2-AMT compounds (Table II). All nine of the 2-AMTs exhibited poor solubility at pH 7.4, but good solubility in the cell-culture media containing proteins. IND44 showed poor solubility in the media. IND22 had high solubility at both pH 2.0 and in FaSSIF. Among the others tested, IND44, IND46, and IND47 had poor solubility at both pH 2.0 and in FaSSIF. Some, like IND81 and IND85, had good solubility at pH 2.0 but poor solubility in FaSSIF, whereas others, like IND24 and IND33, had high solubility in FaSSIF but low solubility at pH 2.0. The pH-dependent solubility is most likely related to the pKa values for each compound and the improved solubility in cell-culture media is most likely related to enhanced solubility in the presence of proteins that bind them.
Table II.
Solubility and Permeability in MDCK-MDR1 Cells of 9 Selected 2-AMT Compounds
| Compound | Solubility (μM)
|
Permeability in MDCK-MDR1 cells
|
|||||
|---|---|---|---|---|---|---|---|
| pH 2.0 | pH 7.4 | FaSSIFa | Mediab | Papp (A−B) × 10−6cm/s | Papp (B−A) × 10−6cm/s | Efflux ratio | |
| IND22 | 12 | <1 | 59 | 20 | 9.5 | 8.4 | 0.9 |
| IND24 | 1 | <1 | 118 | >250 | 5.6 | 7.3 | 1.3 |
| IND33 | 1 | <1 | 70 | 225 | 6.8 | 8.3 | 1.2 |
| IND44 | 1 | <1 | <1 | 3 | 20.1 | 24.4 | 1.2 |
| IND46 | <1 | <1 | 5 | 174 | 13.2 | 10.5 | 0.8 |
| IND47 | <1 | <1 | 5 | 52 | 9.2 | 8.9 | 1 |
| IND52 | 215 | 1 | 2 | 76 | 36.2 | 48.7 | 1.3 |
| IND81 | 78 | <1 | 3 | 207 | 16 | 20.9 | 1.3 |
| IND85 | 196 | <1 | 3 | 116 | 3.03 | 4.44 | 1.5 |
Fasted-state simulated intestinal fluid
Media used in the cell-based assays
Bidirectional MDCK-MDR1 Cell Permeability
Next, we evaluated whether any of the nine 2-AMTs are substrates for P-gp, which is an efflux transporter in the blood–brain barrier and intestinal wall. Permeability of the nine select 2-AMTs was evaluated in MDCK-MDR1 cells (Table II). The apparent permeability (Papp) values ranged from 3 to 49 (×10−6cm/sec). Efflux ratios of the compounds ranged from 0.8 to 1.5, indicating that none are substrates for P-gp.
Stability in Liver Microsomes
The nine 2-AMTs and Compd B were evaluated for hepatic microsomal stability using mouse, rat, and human liver microsomes. As shown in Table III, IND24 appeared to be relatively stable in all 3 species tested, with a t1/2 of >60 min. A few of the 2-AMTs, like IND33, IND46, and IND47, were also relatively stable in the 3 species tested, whereas others like IND85 and IND52 appeared to be rapidly metabolized. Compd B appeared relatively stable in human and rat, but was rapidly metabolized in mouse liver microsomes. There was a good correlation between the hepatic microsomal intrinsic clearance values and in vivo clearance values for IND24 and IND81, for which these data were available (data not shown). This would seem to indicate that in vitro hepatic microsomal assays might help in predicting in vivo clearances for other compounds with the 2-AMT scaffold.
Plasma Protein, Brain Tissue, and Cell Culture Binding
Because we were only advancing IND24 and IND81 to mouse models of prion disease, along with Compd B as a positive control, we evaluated each for binding in plasma, brain homogenates, and in the cell-culture media used for determining EC50 in the ELISA assays, which was supplemented with 10% FBS (Table IV). This was done at a concentration of 1 μM, approximating the EC50 concentration, using rapid equilibrium dialysis (RED). The fraction unbound for IND24, IND81, and Compd B was 6.6, 6.5, and 9% in mouse plasma, and 4.9, 5.8, and 2.1% in human plasma, respectively. Fraction-unbound values for warfarin, a control, were in good agreement with the published literature. Fraction unbound in mouse brain tissue was 9% for IND81, 8% for IND24, and 11% for Compd B. In the cell-culture media, the unbound fraction for IND24, IND81, and Compd B was 26%, 23%, and 37%, respectively. It is important to use the corresponding values when calculating AUC values in plasma and brain tissue and for EC50 values to determine the ratios of AUC/EC50 based on unbound concentrations.
Table IV.
Fraction Unbound of IND24, IND81, and Compd B in Mouse Brain Homogenate, Mouse and Human Plasma, and Cell-Culture Media at 1 μM. Mean Percentage ± SD Based on n=3, Except For Warfarin Where n=2
| Compound | Sample | Unbound (%) |
|---|---|---|
| IND24 | Mouse Plasma | 6.63±1.55 |
| Mouse Brain Homogenate | 8.20±0.35 | |
| Human Plasma | 4.91±1.03 | |
| Cell-culture Media | 25.7±4.93 | |
| IND81 | Mouse Plasma | 6.48±1.10 |
| Mouse Brain Homogenate | 9.43±1.49 | |
| Human Plasma | 5.78±3.21 | |
| Cell-culture Media | 23.2±2.68 | |
| Compd B | Mouse Plasma | 9.06±2.31 |
| Mouse Brain Homogenate | 11.4±2.89 | |
| Human Plasma | 2.10±1.11 | |
| Cell-culture Media | 36.5±3.30 | |
| Warfarin | Mouse Plasma | 10.8±0.0044 |
| Human Plasma | 0.68±0.0023 |
Identification of Metabolites of IND24 and IND81
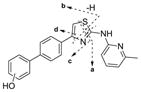
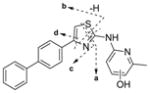
To identify the metabolic profile for IND24 and IND81, we incubated these compounds (10 μM) with human, mouse, rat, and dog liver microsomes, then scanned samples by LC/MS/MS. The structures of the metabolites were characterized using an Orbitrap mass spectrometer and by comparison with the mass spectrum of the parent compound. For IND24, metabolites M1–M3 and M5 were observed in all of the microsomal assays while M4 was only observed in the dog liver microsomal incubation (Fig. 2a). Unchanged compound IND24 eluted at 35.1 min and gave a protonated molecular ion (MH+) at m/z 344.1212 (Fig. 2b). The mass spectrum of MH+ ion at m/z 344.1212 gave fragment ions at m/z 226.0687, 209.0421, 193.0887, and 165.0698 (Fig. 2b, Table V). These fragment ions were most likely formed via cleavage of the thiazole moiety because their masses were similar to the calculated masses (Table V and Fig. 2b). The peaks at 30 (M1), 30.3 (M2), 30.8 (M3), 31 (M4), and 31.5 (M5) min gave a MH+ ion at m/z 360, an addition of 16 amu to 344, suggesting hydroxylation of the parent. The mass spectrum of M1 at m/z 360.1162 showed a major fragment ion at m/z 342.1060 in the MS/MS mass spectrum (Table V). Further fragmentation of the ion m/z 342.1060 in a data-dependent manner (MS3) resulted in major ions at m/z 315.0951, 309.1262, and 161.0167. The ion at m/z 342.1060 resulted from loss of a water molecule from m/z 360.1162; a loss of a water molecule suggests that either the sulfur atom of the thiazole moiety or the methyl group on the pyridine ring was hydroxylated. The exact masses of the fragment ions at m/z 315, 309, and 161 were also in good agreement with the calculated exact masses (Table V). It is well known that the S-oxides formed via oxidation of thiophene rings are generally unstable (40). The same probably applies to the thiazolyl S-oxides. Hence, the site of hydroxylation in metabolite M1 is proposed to be the methyl group on the pyridine ring (Fig. 3).
Fig. 2.

Metabolic profile (UV chromatogram at 268 nm) of IND24 in human, mouse, rat, and dog liver microsomal incubations (a). The parent drug is not shown in the chromatogram. Metabolites M2, M3, and M4 had the same mass spectrum as depicted in Table VI. Extracted ion chromatogram and mass spectrum of unchanged IND24 following liver microsomal incubations (b).
Table V.
Observed (Obs.) and Calculated (Calc.) Molecular Ions And Mass Spectral Fragment Ions of IND24 and its Metabolites (top panel), and IND81 and its Metabolites (bottom panel) Following Incubation With Human, Mouse, Rat, and Dog Liver Microsomes
| Metabolite | Structure | Molecular Ions | Fragment Ions | |||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| MH+ | a | b | c | d | e | |||
| Parent IND24 |
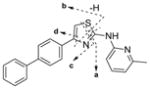
|
Obs. | 344.1212 | 226.0687 | 209.0421 | 193.0887 | 165.0698 | |
| Calc. | 344.1216 | 226.0684 | 209.0420 | 193.0886 | 165.0687 | |||
| M1 |
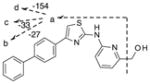
|
Obs. | 360.1162 | 342.1060 | 35.0951 | 309.1262 | 161.0167 | |
| Calc. | 360.1165 | 342.1059 | 315. 0950 | 309.1260 | 6.0167 | |||
| M2/M3/M4 |
|
Obs. | 360.1158 | 242.0634 | 225.0369 | 209.0835 | 181.0647 | |
| Calc. | 360.1165 | 242.0634 | 225.0363 | 209.0835 | 181.0647 | |||
| M5 |
|
Obs. | 360.1160 | 226.0686 | 209.0421 | 193.0886 | 65.0698 | |
| Calc. | 360.1165 | 226.0684 | 209.0420 | 193.0886 | 165.0687 | |||
| Parent IND8 |
|
Obs. | 351.0729 | 243.0048 | 233.0205 | 218.0096 | 206.0095 | |
| Calc. | 351.0732 | 243.0039 | 233.0202 | 218.0092 | 206.0092 | |||
| M1/M3 |
|
Obs. | 367.0680 | 258.9997 | 249.0154 | 234.0045 | 222.0045 | |
| Calc. | 367.0681 | 258.9988 | 249.0150 | 234.0042 | 222.0041 | |||
| M2 |
|
Obs. | 367.0679 | 243.0048 | 233.0204 | 218.0095 | 206.0094 | 349.0576 |
| Calc. | 351.0732 | 243.0039 | 233.0202 | 218.0092 | 206.0092 | 349.0579 | ||
Fig. 3.

Metabolic schemes of IND24 (a) and IND81 (b) following incubation with human, mouse, rat, and dog liver microsomes. For IND24, metabolite M4 was only observed following incubation with dog liver microsomes.
For M2, M3, and M4 of IND24, the mass spectrum of MH+ at m/z 360.1158 resulted in fragment ions at m/z 242.0634 and 225.0369, and m/z 209.0835 and 181.0647 in the MS/MS and MS3 spectra, respectively. An addition of 16 amu indicated an insertion of oxygen into the molecule, but the lack of an ion resulting from loss of a water molecule in the mass spectra suggested that either the biphenyl ring or the pyridine ring of the 2-methylaminopyridine moiety was modified. Modification of the methylpyridine group was ruled out by the ions at m/z 242, 225, 209, and 181, which showed an addition of 16 amu to the masses of the fragment ions observed in the mass spectrum of the parent compound. The observed exact masses of the ions were also in good agreement with the calculated exact masses for the projected fragment ions (Table V). This observation suggests modification of the biphenyl ring in all metabolites (Fig. 3). Although the spectra of the metabolites indicated modification of the biphenyl ring, the exact position of hydroxylation on this moiety could not be determined. Because the mass spectra of M2, M3, and M4 were similar, the metabolites are assumed to be regioisomers. The mass spectrum of M5 at m/z 360.1160 gave fragment ions at m/z 226.0686, 209.0421, 193.0886, and 165.0698 that were consistent with those observed in the mass spectrum of the parent compound. Similarity between the fragment ions from the spectra of M5 and the parent IND24, and the lack of loss of a water molecule for M5, suggest hydroxylation of the pyridine ring of the methylpyridine moiety (Fig. 3).
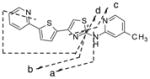
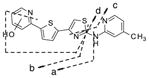
For IND81, metabolites M1–M3 were observed in microsomes of all species evaluated (Fig. 4a). Unchanged compound IND81 eluted at 30.3 min and gave a MH+ ion at m/z 351.0729 (Fig. 4b). The mass spectrum of MH+ ion at m/z 351.0729 gave fragment ions at m/z 243.0048, 233.0205, 218.0096, and 206.0095 (Fig. 4b, Table V). The ion at m/z 243.0048 was a result of the cleavage of the bond between the amino group of the aminomethylpyridine ring and the thiazole ring (Fig. 3b, fragment ion a, Table V). Other fragment ions were most likely generated via cleavage of the thiazole moiety, as their observed masses were similar to the calculated masses of these fragment ions. The peaks at 27.4 (M1), 28.2 (M2), and 28.5 (M3) min gave a molecular ion at m/z 367, an addition of 16 amu to m/z 351, suggesting hydroxylation of the parent IND81. The mass spectra of M1 and M3 at m/z 367.0680 were similar and showed fragment ions at m/z 258.9997, 249.0154, 234.0045, and 222.0045 (Table V). All fragment ions indicated an addition of 16 amu to the masses of the fragment ions observed in the mass spectrum of the parent compound. The observed exact masses of the ions were also in good agreement with the calculated exact masses for the projected fragment ions (Table V). This similarity suggests that the thienylpyridine ring of IND81 is the site of modification (Fig. 3). However, the exact site of oxidation could not be elucidated from this spectral information. Because the mass spectra of M1 and M3 were similar, the metabolites are assumed to be regioisomers. The mass spectrum of M2 at m/z 367.0679 gave fragment ions at m/z 243.0048, 233.0202, 218.0092, 206.0092, and 349.0579 (Table V). While the first four ions were consistent with those observed in the mass spectrum of the parent compound, the ion at m/z 349 indicated a loss of water from the molecule. The observation of the loss of water in the mass spectrum and the consistency of fragmentation of M2 with that of the parent suggest that the methyl group on the pyridine ring is the site of oxidation (Fig. 3b).
Fig. 4.

Extracted ion chromatograms of oxidative metabolites of IND81 at m/z 367 following its incubation with human, mouse, rat, and dog liver microsomes (a). The UV chromatogram of the metabolic profile could not be obtained due to low intensity of the metabolites in the incubation mixture. Extracted ion chromatogram and mass spectrum of IND81 following incubation with liver microsomes (b).
Human P450 Isoforms Involved in Metabolism of IND24 and IND81
To determine which P450 isoforms are involved in the metabolism of IND24 and IND81, different P450 chemical inhibitors were coincubated with the 2-AMTs in human liver microsomes (HLMs). As has been observed previously, there was very little metabolism of IND24 (1 μM) in the HLM incubations, either in the presence or absence of any CYP inhibitors (Table VI). Therefore, the role of any specific P450 isozymes in the metabolism of IND24 could not be determined. In contrast, IND81 was metabolized in HLM with relatively high turnover (Table VI). Based on the change in percentage disappearance, the P450 isozymes most likely to be involved in the metabolism of IND81 are CYP2D6 and CYP3A4; CYP2C19 might be involved to a smaller extent, and CYP1A2 and CYP2C9 are unlikely to be involved.
Table VI.
CYP Phenotyping of IND24 and IND81 at 1 μM in Human Liver Microsomes
| Enzyme | Treatment | IND24
|
IND81
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Measured Conc. (μM) | Disappearance
|
CYP? | Measured Conc. (μM) | Disappearance
|
CYP? | ||||
| % | % Δa | % | % Δa | ||||||
| Irreversible incubation condition | |||||||||
| Negative Control | No NADPH/No Inhibitor | 0.622 | 0.899 | 0 | |||||
| Maximum Metabolism | NADPH/No Inhibitor | 0.733 | 0 (−18) | 0 | 0.143 | 84 | 0 | ||
| CYP1A2 | NADPH + Furafylline | 0.766 | 0 (−23) | 0 | Unlikely | 0.534 | 41 | 43 | Unlikely |
| Reversible incubation condition | |||||||||
| Negative Control | No NADPH/No Inhibitor | 0.654 | 0 | 0.664 | 0 | ||||
| Maximum Metabolism | NADPH/No Inhibitor | 0.655 | 0 | 0 | 0.226 | 66 | 0 | ||
| CYP2C9 | NADPH + Sulphaphenazol | 0.566 | 14 | 0 (−14) | Unlikely | 0.238 | 64 | 1.8 | Unlikely |
| CYP2C19 | NADPH + N-3-B | 0.537 | 18 | 0 (−18) | Unlikely | 0.283 | 57 | 8.5 | Possible |
| CYP2D6 | NADPH + Quinidine | 0.555 | 15 | 0 (−15) | Unlikely | 0.300 | 55 | 11 | Likely |
| CYP3A4 | NADPH + Ketoconazole | 0.557 | 15 | 0 (−15) | Unlikely | 0.324 | 51 | 15 | Likely |
The involvement of the CYP considered unlikely for values <5; possible for values 5–10; and likely for values >10
Single-Dose Pharmacokinetics
Brain and plasma concentrations (AUClast and Cmax values) spanned four orders of magnitude across the 27 2-AMT analogs and Compd B dosed at 40 mg/kg (Fig. 5). IND48 and IND49 had the lowest AUClast values; this observation is not surprising because they were the only two compounds examined that had more than one hydrogen bond donor. Both IND24 and IND81 showed very good brain and plasma exposure, with brain/plasma AUClast ratios of 2.6 and 5.5, respectively, compared to a ratio of 0.5 for Compd B (Table VII). Comparison of the calculated PSA values (Table I) to the observed AUClast values (Fig. 5) showed only a modest correlation. The majority of the 2-AMT analogs had maximal brain and plasma concentrations (Cmax values) and AUC values that far exceeded their in vitro EC50 values (Fig. 6; see Table I for EC50 values). For many analogs at the 40 mg/kg dose, especially IND24 and IND81, the AUClast/EC50 ratio was greater than 50 in brain and greater than 100 in plasma (Fig. 6).
Fig. 5.

Brain (black) and plasma (white) area under the curve (AUClast [μM*h]) for 2-AMTs and Compd B after a single 40 mg/kg oral dose.
Table VII.
Single-Dose Plasma and Brain Pharmacokinetics of IND24, IND81, and Compd B in Female FVB Mice. Intravenous (IV) Dose was 1 mg/kg; Dose by Oral Gavage (PO) was 10 mg/kg. Mean ± SD Shown for Cmax and AUC Value (n = 2)
| Compd | Route/Matrix | Vss (L/kg) | CL (L/h/kg) | t1/2 (h) | Cmax (μM) | tmax (h) | AUClast (μM*h) | %F | Brain/Plasma AUClast Ratio |
|---|---|---|---|---|---|---|---|---|---|
| IND24 | IV/Plasma | 2.46 | 0.92 | 2.16 | 2.49±2.65 | 0.083 | 2.78±1.09 | – | – |
| PO/Plasma | – | – | 4.65 | 1.66±0.11 | 2 | 11.2±1.31 | 40.3 | – | |
| PO/Brain | – | – | nd | 2.45±0.74 | 4 | 29.1±1.24 | – | 2.60 | |
| IND81 | IV/Plasma | 12.6 | 9.05 | 1.18 | 0.52±0.28 | 0.083 | 0.30±0.04 | – | – |
| PO/Plasma | – | – | 0.98 | 0.43±0.03 | 0.5 | 0.82±0.39 | 27.3 | – | |
| PO/Brain | – | – | 1.46 | 1.62±0.54 | 0.5 | 4.54±2.28 | – | 5.53 | |
| Compd B | IV/Plasma | 1.53 | 4.56 | 0.22 | 1.76±0.14 | 0.083 | 0.83±0.12 | – | – |
| PO/Plasma | – | – | 1.01 | 0.83±0.38 | 0.5 | 2.06±0.06 | 24.8 | – | |
| PO/Brain | – | – | nd | 0.46±0.16 | 2 | 1.06±0.10 | – | 0.51 |
Fig. 6.

The ratios of AUC [AUClast (μM*h)] to EC50 in brain (black) and plasma (white) for 2-AMTs and Compd B after a single 40 mg/kg oral dose.
Following 1 mg/kg IV dose, the half-life of IND24 was 2 h, which was 2× and 10× longer than that of IND81 and Compd B, respectively (Table VII). IND24 also had higher oral bioavailability (40%) compared to IND81 (27%) and Compd B (25%).
Multidose Pharmacokinetics
The 2-AMT analogs were administered at 40, 80, 130, and 210 mg/kg/day to FVB mice for 3 days in a liquid diet, then brain and plasma were collected 3 h after the end of the last dark cycle. IND24, IND81, and IND33 achieved the highest concentrations in both brain and plasma for doses greater than 40 mg/kg (Fig. 7). For IND24, both brain and plasma concentrations appeared to approach a plateau at 130 mg/kg, although it was highest at 210 mg/kg. IND81 showed higher but nonlinear increases in brain and plasma with increasing doses. IND33 showed a dose-dependent increase in plasma, but peaked at ~30 μM with the 130 mg/kg dose in brain. IND52 had no measurable brain concentrations at a dose of 40 mg/kg, while exposure was comparable at all other doses. IND22 showed comparable brain and plasma concentrations at all doses. The brain and plasma concentrations of 5 analogs (IND85, IND44, IND46, IND47, IND91) were too low (≤1 μM) to be of therapeutic value. A toleration study, in which mice were dosed with IND24 and IND81 at 210 mg/kg/day for 14 days, indicated that these compounds were well tolerated (data not shown).
Fig. 7.

C3-day concentrations of ten 2-AMT compounds and Compd B in brain (top) and plasma (bottom) following 3-day dosing in liquid diet. Doses were 40, 80, 130, and 210 mg/kg/day for the 2-AMTcompounds, and 25, 50, 100, and 150 mg/kg/day for Compd B. Mean values (n=3) are plotted; mean ± SD values are shown in the Supplementary Material Table. In several instances, concentrations at low doses were below the lower limit of quantification by LC/MS/MS; these brain concentrations appear as missing values.
Additionally, the PK of Compd B was evaluated to determine the suitable dose for use as a positive control in RML-infected mice. Doses of 25, 50, 100, and 150 mg/kg were administered to FVB mice for 3 days in a liquid rodent diet, and then brain and plasma concentrations measured (Fig. 7). Brain and plasma concentrations were below the level of quantification at the 25 and 50 mg/kg/day doses. All animals had measurable levels at 100 mg/kg/day and none showed lethal toxicity at doses of 25, 50, and 100 mg/kg/day. In contrast, two of four mice dosed at 150 mg/kg/day for 8 days died, leaving only two animals to assess drug exposure (Supplementary Material Table).
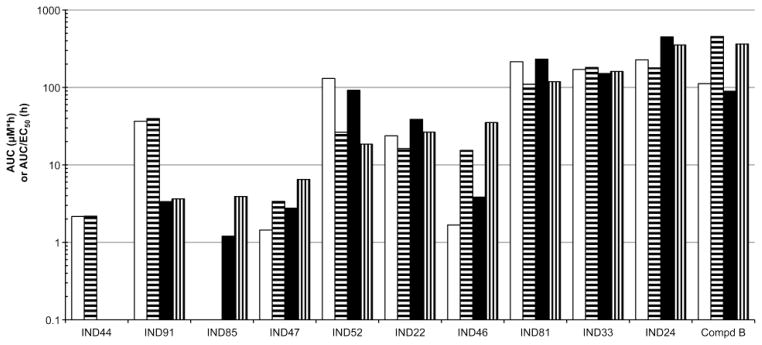
The AUC and AUC/EC50 ratios for plasma and brain after 3-day dosing of IND24 and IND81 at 210 mg/kg/day or 100 mg/kg/day of Compd B were calculated from the C3-day values multiplied by the dosing interval (τ) (Fig. 8). The AUC/EC50 ratios for IND24, IND81, and Compd B were all greater than 100 based on total concentrations in plasma and brain. After correction for binding of the compounds to proteins in brain homogenate, the AUC/EC50 ratios in brain (Fig. 9) were 113, 144, and 48 for IND24, Compd B, and IND81, respectively.
Fig. 8.
AUC values for brain (black) and plasma (white) and AUC/EC50 ratios for brain (vertical stripes) and plasma (horizontal stripes) based on brain and plasma concentrations for ten AMTanalogs and Compd B. Values are based on a dose of 210 and 100 mg/kg/day given for 3 days for the AMTanalogs and Compd B, respectively. AUC values calculated from C3-day × τ, as described in the Methods.
Fig. 9.
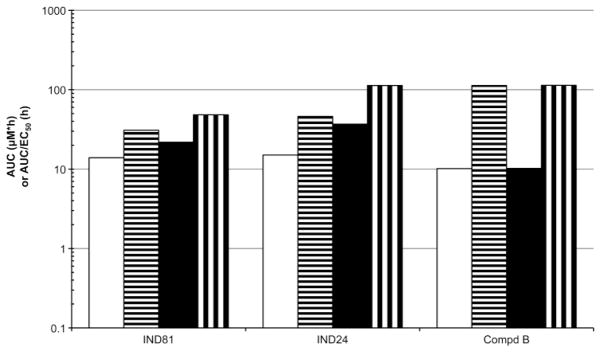
AUC values for brain (black) and plasma (white) and AUC/EC50 ratios for brain (vertical stripes) and plasma (horizontal stripes) based on brain and plasma concentrations and corrected for fraction unbound for IND81, IND24, and Compd B. Values are based on a dose of 210 mg/kg/day (IND81 and IND24) or 100 mg/kg/day (Compd B) given for 3 days. AUC values calculated from C3-day × τ, as described in Methods.
DISCUSSION
No approved drug exists for human use that slows CJD, or any of the other PrP prion disorders. We previously reported the identification of diverse chemical leads that have good potency in reducing PrPSc in prion-infected mouse neuronal cells, including a 2-AMT series (30). Subsequently, we described structure-activity relationships in the 2-AMT series and the identification of more potent and drug-like analogs (31). To date, 235 analogs in the 2-AMT series have been synthesized and evaluated for antiprion potency (EC50).
The goal of the present work was to select two to three compounds for testing in separate experiments using prion-infected mouse models that recapitulate the pathology seen in patients with CJD (10,41). We set out to determine if in vitro potency in the ScN2a-cl3 assay and PK parameters like AUC and the ratio of AUC/EC50 predict in vivo efficacy in the models before testing in patients. Thus far, except for Compd B, no drug has been able to extend survival in these mouse models beyond 150 days (28), depending on the model. Mortality is typically observed at approximately 110–120 days post inoculation (dpi) with RML prions in wt FVB mice expressing normal levels of PrPC (42).
From the first ~100 analogs of 2-AMTs, we evaluated 27 in single-dose PK studies (Table I). From these 27, we selected 10, based primarily on AUC and AUC/EC50 ratios, and also Cmax/EC50 ratios, for multiple-dose PK studies, where we determined concentrations at the end of 3 days of dosing (C3-day), AUC, and the ratio of AUC/EC50 in brain and plasma. We also evaluated drug-like properties for 9 of the 2-AMTs, including solubility, permeability, and P-gp substrate potential, a good predictor of brain delivery limitations due to P-gp (Table II).
IND24 and IND81 showed high AUC and AUC/EC50 ratios in brain and plasma. Because they had some of the highest AUC and AUC/EC50 ratios in brain and plasma after the 210 mg/kg/day dose, and acceptable drug-like properties, including toleration after 14 days of dosing at 210 mg/kg/day, they were selected to advance to in vivo animal studies. Some compounds, like IND85, actually had a better Cmax/EC50 ratio at either dose than IND24 and IND81, but had very low C3-day concentrations, AUC, and AUC/EC50 ratios in brain and plasma on multiple dosing and were not evaluated further.
From the C3-day results, we reasoned that high AUC and AUC/EC50 ratios greater than 10 would support a proof-of-concept experiment in prion-infected mice. Multiples of AUC/MIC ratios, where AUC is based on plasma data, have been associated with successful treatment and avoidance of resistance in the treatment of bacterial infections with many antibiotics (32, 33). Brain and plasma AUC and the AUC/EC50 ratios were greater than 100 for IND24 and IND81 based on total drug concentrations (Fig. 8), and 113 for IND24 and 48 for IND81 based on unbound brain concentrations (Fig. 9). It was important to take into account the binding of the compounds in cell-culture media when using the EC50 values for calculating the AUC/EC50 ratios for unbound drug.
The “drug in liquid diet” approach used in the 3-day studies is practical because it simplifies drug administration during chronic dosing, allowing mice to achieve and maintain drug concentrations for long periods each day as mice are nocturnal and feed/drink predominantly during the dark cycle. It seemed crucial to have a dosing regimen that could be tolerated for up to 300 days or longer without the need to frequently handle mice daily over long periods of drug treatment.
It was important to characterize Compd B in PK studies to determine doses and formulations to be used as a positive control in studies in prion-infected mice. Compd B has been shown to extend the incubation times in prion-infected transgenic mice that overexpress PrPC (28), but may not be acceptable for use in humans because it contains a hydrazone moiety that is metabolically unstable and is likely to lead to reactive intermediates with adverse side effects (34). Our studies with Compd B revealed lethal toxicity when administered chronically in vivo for 8 days at doses >110 mg/kg/day. We found that doses up to 100 mg/kg/day resulted in AUC/EC50 ratios greater than 100 for total and unbound concentrations.
In summary, IND24 and IND81 reduced PrPSc in ScN2a-cl3 cells and have good PK properties in vivo, especially high AUC and AUC/EC50 ratios in brain and plasma at doses of 210 mg/kg/day based on total and unbound compounds. We believe that these compounds are good candidates for testing in prion-infected mouse models.
Supplementary Material
Table III.
Metabolic Stability of 9 Selected 2-AMT Compounds and Compd B
| Compound | t1/2 in min (% remaining after a 60-min incubation)
|
||
|---|---|---|---|
| Mouse | Rat | Human | |
| IND22 | 49 (40) | >60 (64) | >60 (99) |
| IND24 | >60 (89) | >60 (90) | >60 (105) |
| IND33 | >60 (111) | >60 (73) | 54 (42) |
| IND44 | 29 (24) | 45 (39) | 58 (48) |
| IND46 | >60 (58) | >60 (61) | >60 (83) |
| IND47 | >60 (56) | >60 (50) | >60 (113) |
| IND52 | 6 (0) | 16 (8) | 36 (30) |
| IND81 | 19 (10) | 58 (48) | 52 (48) |
| IND85 | 15 (6) | 18 (32) | 53 (66) |
| Compd B | 30 (29) | >60 (77) | >60 (68) |
Acknowledgments
ACKNOWLEDGMENTS AND DISCLOSURES
The authors thank Ms. Ana Serban, Ms. Julia Becker, and Mr. Frederic Letessier for D13 and D18 antibodies; Mr. Phillip Benner and the staff of the Hunter’s Point animal facility for expert animal studies; Dr. Sina Ghaemmaghami for many helpful discussions; and Ms. Hang Nguyen for editorial assistance. This work was supported by grants from the National Institutes of Health (AG002132, AG010770, AG031220, and AG021601) as well as by gifts from the Sherman Fairchild Foundation, Rainwater Charitable Foundation, Lincy Foundation, Fight for Mike Homer Program, and Robert Galvin. MPJ is a consultant to Schrodinger LLC.
ABBREVIATIONS
- 2-AMT
2-aminothiazole scaffold
- AUC
area under the drug concentration time curve
- C3-day
drug concentration after 3 days of dosing
- CJD
Creutzfeldt-Jakob disease
- Cl
clearance (total, intrinsic, hepatic, or otherwise)
- Cmax
maximum drug concentration
- dpi
days postinoculation with prions
- EC50
potency; drug concentration producing 50% of the maximal effect
- FaSSIF
fasted-state simulated intestinal fluid
- HTS
high-throughput screening
- IV
intravenous
- MDCK-MDR1
Madin Darby canine kidney cells transfected with MDR1 gene
- MDR1
multidrug resistance protein 1, ATP-binding cassette sub-family B member 1
- MIC
minimum inhibitory concentration
- P-gp
p-glycoprotein
- PK
pharmacokinetics
- PO
oral
- PrPC
benign normally occurring prion protein on cell surface or inside cell
- PrPSc
abnormal, misfolded, pathogenic form of PrPC
- RML
Rocky Mountain Laboratory
- SAR
structure-activity relationship
- ScN2a-cl3
scrapie (RML)-infected neuroblastoma cells that overexpress PrPC
- V
volume of distribution (steady-state or otherwise)
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s11095-012-0912-4) contains supplementary material, which is available to authorized users.
Contributor Information
B. Michael Silber, Institute for Neurodegenerative Diseases, University of California, San Francisco, California, USA. Department of Neurology, University of California, San Francisco, California, USA. Department of Bioengineering and Therapeutic Sciences, University of California, San Francisco, California, USA.
Satish Rao, Institute for Neurodegenerative Diseases, University of California, San Francisco, California, USA. Department of Neurology, University of California, San Francisco, California, USA.
Kimberly L. Fife, Institute for Neurodegenerative Diseases, University of California, San Francisco, California, USA. Department of Bioengineering and Therapeutic Sciences, University of California, San Francisco, California, USA
Alejandra Gallardo-Godoy, Small Molecule Discovery Center, University of California, San Francisco, California, USA.
Adam R. Renslo, Small Molecule Discovery Center, University of California, San Francisco, California, USA. Department of Pharmaceutical Chemistry, University of California, San Francisco, California, USA
Deepak K. Dalvie, Pfizer Global Research & Development, La Jolla, California, USA
Kurt Giles, Institute for Neurodegenerative Diseases, University of California, San Francisco, California, USA. Department of Neurology, University of California, San Francisco, California, USA.
Yevgeniy Freyman, Institute for Neurodegenerative Diseases, University of California, San Francisco, California, USA.
Manuel Elepano, Institute for Neurodegenerative Diseases, University of California, San Francisco, California, USA.
Joel R. Gever, Institute for Neurodegenerative Diseases, University of California, San Francisco, California, USA. Department of Neurology, University of California, San Francisco, California, USA
Zhe Li, Institute for Neurodegenerative Diseases, University of California, San Francisco, California, USA. Department of Neurology, University of California, San Francisco, California, USA. Department of Bioengineering and Therapeutic Sciences, University of California, San Francisco, California, USA.
Matthew P. Jacobson, Department of Pharmaceutical Chemistry, University of California, San Francisco, California, USA
Yong Huang, Department of Bioengineering and Therapeutic Sciences, University of California, San Francisco, California, USA.
Leslie Z. Benet, Department of Bioengineering and Therapeutic Sciences, University of California, San Francisco, California, USA
Stanley B. Prusiner, Email: stanley@ind.ucsf.edu, Institute for Neurodegenerative Diseases, University of California, San Francisco, California, USA. Department of Neurology, University of California, San Francisco, California, USA. 675 Nelson Rising Ln, Room 318, San Francisco, California 94143-0518, USA
References
- 1.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–44. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 2.Weissmann C. Spongiform encephalopathies - the prion’s progress. Nature. 1991;349:569–71. doi: 10.1038/349569a0. [DOI] [PubMed] [Google Scholar]
- 3.Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–50. doi: 10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- 4.Prusiner SB. Prions and neurodegenerative diseases. N Engl J Med. 1987;317:1571–81. doi: 10.1056/NEJM198712173172505. [DOI] [PubMed] [Google Scholar]
- 5.Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD, et al. Linkage of a prion protein missense variant to Gerstmann-Sträussler syndrome. Nature. 1989;338:342–5. doi: 10.1038/338342a0. [DOI] [PubMed] [Google Scholar]
- 6.Alpers M, Gajdusek DC. Changing patterns of kuru: epidemiological changes in the period of increasing contact of the Fore people with western civilization. Am J Trop Med Hyg. 1965;14:852–79. doi: 10.4269/ajtmh.1965.14.852. [DOI] [PubMed] [Google Scholar]
- 7.Prusiner SB. Shattuck lecture — neurodegenerative diseases and prions. N Engl J Med. 2001;344:1516–26. doi: 10.1056/NEJM200105173442006. [DOI] [PubMed] [Google Scholar]
- 8.Frost B, Diamond MI. Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci. 2010;11:155–9. doi: 10.1038/nrn2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caughey B, Baron GS, Chesebro B, Jeffrey M. Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu Rev Biochem. 2009;78:177–204. doi: 10.1146/annurev.biochem.78.082907.145410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeArmond SJ, McKinley MP, Barry RA, Braunfeld MB, McColloch JR, Prusiner SB. Identification of prion amyloid filaments in scrapie-infected brain. Cell. 1985;41:221–35. doi: 10.1016/0092-8674(85)90076-5. [DOI] [PubMed] [Google Scholar]
- 11.Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science. 2005;307:262–5. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 12.Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14:504–6. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- 13.Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA. 2009;106:13010–5. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nekooki-Machida Y, Kurosawa M, Nukina N, Ito K, Oda T, Tanaka M. Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc Natl Acad Sci USA. 2009;106:9679–84. doi: 10.1073/pnas.0812083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sydow A, Mandelkow EM. ‘Prion-Like’ propagation of mouse and human tau aggregates in an inducible mouse model of tauopathy. Neurodegener Dis. 2010;7:28–31. doi: 10.1159/000283479. [DOI] [PubMed] [Google Scholar]
- 16.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–98. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 17.Novitskaya V, Bocharova OV, Bronstein I, Baskakov IV. Amyloid fibrils of mammalian prion protein are highly toxic to cultured cells and primary neurons. J Biol Chem. 2006;281:13828–36. doi: 10.1074/jbc.M511174200. [DOI] [PubMed] [Google Scholar]
- 18.Race RE, Fadness LH, Chesebro B. Characterization of scrapie infection in mouse neuroblastoma cells. J Gen Virol. 1987;68:1391–9. doi: 10.1099/0022-1317-68-5-1391. [DOI] [PubMed] [Google Scholar]
- 19.Kocisko DA, Baron GS, Rubenstein R, Chen J, Kuizon S, Caughey B. New inhibitors of scrapie-associated prion protein formation in a library of 2000 drugs and natural products. J Virol. 2003;77:10288–94. doi: 10.1128/JVI.77.19.10288-10294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kocisko DA, Caughey B, Morrey JD, Race RE. Enhanced anti-scrapie effect using combination drug treatment. Antimicrob Agents Chemother. 2006;50:3447–9. doi: 10.1128/AAC.00715-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trevitt CR, Collinge J. A systematic review of prion therapeutics in experimental models. Brain. 2006;129:2241–65. doi: 10.1093/brain/awl150. [DOI] [PubMed] [Google Scholar]
- 22.Sim VL, Caughey B. Recent advances in prion chemotherapeutics. Infect Disord Drug Targets. 2009;9:81–91. doi: 10.2174/1871526510909010081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korth C, May BCH, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci USA. 2001;98:9836–41. doi: 10.1073/pnas.161274798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.May BCH, Witkop J, Sherrill J, Anderson MO, Madrid PB, Zorn JA, et al. Structure-activity relationship study of 9-aminoacridine compounds in scrapie-infected neuroblastoma cells. Bioorg Med Chem Lett. 2006;16:4913–6. doi: 10.1016/j.bmcl.2006.06.050. [DOI] [PubMed] [Google Scholar]
- 25.Kempster S, Bate C, Williams A. Simvastatin treatment prolongs the survival of scrapie-infected mice. NeuroReport. 2007;18:479–82. doi: 10.1097/WNR.0b013e328058678d. [DOI] [PubMed] [Google Scholar]
- 26.Kimata A, Nakagawa H, Ohyama R, Fukuuchi T, Ohta S, Dohura K, et al. New series of antiprion compounds: pyrazolone derivatives have the potent activity of inhibiting protease-resistant prion protein accumulation. J Med Chem. 2007;50:5053–6. doi: 10.1021/jm070688r. [DOI] [PubMed] [Google Scholar]
- 27.Thompson MJ, Borsenberger V, Louth JC, Judd KE, Chen B. Design, synthesis, and structure–activity relationship of indole-3-glyoxylamide libraries possessing highly potent activity in a cell line model of prion disease. J Med Chem. 2009;52:7503–11. doi: 10.1021/jm900920x. [DOI] [PubMed] [Google Scholar]
- 28.Kawasaki Y, Kawagoe K, Chen CJ, Teruya K, Sakasegawa Y, Dohura K. Orally administered amyloidophilic compound is effective in prolonging the incubation periods of animals cerebrally infected with prion diseases in a prion strain-dependent manner. J Virol. 2007;81:12889–98. doi: 10.1128/JVI.01563-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Supattapone S, Wille H, Uyechi L, Safar J, Tremblay P, Szoka FC, et al. Branched polyamines cure prion-infected neuroblastoma cells. J Virol. 2001;75:3453–61. doi: 10.1128/JVI.75.7.3453-3461.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghaemmaghami S, May BCH, Renslo AR, Prusiner SB. Discovery of 2-aminothiazoles as potent antiprion compounds. J Virol. 2010;84:3408–12. doi: 10.1128/JVI.02145-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gallardo-Godoy A, Gever J, Fife KL, Silber BM, Prusiner SB, Renslo AR. 2-Aminothiazoles as therapeutic leads for prion diseases. J Med Chem. 2011;54:1010–21. doi: 10.1021/jm101250y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Craig WA. The role of pharmacodynamics in effective treatment of community-acquired pathogens. Johns Hopkins Adv Stud Med. 2002;2:126–34. [Google Scholar]
- 33.Ambrose PG, Bhavnani SM, Rubino CM, Louie A, Gumbo T, Forrest A, et al. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: it’s not just for mice anymore. Clin Infect Dis. 2007;44:79–86. doi: 10.1086/510079. [DOI] [PubMed] [Google Scholar]
- 34.Jonen HG, Werringloer J, Prough RA, Estabrook RW. The reaction of phenylhydrazine with microsomal cytochrome P-450. Catalysis of heme modification. J Biol Chem. 1982;257:4404–11. [PubMed] [Google Scholar]
- 35.Ghaemmaghami S, Ullman J, Ahn M, StMartin S, Prusiner SB. Chemical induction of misfolded prion protein conformers in cell culture. J Biol Chem. 2010;285:10415–23. doi: 10.1074/jbc.M109.045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos. 1999;27:1350–9. [PubMed] [Google Scholar]
- 37.Hilgers AR, Conradi RA, Burton PS. Caco-2 cell monolayers as a model for drug transport across the intestinal mucosa. Pharm Res. 1990;7:902–10. doi: 10.1023/a:1015937605100. [DOI] [PubMed] [Google Scholar]
- 38.Kalvass JC, Maurer TS. Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm Drug Dispos. 2002;23:327–38. doi: 10.1002/bdd.325. [DOI] [PubMed] [Google Scholar]
- 39.Gibaldi M, Perrier D. Pharmacokinetics. 2. New York: Marcel Dekker, Inc; 1982. [Google Scholar]
- 40.Ha-Duong NT, Dijols S, Macherey AC, Goldstein JA, Dansette PM, Mansuy D. Ticlopidine as a selective mechanism-based inhibitor of human cytochrome P450 2C19. Biochemistry. 2001;40:12112–22. doi: 10.1021/bi010254c. [DOI] [PubMed] [Google Scholar]
- 41.Korth C, Kaneko K, Groth D, Heye N, Telling G, Mastrianni J, et al. Abbreviated incubation times for human prions in mice expressing a chimeric mouse–human prion protein transgene. Proc Natl Acad Sci USA. 2003;100:4784–9. doi: 10.1073/pnas.2627989100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghaemmaghami S, Ahn M, Lessard P, Giles K, Legname G, DeArmond SJ, et al. Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog. 2009;5:e1000673. doi: 10.1371/journal.ppat.1000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.