

Abstract
The assembly and deposition of amyloid β-protein (Aβ) in brain is a key pathological feature of Alzheimer’s disease and related disorders. Factors have been identified that can either promote or inhibit Aβ assembly in brain. We previously reported that myelin basic protein (MBP) is a potent inhibitor of Aβ fibrillar assembly [Hoos et al. 2007 J. Biol. Chem. 282:9952–9961; Hoos et al. 2009 Biochemistry 48:4720–4727]. Moreover, the region on MBP responsible for this activity was localized to the N-terminal 64 amino acids (MBP1-64) [Liao et al. 2010 J. Biol. Chem. 285:35590–35598]. In the present study we sought to further define the site on MBP1-64 involved in this activity. Deletion mapping studies showed that the C-terminal region (residues 54–64) is required for the ability of MBP1-64 to bind Aβ and inhibit fibril assembly. Alanine scanning mutagenesis revealed that amino acids K54, R55, G56 and K59 within MBP1-64 are important for both Aβ binding and inhibition of fibril assembly as assessed by solid phase binding, thioflavin T binding and fluorescence, and transmission electron microscopy studies. Strong spectral shifts are observed by solution NMR spectroscopy of specific N-terminal residues (E3, R5, D7, E11 and Q15) of Aβ42 upon the interaction with MBP1-64. Although the C-terminal region of MBP1-64 is required for interactions with Aβ, a synthetic MBP50-64 peptide was itself devoid of activity. These studies identify key residues in MBP and Aβ involved in their interactions and provide structural insight into how MBP regulates Aβ fibrillar assembly.
Extracellular deposition of the amyloid β-protein (Aβ) in brain is a prominent pathological feature of Alzheimer’s disease (AD) and a number of related disorders1,2. Aβ is a 39–43 amino acid peptide that exhibits a high propensity to self-assemble into β sheet-containing soluble oligomeric forms and fibrils3,4. Aβ peptides are proteolytically derived from a large type I integral membrane precursor protein, termed the amyloid β-protein precursor (AβPP)5–8. The amyloidogenic processing of AβPP initially involves a proteolytic cleavage at the amino terminus of the Aβ peptide sequence by β-secretase, an aspartyl proteinase named BACE9–11. Subsequent proteolytic cleavage of the remaining amyloidogenic membrane spanning AβPP carboxyl terminal fragment by γ-secretase liberates the predominant Aβ40 or Aβ42 residue peptides12–14. In AD, cerebral Aβ deposition occurs primarily in the form of parenchymal amyloid plaques1,2. The deposition of Aβ peptides also occurs in cerebral blood vessels, a condition known as cerebral amyloid angiopathy (CAA)15–17.
Aβ42 is considered to be more pathogenic due to its stronger ability to assemble into toxic species compared to Aβ401,2,4,18. Further, specific mutations in Aβ, including Dutch E22Q and Iowa D23N substitutions that are associated with familial forms of CAA19–21, exhibit greatly enhanced the fibrillogenic and pathogenic properties compared to the normal, wild-type (WT) forms of Aβ22–26. Monomeric Aβ peptides initially aggregate as low molecular mass oligomeric species that adopt progressive β-sheet content to assemble into higher oligomeric forms, protofibrils, and ultimately amyloid fibrils that deposit in cerebral tissues4,27–29. It is likely that different assemblies of Aβ can promote various pathogenic responses that collectively contribute to the syndrome of AD. For example, different soluble oligomeric species of Aβ are directly toxic to neurons, can interfere with long-term potentiation, and disrupt the integrity of cell membranes4,29–33. On the other hand, fibrillar assemblies of Aβ are toxic to neuronal and cerebral vascular cells, can activate complement, and can stimulate potent neuroinflammatory responses34–39. Understanding the assembly of Aβ is key to unraveling its pathogenesis in AD and related disorders.
A number of naturally occurring Aβ chaperone molecules have been identified in the CNS that modulate fibrillar assembly of the peptide. For example, apolipoprotein E (apoE) may either promote or inhibit Aβ fibril formation in vitro dependent on the isoform (E3 vs. E4) and/or the extent of lipidation40–43. Further in vivo studies in transgenic mice have demonstrated that endogenous mouse apoE facilitates Aβ fibril formation44,45. Similarly, apolipoprotein J, otherwise known as clusterin, is another chaperone protein that promotes Aβ fibril formation in vitro and in vivo46,47. Some other reported Aβ chaperones include apolipoprotein A-148,49 α1-anti-chymotrypsin50,51, transthyretin52,53 and gangliosides54,55. Earlier, we identified myelin basic protein (MBP), an abundant component of the axonal myelin sheath, as a novel chaperone that can bind both WT forms of Aβ and Dutch/Iowa (D/I) CAA mutant forms of Aβ and potently inhibit their fibrillogenesis56,57. Detailed ultrastructural analysis showed that MBP allows the assembly of soluble oligomeric species but prevents their further maturation into larger protofibrils and amyloid fibrils56,57. Further analysis revealed that the N-terminal residues 1–64 of MBP contained the Aβ binding domain and inhibited Aβ fibrillogenesis in a similar manner as intact MBP58.
Here we report the further characterization of the Aβ binding site on MBP1-64 involved in its fibril inhibiting activity. Deletion mapping studies showed that the C-terminal region of MBP1-64 (residues 54–64) is required for its ability to bind Aβ and inhibit fibril assembly. Alanine mutagenesis scanning revealed that amino acids K54, R55, G56 and K59 within MBP1-64 are important for both Aβ binding and inhibition of fibril assembly. We further compared the interaction of MBP1-64 with Aβ42WT using solution NMR spectroscopy in order to assess the mechanism of the MBP1-64 – Aβ interaction and to compare to small molecule inhibitors of Aβ fibrillization. The analysis reveals specific residues in Aβ42WT that shift in response to its interaction with MBP1-64. Although the region MBP54-64 is required for interactions with Aβ a synthetic MBP50-64 peptide was itself devoid of activity. These studies identify precise residues in MBP that mediate its activities towards Aβ and provide structural insight into how MBP regulates Aβ fibrillogenesis.
MATERIALS AND METHODS
Reagents and Chemicals
Aβ42WT and Aβ40DI peptides were synthesized by solid-phase Fmoc (9-fluorenylmethoxycarbonyl) amino acid chemistry, purified by reverse phase HPLC, and structurally characterized as previously described59. Aβ peptides were initially prepared in hexafluoroisopropanol, lyophilized, and resuspended in either dimethylsulfoxide (Me2SO) or 100 mM NaOH as previously described27. MBP50-64 was synthesized and purified to >95% by reverse phase HPLC (China Peptides, Shanghai, China).
Recombinant MBP Peptide Expression
MBP derived peptide gene sequences (MBP1-64 and MBP1-53) were cloned into a pTYB11 plasmid vector (New England Biolabs, Ipswich, MA) and transformed into competent E. coli BL21 DE3 cells by heat shock. Cells were grown at 37 °C in 1 L cultures of LB broth containing 0.1 mg/ml ampicillin until an optical density of 0.600 AU at 600 nm was reached. Expression of the fusion protein was induced with 0.3 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 26 °C for 18 h. Cells were harvested by centrifugation at 5,000 × g for 30 min at 4 °C and cracked in a French press in 20 mM Tris-HCl, pH 9.0/0.5 M NaCl/1 mM EDTA containing Complete Protease Inhibitor (Roche, Mannheim, Germany). Cell lysate was clarified by centrifugation and passed over Chitin Beads (New England Biolabs, Ipswich, MA) equilibrated with 20 mM Tris-HCl, pH 9.0/0.5 M NaCl/1 mM EDTA (EQ buffer). The column was washed with EQ buffer containing 0.05% Triton X-100, and the peptide cleaved and eluted from the intein fusion protein by incubation of the column in 40 mM dithiothreitol (DTT) as per manufacturers instruction. The eluate was diluted 10 fold into 50 mM glycine, pH 9.0 and passed over a CM52 column equilibrated with 50 mM glycine, pH 9.0/50 mM NaCl (CM EQ). The column was washed in CM EQ and eluted with high salt. Fractions were analyzed by SDS-PAGE, pooled, dialyzed against water, lyophilized, and stored at −70 °C.
Site-Directed Mutagenesis of Human MBP1-64
The MBP1-64 in pTBY11 vector (plasmid DNA template) was used for alanine scanning site-directed mutagenesis, as per manufacturers instruction (Affymetrix, Santa Clara, CA), to produce MBP1-64 with the following mutated residues (K54A, R55A, G56A, S57A, G58A, D60A, S61A, H62A, and H63A). The mutant MBP1-64 peptides were expressed and purified as described above.
Solid Phase Binding Assay
Lyophilized Aβ40D/I and Aβ42WT peptides were resuspended with Me2SO to 2.5 mM, diluted to 12.5 μM in 50 μl μl of PBS, and then coated on flat bottom 96 well plates (Fisher Scientific, Pittsburgh, PA) by incubation at 37 °C for 18h. Each well was blocked in 100 μl of 1% BSA/PBS for 1 h at RT. Then 1.56 μM of purified recombinant MBP peptides in 50 μl PBS was added to each well and incubated at 4 °C overnight. After washing 3 × 5 min with 1% BSA/PBS/0.05% Tween20 (PBS-T), rat monoclonal antibody to MBP (1:1000; AbD Serotec, Raleigh, NC) in PBS-T was added for 1 h at RT. Wells were washed 3 × 5 min with PBS-T. Secondary horseradish peroxidase-conjugated goat anti-rat IgG was then added to each well (1:5000; GE Healthcare, Buckinghamshire, UK), which were then washed 3 × 5 min with 1% BSA/PBS-T. SureBlue TMB microwell peroxidase substrate (KPL, Gaithersburg, MD) was added, developed, and the reaction was terminated by adding 1N HCl. Absorbance of the samples was measured at a wavelength of 450 nm in a SpectraMax spectrofluorometer (Molecular Devices, Sunnyvale, CA) using SoftMax Pro control software.
Thioflavin T Fluorescence Assay
Lyophilized Aβ40D/I peptide was first resuspended with Me2SO to 2.5 mM, diluted to 12.5 μM in PBS, and then incubated at 37 °C with shaking either alone or with 1.56 μM MBP peptides. Control samples containing 0.5% Me2SO and 1.56 μM MBP peptides in PBS were also included. At each time point, 100 μl samples of each reaction were placed in a 96-well microplate in triplicate and 10 μl of 100 μM thioflavin T was added. The plate was mixed for 5 sec and incubated at 25 °C in the dark for 10 min before each reading. Fluorescence was measured at 25 °C at 490 nm using an excitation wavelength of 446 nm in a SpectraMax spectrofluorometer (Molecular Devices, Sunnyvale, CA) using SoftMax Pro control software.
Transmission Electron Microscopy
Sample mixtures were deposited onto carbon-coated copper mesh grids (EM Sciences, Hatfield, PA) and negatively stained with 2% (w/v) uranyl acetate. The samples were viewed with a FEI Tecnai 12 BioTwin transmission electron microscope, and digital images were taken with an AMT camera.
Solution NMR Spectroscopy
15N-labeled Aβ42WT was dissolved in 100 mM NaOH to a concentration of 2 mM. For NMR measurements, aliquots of this stock solution were diluted to 100 μM with low salt buffer containing 10% D2O. The pH was adjusted to 7.4 with dilute HCl, and the sample was adjusted to a total final volume of 400 μL. Lyophilized MBP1-64 and MBP50-64 were dissolved in distilled, deionized water to a concentration of 5 mM. The MBP1-64 and MBP50-64 stocks were diluted to a concentration of 100 μM in low salt buffer with 10% D2O. The pH was adjusted to 7.4 with dilute HCl, and the sample was adjusted to a total final volume of 400 μL.
Solution NMR spectra were obtained at 4 °C on a 700 MHz Bruker Avance spectrometer. 1H spectra of the 100 μM MBP1-64, MBP50-64 and Aβ42WT samples were acquired and compared to ensure that all of the samples were at the same concentration before mixing. 1H-15N HSQC spectra were obtained of 50 μM 15N-Aβ42WT and 50 μM MBP1-64 or 50 μM MBP50-64 after mixing of the 100 μM samples.
RESULTS
Residues 54–64 of MBP1-64 are required for its interactions with Aβ peptides
Previously, we reported that MBP binds Aβ peptides and inhibits their assembly into fibrils56-58. Moreover, we showed that the Aβ binding region on MBP resides within the N-terminal residues MBP1-6458. To further identify the region responsible for its interactions with Aβ peptides we performed deletion analyses on MBP1-64. Recombinant MBP1-64 and MBP1-53 were expressed and purified (Fig. 1A). Since our earlier studies showed that MBP interacts most strongly with highly fibrillogenic peptides such as Aβ40D/I and Aβ42WT we chose to use these in our analyses56,57. Solid-phase binding assays showed that compared to MBP1-64 the C-terminal deleted MBP1-53 was largely devoid of binding to immobilized Aβ40D/I and Aβ42WT peptides (Fig. 1B). Similarly, MBP1-53 was largely ineffective in blocking the fibrillar assembly of Aβ40D/I as assessed by thioflavin T binding and fluorescence (Fig. 1C) and TEM analysis of fibril structure (Fig. 1F). In contrast, similar studies showed that deletion of the first ten N-terminal residues of MBP1-64 (MBP11-64) had minimal impact on its ability to inhibit Aβ fibrillar assembly (data not shown).
FIGURE 1.
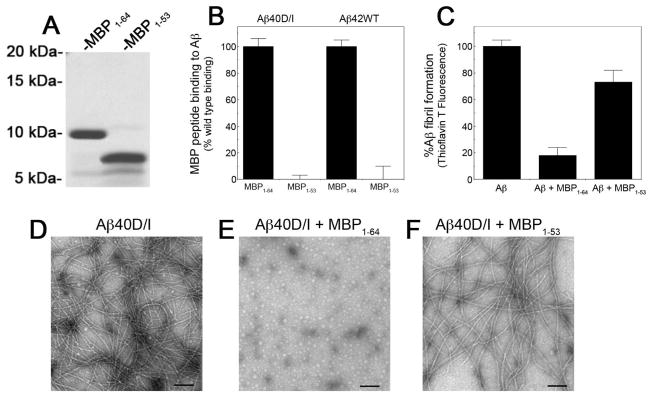
Residues 54–64 of MBP1-64 are required for its interactions with Aβ peptides. A. MBP1-64 (first lane) and MBP1-53 (second lane) were recombinantly expressed, purified, and analyzed by SDS-PAGE. B. The interaction of purified MBP1-64 and MBP1-53 with Aβ40DI or Aβ42WT was analyzed by solid phase binding assay. C. The inhibition of Aβ40DI (12.5 μM) fibrillogenesis by purified MBP1-64 (1.56 μM) and MBP1-53 (1.56 μM) was assessed by thioflavin T binding and fluorescence. D–F. TEM analysis of Aβ40DI in the absence (D) or presence of purified MBP1-64 (E) or purified MBP1-53 (F). Scale bars = 100 nm. MBP1-64, but not MBP1-53, bound to Aβ and inhibited its fibrillar assembly.
Residues K54, R55, G56, and K59 mediate MBP1-64 binding to Aβ and inhibition of fibril assembly
Since the region 54–64 is required for MBP1-64 interactions with Aβ we next performed an alanine scanning mutagenesis analysis of this region to identify specific amino acids that are important for these activities. First we conducted solid phase binding experiments to measure the interactions of MBP1-64 mutants with immobilized Aβ40D/I and Aβ42WT peptides as we previously described58. The binding data show that in MBP1-64 the sequential residues K54A, R55A, and G56A as well as K59A and H63A markedly reduced binding to Aβ40D/I (Fig. 2A). On the other hand, residues S57A, G58A, D60A, S61A, and H62A had much less or no effect on MBP1-64 binding to Aβ40D/I. A similar pattern was observed when analyzing the binding of MBP1-64 mutants to Aβ42WT suggesting that the same residues in MBP are involved with binding to both of these fibrillogenic forms of Aβ.
FIGURE 2.

Alanine scanning mutagenesis analysis of residues 54–64 of MBP1-64. A. The interaction of recombinantly expressed and purified MBP1-64 alanine mutants with Aβ40D/I and Aβ42WT was analyzed by solid phase binding assay. B. The inhibition of Aβ40D/I (12.5 μM) fibrillogenesis by purified MBP1-64 alanine mutants (1.56 μM) was assessed by thioflavin T binding and fluorescence. D–F. TEM analysis of Aβ40D/I in the absence (D) or presence of purified K54A mutant MBP1-64 (E) or purified S57A mutant MBP1-64 (F). Scale bars = 100 nm. Mutation of residues K54, R55, G56 and K59 markedly impaired the ability of MBP1-64 to bind Aβ and inhibit its fibrillar assembly.
We next investigated how each of the specific alanine mutants of MBP1-64 affected the inhibition of Aβ fibril assembly. In this case we focused on the Aβ40D/I peptide as it more rapidly assembles into Aβ fibrils compared to Aβ42WT. Similar to the results obtained in the binding experiments we found that in MBP1-64 the sequential residues K54A, R55A, and G56A as well as K59A substantially reduced its ability to inhibit Aβ fibril assembly (Fig. 2B). Although the H63A mutant showed reduced binding to immobilized Aβ peptides (Fig. 2A), it did not show a significant effect on interfering with Aβ fibril assembly. However, this may be a consequence of the different methodologies using immobilized Aβ for binding and Aβ in solution for fibril assembly.
To directly confirm the fibril inhibition results obtained in the thioflavin T binding and fluorescence experiments, we performed TEM analysis of Aβ40D/I assembly in the absence or presence of select MBP1-64 mutants. The K54A mutant, with markedly reduced Aβ binding and fibril assembly inhibiting activity, allowed for assembly of abundant, mature Aβ fibrils (Fig. 2D). Alternatively, the S57A mutant, with no appreciable effect on either Aβ binding or inhibition of fibril assembly, effectively blocked the assembly of mature Aβ fibrils (Fig. 2E). Similar corresponding TEM results were obtained with other MBP1-64 alanine mutants (data not shown). Together, these results are consistent in that they implicate MBP residues K54, R55, G56, and K59 as important for the ability of MBP to bind Aβ and inhibit fibril assembly.
Specific residues in Aβ42WT shift upon interaction with MBP1-64
Solution NMR spectroscopy was undertaken to localize the positions on Aβ42WT interacting with MBP1-64. Aβ42WT was chosen for this analysis as MBP1-64 binds to this peptide and inhibits it fibrillar assembly57 Aβ42WT can be stabilized at low temperature (4 °C) in a largely monomeric form that associates, as the temperature is increased, to low MW oligomers in the process of forming higher MW oligomers, protofibrils and fibrils60,61. There is a growing body of literature describing the intermediates in the oligomerization pathway of Aβ42WT and the ability of small molecule inhibitors to interact with specific oligomers62. One of the striking features of the MBP1-64 – Aβ interaction is the ability of the MBP protein to inhibit fibril formation at sub-stoichiometric ratios of MBP to Aβ.
Fig. 3 presents the 1H-15N HSQC spectra of 15N-labeled Aβ42WT with (red) and without (black) unlabeled MBP1-64. The largest shifts are observed in the N-terminus and central portion of Aβ42WT. In the N-terminus, shifts are observed in the resonances corresponding to negatively charged residues (E3, D7 and E11), as well as F4 and R5. In the central portion of Aβ42WT, shifts are observed in H14-L17. The shifts in Q15 and L17, as well as R5, are similar to those observed with small molecule inhibitors (curcumin and resveratrol) of Aβ assembly63. Resonances in the hydrophobic C-terminus of Aβ42WT are largely unaffected by binding of MBP1-64. In contrast, the 1H-15N HSQC spectra of 15N-labeled Aβ40WT, which exhibits weak interaction with MBP56,57, produced no appreciable shifts in residue resonances in the presence of unlabeled MBP1-64 (Figure S1 of the Supporting Information).
FIGURE 3.
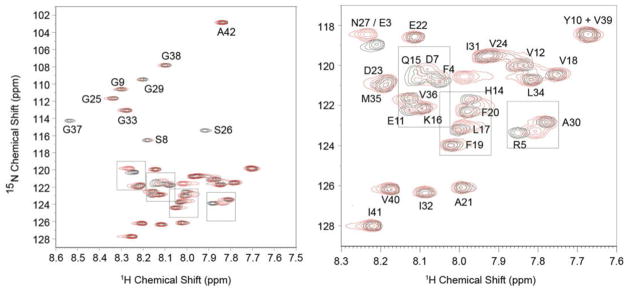
Specific Aβ42WT residue interactions with MBP1-64. 1H-15N HSQC spectra of 15N-labeled Aβ42WT with (red) and without (black) unlabeled MBP1-64. The largest shifts are observed in three regions of Aβ42WT: the N-terminus (E3-D7 and E11) and in two regions within the central portion of the peptide (H14-L17 and S26-G28). In addition, intensity is lost in the resonance to Gly37 in the hydrophobic C-terminus of Aβ42WT.
MBP50-64 is insufficient for Aβ binding and inhibition of fibril assembly
Since MBP residues 54–64 appear to be important for both Aβ binding and inhibition of fibril assembly as shown above we next determined if a small peptide corresponding to this region was sufficient for these activities. A synthetic MBP50-64 peptide was prepared and tested for its ability to interact with Aβ and inhibit Aβ fibril assembly. MBP50-64 induced no appreciable changes in the 15N-HSQC NMR spectrum of Aβ42WT when the two peptides were co-mixed (Fig. 4A). Similarly, MBP50-64 was ineffective at inhibiting Aβ fibril assembly as assessed by thioflavin T binding and fluorescence (Fig. 4B) and TEM analysis (Fig. 4D). Together, these findings indicate that although residues 54–64 are required for MBP1-64 to bind Aβ and inhibit fibril assembly a small peptide encompassing these residues was not sufficient to elicit these activities on its own.
FIGURE 4.

MBP50-64 is insufficient for Aβ binding and inhibition of fibril assembly. A. 1H-15N HSQC spectra of 15N-labeled Aβ42WT with (red) and without (black) unlabeled MBP50-64. No significant spectral shifts are observed. B. The inhibition of Aβ40D/I (12.5 μM) fibrillogenesis by purified MBP1-64 (1.56 μM) or MBP50-64 (1.56 μM) was assessed by thioflavin T binding and fluorescence. C,D. TEM analysis of Aβ40D/I in the absence (C) or presence of purified MBP50-64 (D). Scale bars = 100 nm. MBP50-64 did not bind Aβ or inhibit its fibrillar assembly.
DISCUSSION
The assembly and deposition of Aβ in brain is a key feature of the pathology of AD and related disorders. Thus, endogenous cerebral Aβ chaperones that influence the assembly process can have a marked impact on the pathogenesis of disease affecting both the onset and spatial location of Aβ deposition. For example, the chaperone ApoE4 can decrease the age of onset, increase the severity, and promote cerebral vascular deposition of fibrillar amyloid64. Therefore, elucidating the composition of Aβ chaperones in the brain and their respective mechanisms of action will provide a more complete understanding of how Aβ pathology develops and also offer opportunities for intervention.
Previously, we demonstrated that MBP is a novel brain Aβ chaperone that strongly binds Aβ peptides and potently inhibits their assembly into mature amyloid fibrils56,57. Subsequently, it was shown that its activity as an inhibitor was localized to the N-terminal residues 1–64 of MBP, was independent of MBP post-translational modifications, and protected cultured primary neurons from the toxic effects of Aβ58. Furthermore, both in human brain and in human AβPP transgenic mouse brain regions of white matter, which are rich in MBP, are largely devoid of fibrillar Aβ deposits56,65,66,. Thus, identifying the precise region on MBP responsible for its interactions with Aβ provides insight into its mechanism of action and establishes a framework for comparison to other modulators of Aβ assembly.
In the present study we used deletion mapping to show that the C-terminal region of MBP1-64 (residues 54–64) is required for its ability to bind Aβ and inhibit fibril assembly. Deletion of this region disrupted the interaction with both Aβ42WT and the familial CAA mutant Aβ40D/I, two highly fibrillogenic forms of Aβ (Fig. 1). Subsequent site-directed mutagenesis studies in this region identified residues K54, R55, G56 and K59 as important for the ability of MBP1-64 to bind Aβ and inhibit fibrillar assembly (Fig. 2). On the other hand, solution NMR studies identified several residues, including E3, R5, D7, E11 and Q15, on Aβ42WT that exhibit large spectral shifts upon interacting with MBP1-64 (Fig. 3). Together, these findings reveal specific sites on MBP and Aβ that appear key to their interactions with each other.
As its name implies, MBP is a strongly cationic protein with an isoelectric point of >11.0. Accordingly, MBP1-64 is also highly cationic with an isoelectric point of ≈11.5. This observation suggests that the interaction of MBP and its derived fragments with Aβ peptides may be purely ionic in nature. However, this assumption does not agree with several key findings. First, MBP and MBP1-64 interact most strongly with Dutch (E22Q)/Iowa (D23N) CAA mutant Aβ where there is a loss of two negatively charged amino acids increasing the isoelectric point from about 5.3 to 6.056. Second, the MBP50-64 peptide, which has highly basic net charge (isoelectric point > 11.0) and is required for MBP1-64 to interact with Aβ, is itself incapable of influencing fibril assembly (Fig. 4). Finally, eosinophilic cationic protein (ECP), an unrelated protein with a size and isoelectric point very similar to MBP67, did not inhibit Aβ assembly into fibrils (data not shown).
Nevertheless, at a certain level ionic interactions between MBP and Aβ do appear to be involved since mutation of the positively charged residues K54, R55 and K59 in MBP1-64 markedly impair both binding to Aβ and inhibition of fibril assembly (Fig. 2). Also, the NMR results show interaction of MBP1-64 with the negatively charged N-terminus of the Aβ42WT monomer stabilized at 4 °C (Fig. 3). Our earlier work showed that MBP appears to inhibit Aβ fibrillar assembly at the level of an oligomer56–58. Using atomic force microscopy it was demonstrated that both MBP and MBP1-64 allow the initial assembly of shorter low MW oligomers/protofibrils that are further stunted and capped at a height of ≈2 nm56–58. Importantly, the mechanism in which MBP interacts with the monomer, yet allows formation of low MW oligomers is consistent with the reported substoichiometric inhibition of Aβ assembly by MBP and its active fragments56,57. Although MBP and MBP1-64 are largely unstructured in solution, the present data suggest a model where the positively charged region MBP54-64 interacts with the negatively charged N-terminus of Aβ and wraps around the oligomer allowing another upstream region of MBP to cap further assembly of the oligomer into larger protofibril/fibril structures.
This type of “capping” phenomenon is also observed with designed peptides and small molecule inhibitors of Aβ fibrillar assembly68. Although the C-terminal region (residues 54–64) of MBP1-64 is required for Aβ binding and inhibition of fibril assembly, a small peptide corresponding to this region was essentially inactive (Fig. 4). This observation supports the likely need for upstream elements of MBP for these activities and suggests that the future design of peptides or other molecules with various linkers may be key to development of effective inhibitors of pathogenic Aβ assembly.
Supplementary Material
Acknowledgments
This work was supported by grants from the Cure Alzheimer’s Fund, a Collaborative MS Research Award and the National Institutes of Health (RO1-NS035781, R21-AG039215 and R21-NS079951) to WEVN and (RO1-AG027317) to SOS.
Abbreviations
- Aβ
amyloid β-protein
- AD
Alzheimer’s disease
- MBP
myelin basic protein
- AβPP
amyloid β-protein precursor
- CAA
cerebral amyloid angiopathy
- Aβ42WT
wild-type Aβ42 peptide
- Aβ40D/I
Dutch/Iowa CAA double mutant Aβ40 peptide
- PBS
phosphate-buffered saline
- BSA
bovine serum albumin
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- TEM
transmission electron microscopy, NMR, nuclear magnetic resonance
Footnotes
1H-15N HSQC spectra of 15N-labeled Aβ40WT with and without unlabeled MBP1-64 demonstrating no changes in the resonances of Aβ40WT upon addition of MBP1-64. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 3.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–4269. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Felice FG, Viera MNN, Saraiva LM, Figueroa-Villar JD, Garci-Abreu J, Liu R, Chang L, Klein WL, Ferreira ST. Targeting the neurotoxic species in Alzheimer’s disease: inhibitors of Aβ oligomerization. FASEB J. 2004;18:1366–1372. doi: 10.1096/fj.04-1764com. [DOI] [PubMed] [Google Scholar]
- 5.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 6.Goldgaber D, Lerman MI, McBride OW, Saffiotti U, Gajdusek DC. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science. 1987;235:877–880. doi: 10.1126/science.3810169. [DOI] [PubMed] [Google Scholar]
- 7.Tanzi RE, Gusella JF, Watkins PC, Bruns GA, St George-Hyslop P, Van Keuren ML, Patterson D, Pagan S, Kurnit DM, Neve RL. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science. 1987;235:880–884. doi: 10.1126/science.2949367. [DOI] [PubMed] [Google Scholar]
- 8.Robakis NK, Ramakrishna N, Wolfe G, Wisniewski HM. Molecular cloning and characterization of a cDNA encoding the cerebrovascular and the neuritic plaque amyloid peptides. Proc Natl Acad Sci U S A. 1987;84:4190–4194. doi: 10.1073/pnas.84.12.4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vassar R, Bennett BD, Babu-Khan S, Khan S, Mendiaz E, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. β-secretase cleavage of the Alzheimer’s precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 10.Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberberg I, Power M, Tanh H, Tatsuno G, Ting J, Schenk D, Suomensaari SM, Wang S, Walker D, John V, et al. Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature. 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- 11.Beck JP, Bienkowski MJ, Sinha S, Heinrikson RL. Human beta-secretase (BACE) and BACE inhibitors. J Med Chem. 2003;46:4625–4630. doi: 10.1021/jm030247h. [DOI] [PubMed] [Google Scholar]
- 12.De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- 13.Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 14.Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of gamma-secretase activity. Nat Cell Biol. 2003;5:486–488. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- 15.Jellinger KA. Alzheimer’s disease and cerebrovascular pathology: an update. J Neural Trans. 2002;109:813–836. doi: 10.1007/s007020200068. [DOI] [PubMed] [Google Scholar]
- 16.Rensink AA, de Waal RM, Kremer B, Verbeek MM. Pathogenesis of cerebral amyloid angiopathy. Brain Res Brain Res Rev. 2003;43:207–223. doi: 10.1016/j.brainresrev.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 17.Attems J, Jellinger K, Thal DR, Van Nostrand W. Sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol. 2011;37:75–93. doi: 10.1111/j.1365-2990.2010.01137.x. [DOI] [PubMed] [Google Scholar]
- 18.Findeis MA. The role of amyloid beta peptide 42 in Alzheimer’s disease. Pharmacol Ther. 2007;116:266–286. doi: 10.1016/j.pharmthera.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 19.Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GT, Luyendijk W, Frangione B. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 20.Van Broeckhoven C, Haan J, Bakker E, Hardy JA, Van Hul W, Wehnert A, Vegter-Van der Vlis M, Roos RA. Amyloid beta protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch) Science. 1990;248:1120–1122. doi: 10.1126/science.1971458. [DOI] [PubMed] [Google Scholar]
- 21.Grabowski TJ, Cho HS, Vonsattel JP, Rebeck GW, Greenberg SM. Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann Neurol. 2001;49:697–705. doi: 10.1002/ana.1009. [DOI] [PubMed] [Google Scholar]
- 22.Davis J, Van Nostrand WE. Enhanced pathologic properties of Dutch-type mutant amyloid beta-protein. Proc Natl Acad Sci USA. 1996;93:2996–3000. doi: 10.1073/pnas.93.7.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verbeek MM, de Waal RM, Schipper JJ, Van Nostrand WE. Rapid degeneration of cultured human brain pericytes by amyloid beta protein. J Neurochem. 1997;68:1135–1141. doi: 10.1046/j.1471-4159.1997.68031135.x. [DOI] [PubMed] [Google Scholar]
- 24.Miravalle L, Tokuda T, Chiarle R, Giaccone G, Bugiani O, Tagliavini F, Frangione B, Ghiso J. Substitutions at codon 22 of Alzheimer’s abeta peptide induce diverse conformational changes and apoptotic effects in human cerebral endothelial cells. J Biol Chem. 2000;275:27110–27116. doi: 10.1074/jbc.M003154200. [DOI] [PubMed] [Google Scholar]
- 25.Melchor JP, McVoy L, Van Nostrand WE. Charge alterations of E22 enhance the pathogenic properties of the amyloid beta-protein. J Neurochem. 2000;74:2209–2212. doi: 10.1046/j.1471-4159.2000.0742209.x. [DOI] [PubMed] [Google Scholar]
- 26.Van Nostrand WE, Melchor JP, Cho HS, Greenberg SM, Rebeck GW. Pathogenic effects of D23N Iowa mutant amyloid beta -protein. J Biol Chem. 2001;276:32860–32866. doi: 10.1074/jbc.M104135200. [DOI] [PubMed] [Google Scholar]
- 27.Stine WB, Jr, Dahlgren KN, Krafft GA, LaDu MJ. In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J Biol Chem. 2003;278:11612–11622. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- 28.Paravastua AK, Leapman RD, Yau WM, Tycko R. Molecular structural basis for polymorphism in Alzheimer’s β-amyloid fibrils. Proc Natl Acad Sci USA. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Van Nostrand WE, Smith SO. Structural conversion of neurotoxic amyloid-β(1-42) oligomers to fibrils. Nat Struct Mol Biol. 2010;17:561–567. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ono K, Condron MM, Teplow DB. Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc Natl Acad Sci USA. 2009;106:14745–14750. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 32.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837– 842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glabe CG. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol Aging. 2006;27:570–575. doi: 10.1016/j.neurobiolaging.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 34.Urbanc B, Cruz J, Le R, Sanders J, Hsiao Ashe K, Duff K, Stanley HE, Irizarry MC, Hyman BT. Neurotoxic effects of thioflavin S-positive amyloid deposits in transgenic mice and Alzheimer’s disease. Proc Natl Acad Sci USA. 2002;99:13990–13995. doi: 10.1073/pnas.222433299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Nostrand WE, Melchor J, Ruffini L. Pathogenic cell surface amyloid β-protein fibril assembly in cultured human cerebrovascular smooth muscle cells. J Neurochem. 1998;70:216–223. doi: 10.1046/j.1471-4159.1998.70010216.x. [DOI] [PubMed] [Google Scholar]
- 36.Rogers J, Cooper NR, Webster S, Schulz J, McGeer PL, Styren SD, Civin WH, Brachova L, Bradt B, Lieberburg I. Complement activation by β-amyloid in Alzheimer’s disease. Proc Natl Acad Sci USA. 1992;89:10016–10020. doi: 10.1073/pnas.89.21.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tacnet-Delorme P, Chevallier S, Arlaud GJ. β-amyloid fibrils activate the C1 complex of complement under physiological conditions: Evidence for a binding site for Aβ on the C1q globular regions. J Immunol. 2001;167:6374–6381. doi: 10.4049/jimmunol.167.11.6374. [DOI] [PubMed] [Google Scholar]
- 38.Rozemuller AJ, van Gool WA, Eikelenboom P. The neuroinflammatory response in plaques and amyloid angiopathy in Alzheimer’s disease: therapeutic implications. Curr Drug Targets CNS Neurol Disord. 2005;4:223–233. doi: 10.2174/1568007054038229. [DOI] [PubMed] [Google Scholar]
- 39.Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and are required for fibrillar Aβ-stimulated microglial activation. J Neurosci. 2009;29:11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schemchel D, Saunders AM, Goldgaber D, Roses AD. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:8098–8102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE. Isoform-specific binding of apolipoprotein E to β-amyloid. J Biol Chem. 1994;269:23403–23406. [PubMed] [Google Scholar]
- 42.Wisniewski T, Castano EM, Golabek A, Vogel T, Frangione B. Acceleration of Alzheimer’s fibril formation by apolipoprotein-E in vitro. Am J Pathol. 1994;145:1030–1035. [PMC free article] [PubMed] [Google Scholar]
- 43.Manelli AM, Stine WB, van Eldik LJ, LaDu MJ. ApoE and Aβ42 interactions: effects of isoform and conformation on structure and function. J Mol Neurosci. 2004;23:235–246. doi: 10.1385/JMN:23:3:235. [DOI] [PubMed] [Google Scholar]
- 44.Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, Fishman CE, DeLong CA, Piccardo P, Petegnief V, Ghetti B, Paul SM. Apolipoprotein E is essential for amyloid deposition in the APPV717F transgenic mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 1999;96:15233–15238. doi: 10.1073/pnas.96.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Costa DA, Nilsson LN, Bales KR, Paul SM, Potter H. Apolipoprotein E is required for the formation of filamentous amyloid, but not for amorphous Aβ deposition, in an AβPP/PS double transgenic mouse model of Alzheimer’s disease. J Alz Dis. 2004;6:509–514. doi: 10.3233/jad-2004-6508. [DOI] [PubMed] [Google Scholar]
- 46.Matsubara E, Fragnione B, Ghiso J. Characterization of Apolipoprotein J-Alzheimers Aβ interaction. J Biol Chem. 1995;270:7563–7567. doi: 10.1074/jbc.270.13.7563. [DOI] [PubMed] [Google Scholar]
- 47.DeMattos RB, O’dell MA, Parsadanian M, Taylor JW, Harmony JA, Bales KR, Paul SM, Aronow BJ, Holtzman DM. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2002;99:10843–10848. doi: 10.1073/pnas.162228299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lefterov I, Fitz NF, Cronican AA, Fogg A, Lefterov P, Kodali R, Wetzel R, Koldamova R. Apolipoprotein A-1 deficiency increases cerebral amyloid angiopathy and cognitive deficits in APP/PS1dE9 mice. J Biol Chem. 2010;285:36945–36957. doi: 10.1074/jbc.M110.127738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lewis TL, Cao D, Lu H, Mans RA, Su YR, Jungbauer L, Linton MF, Fazio S, LaDu MJ, Li L. Overexpression of human apolipoprotein A-I preserves cognition and attenuates neuroinflammation and cerebral amyloid angiopathy in a mouse model of Alzheimer’s disease. J Biol Chem. 2010;285:36958–36968. doi: 10.1074/jbc.M110.127829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aksenova MV, Aksenova MY, Butterfield DA, Carney JM. Alpha-1-antichymotrypsin interaction with Aβ(1-40) inhibits fibril formation but does not affect the peptide toxicity. Neurosci Lett. 1996;211:45–48. doi: 10.1016/0304-3940(96)12717-8. [DOI] [PubMed] [Google Scholar]
- 51.Potter H, Wefes IM, Nilsson LN. The inflammation-induced pathological chaperones ACT and apoE are necessary catalysts of Alzheimer amyloid formation. Neurobiol Aging. 2001;22:923–930. doi: 10.1016/s0197-4580(01)00308-6. [DOI] [PubMed] [Google Scholar]
- 52.Schwarzman AL, Tsiper M, Wente H, Wang A, Vitek MP, Vasiliev V, Goldgaber D. Amyloidogenic and anti-amyloidogenic properties of recombinant transthyretin variants. Amyloid. 2004;11:1–9. doi: 10.1080/13506120410001667458. [DOI] [PubMed] [Google Scholar]
- 53.Buxbaum JN, Ye Z, Reixach N, Friske L, Levy C, Das P, Golde T, Masliah E, Roberts AR, Bartfai T. Transthyretin protects Alzheimer’s mice from the behavioral and biochemical effects of Abeta toxicity. Proc Natl Acad Sci USA. 2008;105:2681–2686. doi: 10.1073/pnas.0712197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kakio A, Nishimoto SI, Yanagisawa K, Kozutsumi Y, Matsuzaki K. Cholesterol-dependent formation of GM1 ganglioside-bound amyloid β-protein, an endogenous seed for Alzheimer amyloid. J Biol Chem. 2001;276:24985–24990. doi: 10.1074/jbc.M100252200. [DOI] [PubMed] [Google Scholar]
- 55.Yamamoto N, Hirabayashi Y, Amari M, Yamaguchi H, Romanov G, Van Nostrand WE, Yanagisawa K. Assembly of hereditary amyloid β-protein variants in the presence of favorable gangliosides. FEBS Lett. 2005;579:2185–2190. doi: 10.1016/j.febslet.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 56.Hoos MD, Ahmed M, Smith SO, Van Nostrand WE. Inhibition of familial cerebral amyloid angiopathy mutant amyloid β-protein fibril assembly by myelin basic protein. J Biol Chem. 2007;282:9952–9961. doi: 10.1074/jbc.M603494200. [DOI] [PubMed] [Google Scholar]
- 57.Hoos MD, Ahmed M, Smith SO, Van Nostrand WE. Myelin basic protein binds to and inhibits the fibrillar assembly of Aβ42 in vitro. Biochemistry. 2009;48:4720–4727. doi: 10.1021/bi900037s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liao MC, Hoos MD, Aucoin D, Ahmed M, Davis J, Smith SO, Van Nostrand WE. Amino terminal domain of myelin basic protein inhibits amyloid β-protein fibril assembly. J Biol Chem. 2010;285:35590–35598. doi: 10.1074/jbc.M110.169599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A, Yates J, Cotman C, Glabe C. Assembly and aggregation properties of synthetic Alzheimer’s A4/beta amyloid peptide analogs. J Biol Chem. 1992;267:546–554. [PubMed] [Google Scholar]
- 60.Hou LM, Shao H, Zhang Y, Li H, Menon NK, Neuhaus EB, Brewer JM, Byeon IJL, Ray DG, Vitek MP, Iwashita T, Makula RA, Przybyla AB, Zagorski MG. Solution NMR studies of the Aβ(1-40) and Aβ(1-42) peptides establish that the met35 oxidation state affects the mechanism of amyloid formation. J Am Chem Soc. 2004;126:1992–2005. doi: 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- 61.Fawzi NL, Ying J, Ghirlando R, Torchia DA, Clore GM. Atomic-resolution dynamics on the surface of amyloid-β protofibrils probed by solution NMR. Nature. 2011;480:268–272. doi: 10.1038/nature10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ladiwala ARA, Lin JC, Bale SS, Marcelino-Cruz AM, Bhattacharya M, Dordick JS, Tessier PM. Resveratrol selectively remodels soluble oligomers and fibrils of amyloid Aβ into off-pathway conformers. J Biol Chem. 2010;285:24228–24237. doi: 10.1074/jbc.M110.133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ono K, Li L, Takamura Y, Yoshiike Y, Zhu L, Han F, Mao X, Ikeda T, Takasaki J, Nishijo H, Takashima A, Teplow D, Zagorski M, Yamada M. Phenolic compounds prevent amyloid β-protein oligomerization and synaptic dysfunction by site specific binding. J Biol Chem. 2012;287:14631–14643. doi: 10.1074/jbc.M111.325456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer’s disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Behrouz N, Defossez A, Delacourte A, Mazzuca M. The immunohistochemical evidence of amyloid diffuse deposits as a pathological hallmark in Alzheimer’s disease. J Gerontol. 1991;46:B209–212. doi: 10.1093/geronj/46.6.b209. [DOI] [PubMed] [Google Scholar]
- 66.Wisniewski HM, Bancher C, Barcikowska M, Wen GY, Currie J. Spectrum of morphological appearance of amyloid deposits in Alzheimer’s disease. Acta Neuropathol. 1989;78:337–347. doi: 10.1007/BF00688170. [DOI] [PubMed] [Google Scholar]
- 67.Barker RL, Loegering DA, Ten RM, Hamann KJ, Pease LR, Gleich GJ. Eosinophil cationic protein cDNA. Comparison with other toxic cationic proteins and ribonucleases. J Immunol. 1989;143:952–959. [PubMed] [Google Scholar]
- 68.Mastrangelo IA, Ahmed M, Sato T, Liu W, Wang C, Hough P, Smith SO. High-resolution atomic force microscopy of soluble Aβ42 oligomers. J Mol Biol. 2006;358:106–119. doi: 10.1016/j.jmb.2006.01.042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.