

Abstract
Amyloid binding alcohol dehydrogenase (ABAD), a mitochondrial protein, is a cofactor facilitating amyloid-β peptide (Aβ) induced cell stress. Antagonizing Aβ-ABAD interaction protects against aberrant mitochondrial and neuronal function and improves learning memory in the Alzheimer’s disease (AD) mouse model. Therefore, it offers a potential target for Alzheimer’s drug design, by identifying potential inhibitors of Aβ-ABAD interaction. 2D QSAR methods were applied to novel compounds with known IC50 values, which formed a training set. A correlation analysis was carried out comparing the statistics of the measured IC50 with predicted values. These selectivity-determining descriptors were interpreted graphically in terms of principle component analyses, which are highly informative for the lead optimization process with respect to activity enhancement. A 3D pharmacophore model also was created. The 2D QSAR and 3D pharmacophore models will assist in hi-throughput screening. In addition, ADME descriptors were also determined to study their pharmacokinetic properties. Finally, ABAD molecular docking study of these novel molecules was undertaken to determine whether these compounds exhibit significant binding affinity with the binding site. We have synthesized only the compounds that have shown the best drug like properties as candidates for further studies.
Keywords: Molecular Docking, Quantitative Structure Activity Relationship, ADME Prediction, ABAD Inhibitors, Benzothiazole Aminophosphonates
Introduction
Alzheimer’s disease (AD) is one of the most common dementias showing slowly progressive cognitive decline. Alzheimer’s brain is characterized by accumulation of amyloid beta peptide (Aβ) and the formation of neurofibrillary tangles. Aβ plays a central role in the development of AD pathology and contributes to neuronal, synaptic, and cognitive malfunction. Mitochondrial and synaptic dysfunction is an early pathological feature of AD (1–6). Recent studies have highlighted the significance of mitochondrial Aβ accumulation and synaptic mitochondrial dysfunction. Aβ progressively accumulates in synaptic mitochondria and impairs mitochondrial structure and function including membrane potential, membrane permeability transition pore, respiration, energy metabolism, oxidative stress, mitochondrial dynamics, and calcium homeostasis (4, 7–19). Thus, strategies that suppress/attenuate Aβ-induced mitochondrial toxicity in addition to Aβ levels in the brain and improve cognitive function are critical for preventing and/or halting at an early stage of AD.
Amyloid binding alcohol dehydrogenase (ABAD), a mitochondrial enzyme, plays a key role in mitochondrial dysfunction and in the pathogenesis of AD. This enzyme has attracted considerable interest because of its ability to interact with Aβ. Importantly, the interaction of ABAD with Aβ mediates mitochondrial and synaptic dysfunction (9, 20). Antagonizing Aβ-ABAD interaction with the ABAD decoy peptide that encompasses the amino residues responsible for Aβ binding to ABAD protects against aberrant mitochondrial and neuronal function and improves learning memory in AD transgenic mice (9, 17, 20). Furthermore, interception of Aβ-ABAD interaction also significantly reduces mitochondrial and cerebral Aβ accumulation (17). These data support that Aβ-ABAD interaction is a potential target of the drug development for treatment of AD.
In this way, the search for inhibitors of Aβ-ABAD interaction has started and using an ELISA-based screening assay, frentizole, an FDA-approved immunosuppressive drug, was identified as a novel inhibitor of Aβ–ABAD interaction. Analyzing the frentizole SAR studies, we have developed novel benzothiazole ureas with a 30-fold improvement in potency(21). Recently, AG18051 (1-azepan-1-yl-2-phenyl-2-(4-thioxo-1,4-dihydropyrazolo[3,4-d]pyrimidin-5-yl)-ethanone) was also identified as a potent inhibitor of ABAD.(22) However, currently available inhibitors of Aβ-ABAD interaction have the disadvantages of low solubility, poorly crossing the blood brain barrier (BBB), high toxicity, and low cell permeability. Current efforts to design Aβ-ABAD inhibitors have been unsuccessful due largely to poor ADME (Absorption, Distribution, Metabolism and Excretion) properties. To overcome the limitation of currently available ABAD inhibitors, we have designed a new class of small molecular inhibitors of Aβ-ABAD interaction via phosphonate derivatives. The goal of this study is to identify the potential blockers of Aβ-ABAD interaction as therapeutic targets of AD.
The quantitative structure–activity relationship (QSAR) analysis helps to derive highly applicable models that allow designing novel and reactive molecules. Any statistical tool and/or graphical model can predict physicochemical properties to increase the selectivity and affinity of new compounds.(23) These properties may be measured by a variety of calculated descriptors, i.e., potential hydrogen bond donors or acceptors, molar refractivity, hydrophobicity, SlogP. We used descriptors to evaluate the drug-like properties of our compounds. In this present study, we also applied 2D QSAR and 3D ligand based pharmacophore methods to predict the inhibitory activity of a test set of compounds by constructing a model from a training set. For the QSAR model, linearity of the correlation and root mean square error (RSME) and correlation factor (R2) indicate the robustness of the model. The accuracy of the pharmacophore model was evaluated by selecting hits from a test set. Docking studies to ABAD were carried out to determine the binding affinity differences of our compounds. Reliable models were obtained that provided interesting information with respect to binding affinity and compound selectivity discrimination.
In our previous work, we have described the synthesis and evaluation of a novel class of benzothiazole urea derivatives as potent Aβ-ABAD inhibitors.(24) Based on benzothiazole urea and frentizole structure activities studies, we have designed benzothiazole amino and frentizole phosphonate derivatives. Molecular docking, QSAR studies and pharmacokinetics/absorption, distribution, metabolism, and excretion (ADME) prediction for any given scaffold of interest are the most popular methods of computer aided drug design.
With respect to ADME prediction, we have predicted the cell permeability of Caco2 (human colon adenocarcinoma) cell permeability, MDCK (Madin-Darby canine kidney), human intestinal absorption, BBB penetration and plasma protein binding. Finally, the compounds with satisfactory properties were chosen for molecular docking against ABAD protein to find out whether they are able to bind to the protein and inhibit. The results from the molecular docking can be used to interpret the efficacy of the novel molecules to inhibit ABAD and in turn they may be useful as drugs for the treatment and management of AD. Based on the results of these studies, we have synthesized the compounds that have shown the best drug-like properties.
Materials and Methods
General procedure for the synthesis of small molecular weight benzothiazole phosphonate derivatives (Scheme 1)
Scheme 1.

Representative classes of compounds containing the N-C-P scaffold and selected molecular targets of ABAD
To a stirred solution of aromatic/heterocyclic aldehydes (2) in 10 mL of toluene, Mg(ClO4)2 (5 mol %) was added. The mixture was stirred magnetically for 10–15 minutes, later substituted 2-amino benzothiazoles (1) and dimethyl/diethyl phosphite (3) in anhydrous toluene (5 mL) were added drop-wise with stirring at room temperature for 30 minutes. The reaction mixture was refluxed with stirring for 5–12 hours. After completion of the reaction, as determined by TLC (ethyl acetate:hexane), the solvent was removed in a rotary-evaporator. The reaction mixture was extracted with EtOAc (3 × 10 mL). The combined EtOAc extracts were dried (Na2SO4) and concentrated under reduced pressure to afford products 4a-j, which were passed through a column of silica gel and eluted with EtOAc-hexane (1:1).
General procedure for the synthesis of urea/thiourea phosphonate derivatives (Scheme 2)
Scheme 2.

Alternative synthetic route of urea/thiourea phosphonates derivatives
A mixture of urea/thiourea (1 mmol) and methyl 5-formyl-2-hydroxybenzoate (2 mmol) in ethanol (15 mL) was refluxed for the appropriate reaction (TLC) time. The solvent was evaporated in vacuo and the resulting crude material was purified by chromatography on a short column of silica gel (EtOAc: petroleum ether, 1:3) and then recrystallized from ethanol/dichloromethane (4:1) to afford the urea derivatives.
A mixture of urea derivatives (1 mmol), toluene (20 mL), and dimethyl phosphite (DMP)/diethyl phosphate (DEP) (2 mmol) was heated at 110°C for 15 hours. The solvent was evaporated in vacuo and the resulting crude material was purified by chromatography on a short column of silica gel (EtOAc: dichloromethane, 1:3) and then recrystallized from ethanol (4:1) to afford the target molecules.
Construction of models and Molecular Dynamics of the novel leads (25)
The 3D structure of all ligands were constructed in MOE (Molecular Operating Environment) working environment and subjected to energy minimization.(26) The Merck Molecular Force Field (MMFF94x) and parameter set was employed and the related potential energy terms were enabled for all bonded interactions, Van der Waals interactions, electrostatic interactions and restraints. The non-bonded cut off value was enabled between 8–10 Ang. The Generalized Born implicit salvation model was enabled. The gradient was set to 0.05 and force filed partial charges were enabled to calculate during minimization process. The dynamics simulations were carried out with the initial temperature set to 30K and increased to a 300K run time temperature. Heat time and cool time were set to 0 pico seconds. The final stabilized conformations were used for the construction of a local data base of present novel leads and chosen to determine 2D and 3D descriptors, including Lipinski rules that define drug-like properties.
Construction of a 2D QSAR model (25)
The QSAR suite of applications in MOE was used to calculate and analyze the data and build numerical models of the data for prediction and interpretation purposes. Any QSAR model for a given set of molecules correlates the activities with properties inherent to each molecule in the set itself. A database of 45 compounds was used to generate independent training and test data sets. Initially, the QSAR descriptors SlogP, Density, Molar refractivity, Molecular weight, atomic polarizability, logP(o/w), logS, Polar surface area, Van der Waals volume and radius of gyration were calculated for a limited 50 compound training set of moleculesto determine and select compounds from our larger set with drug-like properties. A comparison of these descriptors suggested important properties for drug-like compound selection.
Fitting the experimental data
$PRED_ was chosen as dependent variable and the remaining descriptors (SlogP_VSA) and xsurf_CW) as independent variables of the database. A QSAR model was constructed choosing $PRED as activity filed and the remaining descriptors as model fields. Regression analysis was performed for the training data set and RMSE and r2 values of the fit were reported. This fit model was saved as the QSAR model and used for the prediction of activities of compounds of test data set.
Cross-Validating the Model
The above QSAR fit was used for both model validation and cross validation. This validation procedure will evaluate the predicted activities and the residuals for the training set molecules. The predicted, residual and Z-score values were calculated for both model and cross validations.
Graphical Analysis
The predictive ability of the model was assessed using a correlation plot by plotting the predicted ($PRED) values (X-axis) versus the predicted IC50 activities (Y-axis). This correlation plot was used to find out the outliers that have a Z-score beyond the range of 1.5.
Estimation and validation of predicted activities of test set
The QSAR model fit obtained above was used to evaluate the predicted ($PRED) values of 21 test set compounds. Regression analysis was performed for the test data set and RMSE and r2 values of the fit were reported.
Pruning the Descriptors
Pruning the descriptors is necessary to select the optimum set of molecules under consideration. ‘QuaSAR-Contingency,’ a statistical application in MOE was used to describe the best molecules in the data set. The results were analyzed using Principle Component Analysis (PCA) and the purpose of which is to reduce the dimensionality of set of molecular descriptors by linearly transforming the data or defying a property that would be important to drug design. A Three-dimensional scatter graphical plot was generated using the first three Principal Components (PCA1, PCA2 and PCA3).
ADMET prediction (27, 28)
Absorption, Distribution, Metabolism, Excretion, & Toxicity (ADMET) properties of the 20 novel compounds were calculated using the preADMET online server (http://preadmet.bmdrc.org/). The ADMET properties, human intestinal absorption, in vitro Caco-2 cell permeability, in vitro Maden Darby Canine Kidney (MDCK) cell permeability, in vitro plasma protein binding and in vivo BBB penetration were predicted using this program.
Molecular Docking (29, 30)
Preparation of ABAD protein
The three-dimensional structure of ABAD was retrieved from Protein Data Bank (PDB:http://www.rcsb.org/pdb, PDB ID: 1SO8). To relieve any close contacts, the protein structure was loaded into MOE molecular modeling software and all the water molecules and hetero atoms were removed and polar hydrogen’s were added. Protonation of 3D structure was done for all the atoms in implicit solvated environment (Born salvation model) at a specified temperature of 300K, pH of 7 and with a salt concentration value of 0.1. A non-bonded cut off value of 8 – 10 Å was applied to the Leonard-Jones terms. After the protonation, the complete structure was Energy minimized in MMFF94x force field at a gradient cut off value of 0.05. Molecular dynamics simulations were carried out at a constant temperature of 300 deg K for a heat time of 10 pico seconds. The total simulations were carried out for a total period of 10 nano seconds. The time step was considered as 0.001 and the temperature relaxation time was set to 0.2 pico seconds. The position, velocity and acceleration were saved per every 0.5 pico seconds.
Prediction of Binding site for Ligands
The binding site for ABAD was predicted through PDBSum (http://www.ebi.ac.uk/pdbsum/). The protein structural information was analyzed at PDBSum and its link to Catalytic Site Atlas (CAS) was followed (http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/CSA/CSA_Site-_Wrapper.pl?pdb=1so8). The catalytic site residues were Asn121, Ser155, Tyr168 and Lys172, which were based on the structure of Trihydroxynaphthalene Reductase (1YBV).
Molecular Docking(31)
The ligand data base generated from the list of all novel ligand molecules was docked into the specified binding domain of the ABAD receptor. A total of 30 conformations were generated for each Ligand-Receptor complex and among them, the conformation with least docking score was considered for further analysis. The interaction of all ligand molecules in the binding domain cavity was analyzed from ligand interaction study of MOE.(32) The ligand-receptor complexes were analyzed by both London ΔG free energy approximations and interaction energies, ΔE.
Results and Discussion
ABAD inhibitor design
A total of 20 compounds were designed, and their capacity to inhibit Aβ–ABAD interaction was predicted by using, quantitative structure activity relationship studies, preADME properties, docking studies. The preliminary SAR study indicated that the benzothiazole amine moiety is required for the inhibition of Aβ–ABAD interaction.
Based on the frentizole SAR study and the previous report, we have developed novel benzothiazole ureas with a 30-fold improvement in potency;(21) benzothiazole urea and frentizole analogs provide remarkable enhancements of permeation across biological membranes and of oral bioavailability. Based on frentizole and benzothiazole urea derivatives SAR studies, we have designed and synthesized novel small drug molecules as urea and frentizole phosphonate derivatives (Schemes 1 & Scheme 2), which might have the capacity to cross the BBB and inhibit Aβ-ABAD interaction. While for frentizole a wide range of analogues have been prepared and investigated, up to now there are no reports dealing with derivatives of urea and frentizole phosphonates or their related analogues. Compounds 1-12 and 16-20 are substituted benzothiazole amines bearing a methoxy- or fluoro and dimethyl/diethyl phosphonates, substituted aromatic/heterocyclic aldehydes, respectively. Compounds 13-15 are the urea/thiourea phosphonates moieties. The rational design of 1-20 had also been strongly supported by known phosphonate prodrugs and docking studies showing that Tyr652 and Phe656 play a pivotal role in the ABAD drug binding, by promoting cation-π, π-π, and hydrophobic interactions with the basic nitrogen and aromatic rings of drugs. This might confer the capacity to cross the BBB and to inhibit Aβ-ABAD interaction.
The major novelty of the present approach is the use of a benthiazole phosphonate moiety, which readily penetrates biological membranes such as the blood-brain barrier (BBB) and enters the target organ.(33–35) According, phosphate esters are frequently used as a prodrug strategy, especially for water insoluble compounds, since the phosphate group confers the following characteristic features to the xenobiotic: 1) decreases the adverse effects of the drugs, 2) help in readily crossing the blood-brain barrier (BBB) and enters the target organ, 3) increases water solubility and thereby enables delivery of the drug parenterally, 4) cleavage of the phosphonate carrier/drug entity in vivo provides a hydrophilic, negatively charged intermediate, which is “locked” in the brain or other organ and which provides significant and sustained delivery of the active drug species to the target organ, 5) as phosphonate moiety induces polar nature to the derivatives, bio transformation ( by Phase I enzymes) is not necessary for drug disposition, thereby reducing drug-drug interaction.(33, 34, 36, 37)
Chemistry
To decrease adverse effects, facilitate passage across the BBB, and increase water solubility, phosphonate derivatives were synthesized for use as potential inhibitors of Aβ-ABAD interactions. A series of analogs were synthesized with variations at the aromatic rings and their linking group. As shown in Scheme 1, various aromatic/heterocyclic aldehydes (2) were individually mixed with Mg (ClO4)2 (5 mol %) in anhydrous toluene (5 mL). The mixture was stirred magnetically for 10–15 minutes, after which time substituted benzothiazole amines (1) and dimethyl/diethyl phosphite (3) in anhydrous toluene (5 mL) were added dropwise with stirring at room temperature for 30 minutes. The reaction mixture was refluxed with stirring for 5–12 hours. Herein, we report that Mg (ClO4)2 is an extremely efficient catalyst for the formation of benthiazole aminophosphonates by a one pot, three-component reaction of aromatic/heterocyclic aldehydes, substituted benzothiazole amines, and a dimethyl/diethyl phosphite in dry toluene (Scheme 1). The synthetic route utilized to make our target “ABAD inhibitors” compounds 4a-4n is shown in Scheme 1.
To determine the best experimental conditions, the reaction of 6-fluorobenthiazole amine, methyl 5-formyl-2-hydroxybenzoate, and DMP was considered a model (spectral data has been provided in the supporting information section). The progress of the reaction was monitored by TLC (ethyl acetate:hexane = 1:4) analysis. After completion of the reaction, the solvent was removed under reduced pressure. The reaction mixture was extracted with EtOAc (3×10 mL). The combined EtOAc extracts were dried (Na2SO4) and concentrated under reduced pressure to afford a white solid, which on passing through a column of silica gel and on elution with EtOAc-hexane (80:20) afforded compound (4a). The best results were obtained in the presence of Mg(ClO4)2 at reflux for 6 h, affording the desired methyl 5-((dimethoxyphosphoryl) ((6-fluorobenzo[d]thiazol-2-yl) amino)methyl)-2-hydroxybenzoate in 81% yield (4a). The remaining reactions were carried out following this general procedure. On each occasion, the spectral data (IR, NMR, and MS) of prepared known compounds were identical with those reported in the literature. We have synthesized few representative examples using DMP, DEP, methoxy, fluoro substituted amines, and different aromatic/heterocyclic aldehydes, respectively.
The alternative synthetic route for the preparation of, urea/thiourea phosphonates derivatives uses a mixture of urea/thiourea and methyl 5-formyl-2-hydroxybenzoate in ethanol under reflux to form an imine intermediate, which is further reacted with dialkyl/aryl phosphates to generate the phosphonate derivatives (Scheme 2).(38) The remaining title compounds will be synthesized for further study Aβ-ABAD interaction to evaluate the inhibitory activity.
A total of 20 compounds have been designed and tested as of date, and their capacity to inhibit Aβ–ABAD interaction was predicted by using, QSAR and 3D pharmacophore methods, preADME properties, docking studies and drug-like analysis. A preliminary SAR through our docking studies of these compounds indicated that the benzothiazole amine moiety is required for inhibition of Aβ–ABAD interaction. Urea/thiourea compounds 11, 12, 13, 14, 15 and 16 are not quite as effective because their molecular weight is too big. Substitutions on the benzothiazole ring and phenyl rings dramatically affected potency. Small electron-withdrawing groups were preferred at the benzothiazole ring with F particularly favored. Further, compounds with a hydroxyl group at the para position of the phenyl ring are predicted to more potent.
Molecular dynamics and 2D QSAR study of ligand molecules
The stabilized conformations obtained at the end of the molecular dynamics simulations were used to construct the molecular compound database. For the entire database, molecular QSAR descriptors were calculated and graphs were plotted to analyze whether they are in the optimum range or not. The compounds 11, 12, 13, 14, 15 and 16 are violating the drug like properties especially with respect to molecular weight (Figures.1 to Fig. 13). The remaining compounds all show the satisfactory values, which make them behave as good drugs.
Figure 1.
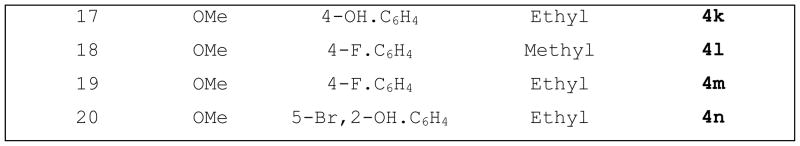
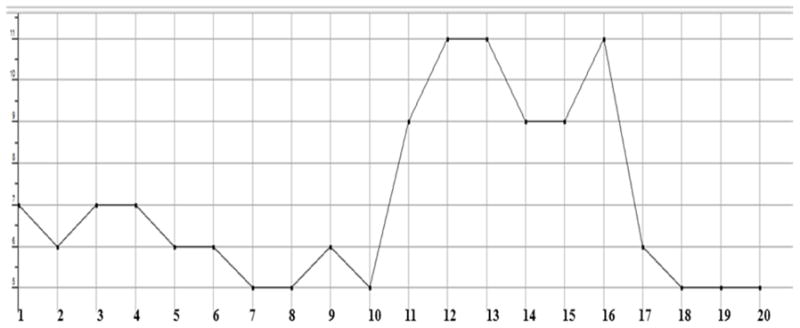
Plot showing the Log of octanol/water partition coefficient of test compounds including implicit hydrogens (SlogP). This property is an atomic contribution model that calculates logP from the given structure; i.e., the correct protonation state.
Figure 13.
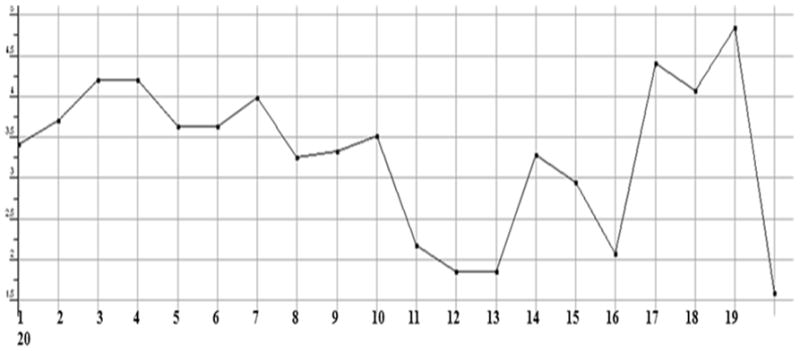
Plot showing the count of hydrogen bonds acceptors in each test compound. All of them showing a count of less than 10, except the compounds 12, 13 and 16, which means that they are violating the desired properties of Lipinski rule.
2D QSAR model descriptors
| SlogP_VSA(i,p) | 2D Subdivided surface areas. Each descriptor in a series is defined to be the sum of the vi over all atoms i such that pi is in a specified range (a, b) |
| vsurf_CW(i=1,8) | 3D volume capacity descriptors |
| SlogP | Log of the octanol/water partition coefficient |
| Density | Molecular mass density |
| MW/Vol (VDW) | Weight divided by vdw_vol (amu/Å3) |
| SMR | Molecular refractivity, (including implicit hydrogens) |
| MW | Molecular weight (including implicit hydrogens) in atomic mass units |
| TPSA | Total polar surface area |
| logs | Log of the aqueous solubility (mol/L) |
The correlation plot generated form the regression analysis showed a linear relationship among the training set of 19 compounds (Fig. 14). The model exhibited excellent linearity [$PRES = 0.9(IC50) + 9.0], with a RMSE = 0.9, R2 = 0.97 and a cross-validated R2 = 0.61. The reliability of the QSAR model was further established by applying this model on an independent set of 21 test compounds and its predictive ability was evaluated. The correlation plot of the test compounds was reasonable and expected based on a limit set of compounds in the training set. The resultant correlation plot of regression analysis showed a linear relationship for the final test data set [$PRES = 0.6(IC50) + 54.0], with a RMSE = 0.8, R2 = 0.6. (Fig 15). As new compound data is added to the training set, we would expect the accuracy for prediction of our QSAR model to improve (Figures 14 & 15).
Figure 14.

Linear correlation graph comparing a independent test set of measured IC50’s with predicted values based on the calculated 2D QSAR model. The linearity of the test model is shown with the values of the error (RMSE) and correlation factor (R2).
Figure 15.
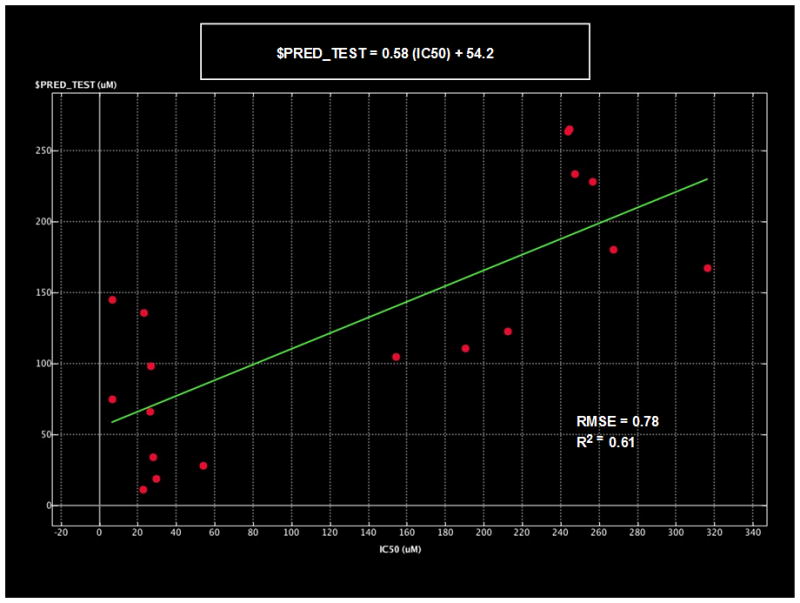
Linear correlation graph comparing a independent test set of measured IC50’s with predicted values based on the calculated 2D QSAR model. The linearity of the test model is shown with the values of the error (RMSE) and correlation factor (R2).
Principal Component Analysis
A principal component analysis using the QSAR descriptors showed that the first three PCA eigenvectors included 63% of the variance. All the data values found to lie in the range of −3 to +3, each spot in the plot represents a molecule and color coded by IC50 activity. Most interestingly the top most active compounds in our data set, as shown in magenta are isolated at PCA1 = 2 and PCA = 2 (Figure 16). This could provide an addition criterion for compound selection.
Figure 16.

PCA plot of the complete set of ABAD compounds. The first three eigenvectors are shown (PCA1, PCA2 and PCA3), which constituted 63 % of the variance. The spheres indicate the position of each of the 45 compounds. Based on SlogP a clear separation is observed. Those compounds with calculated SlogP (<2 ), are shown as magenta spheres) are clustered about PCA1 = 1, PCA2 = 2, PCA3 = 2). Those spheres in red are calculated >4, and green designate between these extremes.
3D Pharmacophore model of ligand molecules (Figure 17)
Figure 17.
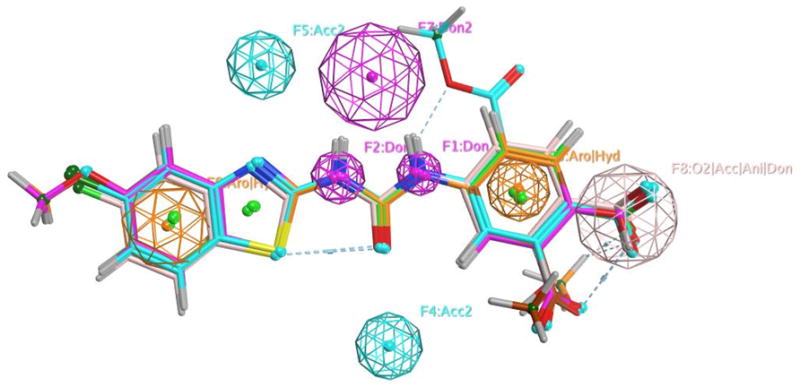
Showing the resulting pharmacophore model (shown as volume sheres) based on the activities (IC50) of 20 compounds in the training set. The various pharmacophore sites are labeled as to the features or properties (H-bond donor, H-bond acceptor, hydrophilic/aromatic and carboxyl acceptor/anion donor). The 6 top most active ABAD inhibitors from the test set are overlayed with the pharmacophore model.
ADME predictions (39, 40)
ADME properties are important conditions and major parts of pharmacokinetics. Whatever the compound that is going to be a drug should have the perfect ADME properties, and then only it will be approved as a drug in clinical tests. The ADME predictions of present 20 compounds are showing satisfactory results. Among the 20, the compounds 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 14, 17, 18, 19 and 20 are showing well intestinal absorption and the compounds 12, 13, 15 and 16 are showing moderate absorption. All of them are showing middle permeability for in vitro Caco-2 cells and low permeability for in vitro MDCK cells. in vivo blood-brain barrier penetration capacity was predicted to be having middle absorption to CNS (Central Nervous System) for the compounds 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 17, 18, 19 and 20 where as low absorption to CNS was observed for the compounds 11, 12, 13, 14, 15 and 16. BBB penetration is a crucial pharmacokinetic property because CNS-active compounds must pass across it and CNS-inactive compounds must not pass across it to avoid CNS side effects. Generally the degree of plasma protein binding of any drug influences not only the drug action but also its disposition and efficacy. Usually, the drug that is unbound to plasma proteins will be available for diffusion or transport across cell membranes and finally interacting with the target. Here in with respect to ADME, the percent of drug bound with plasma proteins was predicted and the compounds 2, 5, 6, 7, 8, 9, 10, 14, 17, 18, 19 and 20 are predicted to bound strongly and the compounds 1, 3, 4, 11, 12, 13, 15 and 16 are predicted to bound weakly to plasma proteins. The predicted ADME properties and their values are presented in the Table 1.
Table 1.
ADME properties predicted for 20 novel compounds.
| Compound | a Human intestinal absorption (%) | bin vitro Caco-2 cell permeability (nm/sec) | cin vitro MDCK cell permeability (nm/sec) | din vitro plasma protein binding (%) | ein vivo blood-brain barrier penetration (C.brain/C.blood) |
|---|---|---|---|---|---|
| 1 | 90.635 | 21.557 | 0.2484 | 83.69 | 0.2324 |
| 2 | 92.359 | 21.480 | 0.4673 | 91.89 | 0.4803 |
| 3 | 92.583 | 21.655 | 0.0732 | 88.53 | 0.4080 |
| 4 | 92.583 | 21.655 | 0.0732 | 88.12 | 0.4082 |
| 5 | 94.664 | 21.654 | 4.4672 | 97.01 | 0.2629 |
| 6 | 94.663 | 21.654 | 4.2637 | 100.00 | 0.2434 |
| 7 | 95.914 | 21.714 | 14.2191 | 100.00 | 0.3311 |
| 8 | 89.279 | 21.694 | 25.7699 | 100.00 | 0.1876 |
| 9 | 96.488 | 21.710 | 6.4507 | 100.00 | 0.1989 |
| 10 | 94.924 | 21.713 | 25.2219 | 100.00 | 0.3009 |
| 11 | 79.779 | 11.035 | 0.0436 | 87.56 | 0.0110 |
| 12 | 49.368 | 17.055 | 0.0434 | 78.52 | 0.0258 |
| 13 | 21.008 | 19.226 | 0.0434 | 60.27 | 0.0306 |
| 14 | 83.536 | 16.043 | 0.0436 | 90.71 | 0.0126 |
| 15 | 62.784 | 19.787 | 0.0447 | 81.16 | 0.0139 |
| 16 | 26.739 | 20.563 | 0.0434 | 66.55 | 0.0325 |
| 17 | 95.453 | 21.697 | 0.6218 | 96.40 | 0.3688 |
| 18 | 97.777 | 21.719 | 4.7661 | 100.00 | 0.6795 |
| 19 | 97.876 | 21.723 | 0.8251 | 98.20 | 0.9963 |
| 20 | 95.990 | 19.217 | 0.0218 | 100.00 | 0.3767 |
Human intestinal absorption is the sum of bioavailability and absorption evaluated from ratio of excretion or cumulative excretion in urine, bile and feces. The value between 0%–20% indicates poor absorption, 20%–70% moderate absorption and 70%–100% well absorption.
Caco-2 cells are derived from human colon adenocarcinoma and possess multiple drug transport pathways through the intestinal epithelium. The value <4 indicates low permeability, 4–70 middle permeability and >70 high permeability.
MDCK cell system may use as good tool for rapid permeability screening. The value <25 indicates low permeability, 25–500 middle permeability and >500 high permeability.
The percent of drug binds to plasma protein. The value <90% indicates weak binding and >90% indicates strong binding to plasma proteins.
Blood-Brain Barrier (BBB) penetration is represented as BB = [Brain]/[Blood]. The value <0.1 indicates low absorption, 0.1 – 2.0 middle absorption and >2.0 higher absorption to CNS.
Molecular Docking
Molecular docking of the 20 novel compounds against ABAD active site revealed that all of them with better IC50’s showed good interaction with good docking scores dominated by hydrogen bonding and phosphonate salt-bridge formation with the binding domain of ABAD. Hydrophobic interactions also were observed to play a contributing role. The least and highest docking scores were found with the compounds 14 and 16, respectively, but both of them are not showing satisfactory QSAR descriptions and violating drug like properties. All of the compounds finally had better interactions (supporting information document). The residues Ser 155 and Val 156 of ABAD active sites are found to be playing a predominant role in interaction with all the compounds. Lys 172 is showing arene interactions, which is because of hydrophobicity of the ring structures with the compounds 4, 6, 11 and 19, which in turn explains strong interaction with the ABAD (Figure 18, 19 & Table 3 in supplemental file). The docking scores and the bonding information with ABAD residues are tabulated and shown in Table 2.
Figure 18.
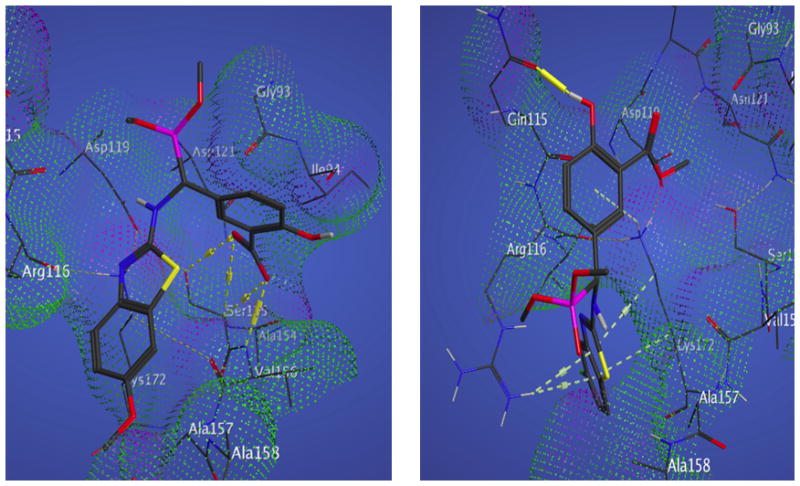
Binding site for active IC50 compound 1 &2. Compound 1 (left) binding affinity is contributed to 4 H-bonds to Ser 155 Ala 156 and Arg 116, as shown by the dotted yellow interactions. Compound 2 (right) binding affinity is contributed to Π-stacking interactions between Lys 172, Arg 116 and a H-bond to Gln 115.
Figure 19.
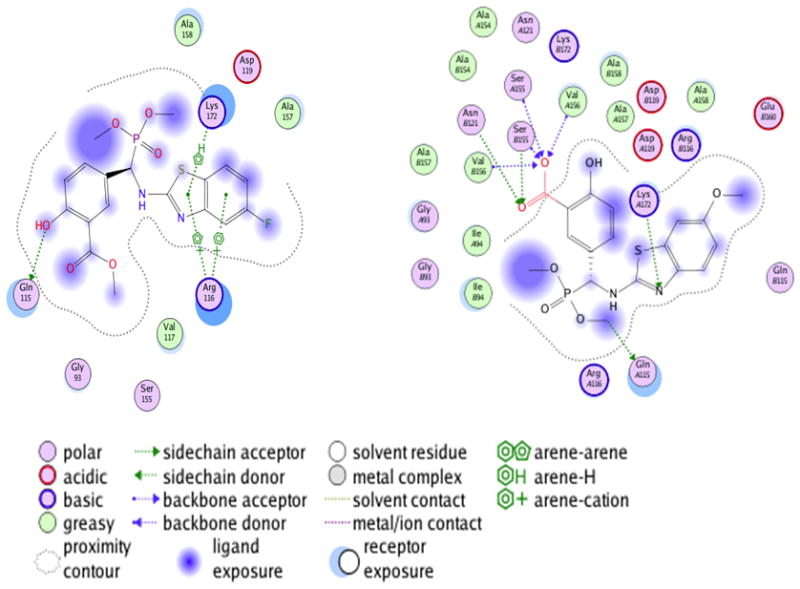
2D Ligand interaction maps of compound 1 (left) compound 2 (right) binding to ABAD.
Table 2.
Molecualr docking interaction of 20 novel compounds against ABAD active site.
| a Compound | b Docking Score | c No. Hydrogen Bonds | d Interacting Residues of ABAD |
|---|---|---|---|
| 1 | −13.203 | 5 | Gln 115, Asn 121, Ser 155, Ala 158, Lys 172 |
| 2 | −11.741 | 2 | Gln 115, Asp 119 |
| 3 | −10.757 | 3 | Phe 114, Gln 115, Ser 155 |
| 4 | −11.160 | Arene interaction | Lys 172 |
| 5 | −10.927 | 2 | Ser 155, Val 156 |
| 6 | −12.043 | 4 | Asn 121, Ser 155, Ser 155, Val 156 |
| Arene interaction | Lys 172 | ||
| 7 | −11.906 | 5 | Asn 121, Ser 155, Ser 155, Val 156, Lys 172 |
| 8 | −11.423 | 4 | Ser 155, Val 156, Lys 172, Lys 172 |
| 9 | −10.727 | 4 | Asn 121, Ser 155, Ser 155, Val 156 |
| 10 | −11.557 | 2 | Ser 155, Val 156 |
| 11 | −11.616 | 2 | Gly 95, Ser 155 |
| Arene interaction | Lys 172 | ||
| 12 | −10.358 | 1 | Glu 160 |
| 13 | −14.813 | 2 | Lys 172, Glu 160 |
| 14 | −11.626 | -- | -- |
| 15 | −11.785 | 5 | Gln 115, Asp 119, Asn 121, Ser 155, Lys 172 |
| 16 | −10.190 | 2 | Gln 115, Ser 155 |
| 17 | −10.623 | 1 | Gln 115 |
| 18 | −10.695 | 1 | Lys 172 |
| 19 | −10.990 | 2 | Ser 155, Val 156 |
| Arene interaction | Lys 172 | ||
| 20 | −12.774 | 3 | Asp 119, Ser 155, Lys 172 |
The novel ABAD inhibitors.
Docking scores generated during MOE docking between the novel leads and ABAD binding domain.
Number of hydrogen bonds formed between the ABAD binding domain and the novel leads.
The interacting active site residues of ABAD protein with the novel inhibitors in the ligand-receptor complex.
Conclusions and Future Directions
From the docking, 2D QSAR and pharmacophore studies, it is concluded that most of these compounds are reasonable inhibitors of ABAD. The exceptions are compounds 11, 12, 13, 14, 15 and 16 as they are not showing proper drug-like QSAR and satisfactory ADMET properties. The ligand – complexes generated from molecular docking process suggest that the molecules are good ABAD inhibitors as they are showing good binding affinity with the ABAD receptor. This molecular docking study will not be used to screen the molecules that are having inhibitory activity against ABAD, since it is an inefficient. However, our demonstration of 2D QSAR and 3D pharmacophore models to predict activity should be very useful to screen a huge number of new compounds. We hope to combine these models to give us greater accuracy in the prediction of active candidates. We would like to point out that only the most active compounds that we have synthesized are going to be used in upcoming studies to discover even more potent analogues for the treatment of the Alzheimer disease.
Supplementary Material
Figure 2.
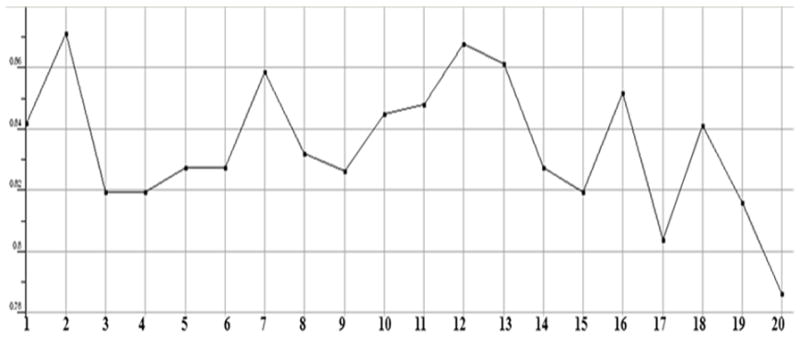
Plot showing the Molecular mass density of test compounds (Weight divided by vdw_vol (amu/Å3)).
Figure 3.
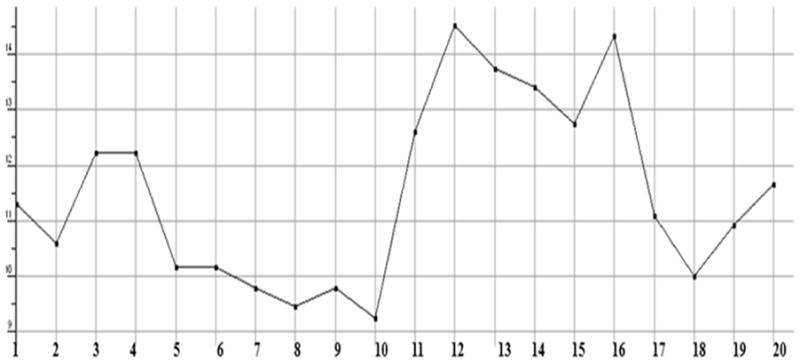
Plot showing the Molecular refractivity of test compounds (including implicit hydrogens). This property is an atomic contribution model that assumes the correct protonation state.
Figure 4.
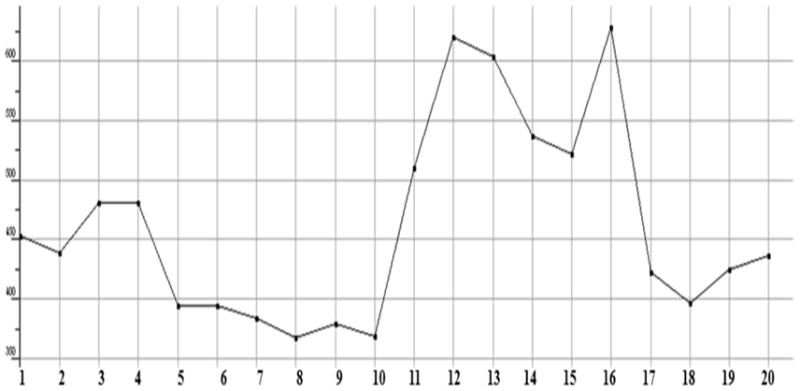
Plot showing the Molecular weight (including implicit hydrogens) of test compounds in atomic mass units.
Figure 5.
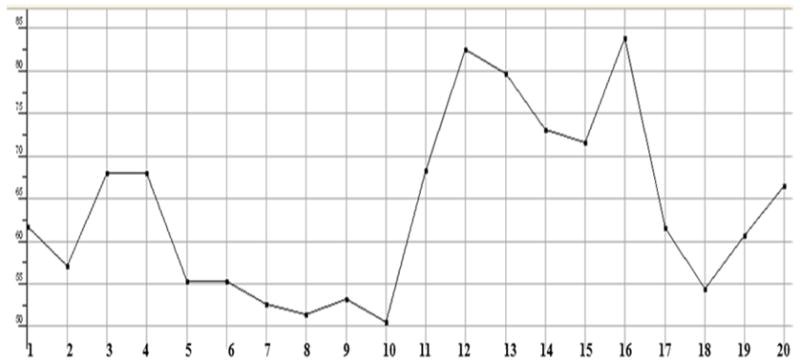
Plot showing the Sum of the atomic polarizabilities of test compounds including implicit hydrogens.
Figure 6.
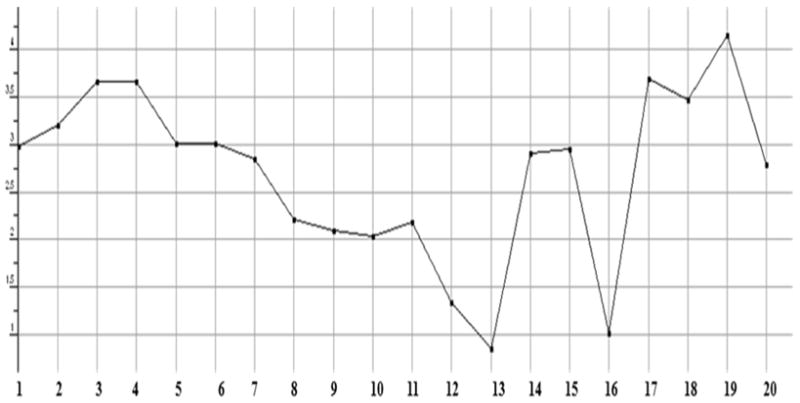
Plot showing the log of the octanol/water partition coefficient (logP(o/w)) of test compounds including implicit hydrogens. This property is calculated from a linear atom type model with r2 = 0.931 and RMSE=0.393.
Figure 7.
Plot showing the log of the aqueous solubility (logs) (mol/L) of test compounds. This property is calculated from an atom contribution linear atom type model.
Figure 8.

Plot showing the polar surface area (Å2) of test compounds calculated using group contributions to approximate the polar surface area.
Figure 9.
Plot showing the van der Waals volume (Å3) of test compounds.
Figure 10.
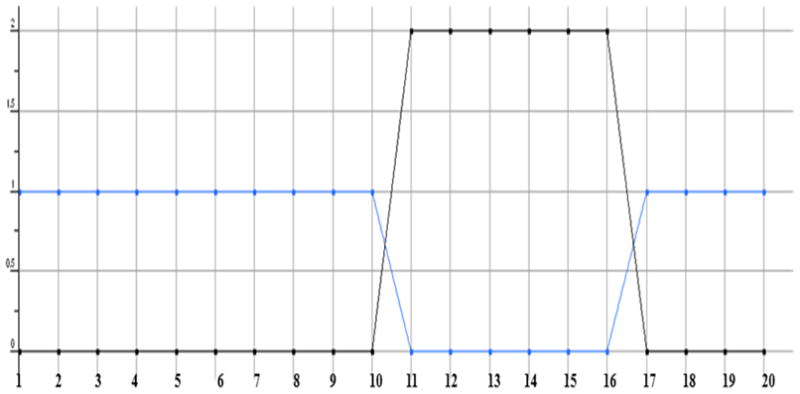
Plot showing the Lipinski rule drug like (blue) and violation (black) of test compounds. The blue colored line with a value of 1 shows the drug likeliness and value of beyond 0 indicates the Lipinski rule violation. The compounds 11, 12, 13, 14, 15 and 16 are showing violations of 2 each.
Figure 11.
Plot showing the radius of gyration of test compounds.
Figure 12.
Plot showing the count of hydrogen bonds donors in each test compound. All of them showing a count of less than 5, which is one of the desired properties of Lipinski rule.
Acknowledgments
This study was supported by grant awards (RO1GM95355 and R37AG037319) from the national Institute of General Medical Sciences and the National Institute on Aging.
Abbreviations
- ABAD
Amyloid binding alcohol dehydrogenase
- Aβ
amyloid beta
- AD
Alzheimer’s disease
- QSAR
quantitative structure–activity relationship
- CoMFA
comparative molecular field analysis
- ADMET
absorption, distribution, metabolism and excretion and toxicity
- FDA
Food and Drug Administration
- BBB
blood brain barrier
- Caco2
human colon adenocarcinoma
- MDCK
Madin-Darby canine kidney
- SAR
structure–activity relationship
- DMP
dimethyl phosphite
- DEP
diethyl phosphite
- SlogP
Log of the octanol/water partition coefficient
- CNS
central nervous system
- MOE
molecular operating environment
- RMSE
Root mean square error
- PCA
Principle Component Analysis
- PDB
Protein Data Bank
- MMFF94x
The Merck Molecular Force Field
Footnotes
Supporting Information Available: Experimental details and spectroscopic data of synthesized compounds and Interaction of remaining 18 novel inhibitors with ABAD active site. Two-dimensional linear representations of ligand-receptor complexes and three-dimensional graphical representations of ligand-receptor complexes were presented.
References
- 1.Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:18670–5. doi: 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Du H, Guo L, Yan SS. Synaptic Mitochondrial Pathology in Alzheimer’s Disease. Antioxidants & redox signaling. 2011 doi: 10.1089/ars.2011.4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen JX, Yan SS. Role of mitochondrial amyloid-beta in Alzheimer’s disease. Journal of Alzheimer’s disease: JAD. 2010;20(Suppl 2):S569–78. doi: 10.3233/JAD-2010-100357. [DOI] [PubMed] [Google Scholar]
- 4.Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, et al. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005;19:2040–1. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 5.Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends in molecular medicine. 2008;14:45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin MT, Beal MF. Alzheimer’s APP mangles mitochondria. Nature medicine. 2006;12:1241–3. doi: 10.1038/nm1106-1241. [DOI] [PubMed] [Google Scholar]
- 7.Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nature medicine. 2008;14:1097–105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Human molecular genetics. 2006;15:1437–49. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 9.Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304:448–52. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 10.Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A. 2008;105:13145–50. doi: 10.1073/pnas.0806192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:9057–68. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eckert A, Hauptmann S, Scherping I, Rhein V, Muller-Spahn F, Gotz J, et al. Soluble beta-amyloid leads to mitochondrial defects in amyloid precursor protein and tau transgenic mice. Neuro-degenerative diseases. 2008;5:157–9. doi: 10.1159/000113689. [DOI] [PubMed] [Google Scholar]
- 13.Hauptmann S, Scherping I, Drose S, Brandt U, Schulz KL, Jendrach M, et al. Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 14.Du H, Guo L, Zhang W, Rydzewska M, Yan S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiology of aging. 2009 doi: 10.1016/j.neurobiolaging.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takuma K, Fang F, Zhang W, Yan S, Fukuzaki E, Du H, et al. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proc Natl Acad Sci U S A. 2009;106:20021–6. doi: 10.1073/pnas.0905686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao J, Du H, Yan S, Fang F, Wang C, Lue LF, et al. Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J Neurosci. 2011;31:2313–20. doi: 10.1523/JNEUROSCI.4717-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Human molecular genetics. 2011;20:2495–509. doi: 10.1093/hmg/ddr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, et al. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:19318–23. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takuma K, Yao J, Huang J, Xu H, Chen X, Luddy J, et al. ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. Faseb J. 2005;19:597–8. doi: 10.1096/fj.04-2582fje. [DOI] [PubMed] [Google Scholar]
- 21.Xie Y, Deng S, Chen Z, Yan S, Landry DW. Identification of small-molecule inhibitors of the Abeta-ABAD interaction. Bioorganic & medicinal chemistry letters. 2006;16:4657–60. doi: 10.1016/j.bmcl.2006.05.099. [DOI] [PubMed] [Google Scholar]
- 22.Marques AT, Fernandes PA, Ramos MJ. Molecular dynamics simulations of the amyloid-beta binding alcohol dehydrogenase (ABAD) enzyme. Bioorgan Med Chem. 2008;16:9511–8. doi: 10.1016/j.bmc.2008.09.043. [DOI] [PubMed] [Google Scholar]
- 23.Srinivas E, Murthy JN, Rao AR, Sastry GN. Recent advances in molecular modeling and medicinal chemistry aspects of phospho-glycoprotein. Current drug metabolism. 2006;7:205–17. doi: 10.2174/138920006775541543. [DOI] [PubMed] [Google Scholar]
- 24.Xie Y, Deng S, Chen Z, Yan S, Landry DW. Identification of small-molecule inhibitors of the Abeta-ABAD interaction. Bioorg Med Chem Lett. 2006;16:4657–60. doi: 10.1016/j.bmcl.2006.05.099. [DOI] [PubMed] [Google Scholar]
- 25.Vilar S, Cozza G, Moro S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr Top Med Chem. 2008;8:1555–72. doi: 10.2174/156802608786786624. [DOI] [PubMed] [Google Scholar]
- 26.Molecular Operating Environment (MOE), 2011.10. Chemical Computing Group Inc; 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada: 2011. [Google Scholar]
- 27.Irvine JD, Takahashi L, Lockhart K, Cheong J, Tolan JW, Selick HE, et al. MDCK (Madin-Darby canine kidney) cells: A tool for membrane permeability screening. Journal of pharmaceutical sciences. 1999;88:28–33. doi: 10.1021/js9803205. [DOI] [PubMed] [Google Scholar]
- 28.Zhao YH, Le J, Abraham MH, Hersey A, Eddershaw PJ, Luscombe CN, et al. Evaluation of human intestinal absorption data and subsequent derivation of a quantitative structure-activity relationship (QSAR) with the Abraham descriptors. Journal of pharmaceutical sciences. 2001;90:749–84. doi: 10.1002/jps.1031. [DOI] [PubMed] [Google Scholar]
- 29.Vilar S, Cozza G, Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr Top Med Chem. 2008;8:1555–72. doi: 10.2174/156802608786786624. [DOI] [PubMed] [Google Scholar]
- 30.Vilar S, Gonzalez-Diaz H, Santana L, Uriarte E. QSAR model for alignment-free prediction of human breast cancer biomarkers based on electrostatic potentials of protein pseudofolding HP-lattice networks. J Comput Chem. 2008;29:2613–22. doi: 10.1002/jcc.21016. [DOI] [PubMed] [Google Scholar]
- 31.Damu AG, Jayaprakasam B, Rao KV, Gunasekar D. A flavone glycoside from Andrographis alata. Phytochemistry. 1998;49:1811–3. doi: 10.1016/s0031-9422(98)00342-2. [DOI] [PubMed] [Google Scholar]
- 32.Sreeramulu K, Rao KV, Rao CV, Gunasekar D. A new naphthoquinone from Bombax malabaricum. J Asian Nat Prod Res. 2001;3:261–5. doi: 10.1080/10286020108040365. [DOI] [PubMed] [Google Scholar]
- 33.Somogyi G, Nishitani S, Nomi D, Buchwald P, Prokai L, Bodor N. Targeted drug delivery to the brain via phosphonate derivatives - I. Design, synthesis and evaluation of an anionic chemical delivery system for testosterone. Int J Pharm. 1998;166:15–26. [Google Scholar]
- 34.Somogyi G, Buchwald P, Nomi D, Prokai L, Bodor N. Targeted drug delivery to the brain via phosphonate derivatives -II. Anionic chemical delivery system for zidovudine (AZT) Int J Pharm. 1998;166:27–35. [Google Scholar]
- 35.Levy D, Ashani Y. Synthesis and Invitro Properties of a Powerful Quaternary Methylphosphonate Inhibitor of Acetylcholinesterase - a New Marker in Blood-Brain-Barrier Research. Biochem Pharmacol. 1986;35:1079–85. doi: 10.1016/0006-2952(86)90142-5. [DOI] [PubMed] [Google Scholar]
- 36.Chen HX, Noble F, Roques BP, Fournie-Zaluski MC. Long lasting antinociceptive properties of enkephalin degrading enzyme (NEP and APN) inhibitor prodrugs. J Med Chem. 2001;44:3523–30. doi: 10.1021/jm0102248. [DOI] [PubMed] [Google Scholar]
- 37.Somogyi G, Buchwald P, Bodor N. Targeted drug delivery to the central nervous system via phosphonate derivatives (Anionic delivery system for testosterone) Pharmazie. 2002;57:135–7. [PubMed] [Google Scholar]
- 38.Qu ZB, Chen XL, Yuan JW, Bai YL, Chen T, Qu LB, et al. New synthetic methodology leading to a series of novel heterocyclic alpha-aminophosphonates: a very attractive expansion of Kabachnik-Fields reaction. Tetrahedron. 2012;68:3156–9. [Google Scholar]
- 39.Crivori P, Cruciani G, Carrupt PA, Testa B. Predicting blood-brain barrier permeation from three-dimensional molecular structure. J Med Chem. 2000;43:2204–16. doi: 10.1021/jm990968+. [DOI] [PubMed] [Google Scholar]
- 40.Nielsen PA, Andersson O, Hansen SH, Simonsen KB, Andersson G. Models for predicting blood-brain barrier permeation. Drug Discov Today. 2011;16:472–5. doi: 10.1016/j.drudis.2011.04.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.