

Abstract
The native function of α-synuclein is thought to involve regulation of synaptic vesicle trafficking. Recent work has also implicated a role in neurotransmission, possibly through interactions with the proteins involved in synaptic vesicle fusion. Here, we demonstrate that α-synuclein inhibits SNARE-mediated vesicle fusion through binding the membrane, without a direct interaction between α-synuclein and any of the SNARE proteins. This work supports a model where the role of α-synuclein in the regulation of vesicle fusion is through modulating properties of the lipid bilayer.
α-Synuclein (αS) is a 14.4 kDa neuronal protein implicated in Parkinson’s Disease (PD), where it is the major component of the Lewy Body plaques found in affected brain tissue. αS localizes to synaptic termini, existing in equilibrium between the cytosol and synaptic membrane (1). In vitro, the ability of αS to bind synthetic lipid bilayers is well-established, and preferences for both specific lipids and highly curved membranes have been observed (2). Binding of αS significantly alters membrane properties; it can induce curvature, thinning of the lipid bilayer and tubulation (3-5). These varying effects of αS upon binding to lipid bilayers allude to potential roles for αS in processes such as membrane fusion that depend on transitions through states of high membrane curvature.
There is also a growing body of work which supports a role for αS in regulation of synaptic vesicle fusion (Reviewed in 6). Soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins are responsible for synaptic vesicle fusion as well as most other fusion events within cells (7). In neurons, v-SNAREs (VAMP2 or synaptobrevin) associate with vesicle membranes while t-SNAREs (syntaxin and SNAP-25) are found in target membranes (8). In a vesicular fusion event, the t- and v-SNAREs assemble into a four-helix bundle pulling the two membranes together to cause fusion (9). Multiple animal models show a decrease in neurotransmitter release upon overexpression of αS (10-12), suggesting that αS may act as a regulator of neurotransmission, altering or disrupting the SNARE driven fusion of synaptic vesicles. An increase in induced dopamine release is found in mice lacking all three (α, β, and γ) synuclein proteins (13).
Here we investigate the role of αS in regulating SNARE-mediated vesicle fusion. To directly assess the effects of αS, we used an in vitro SNARE fusion assay. Vesicles were prepared containing either v- or t-SNARE components (see Supporting Information). Fusion was initiated by mixing the two types of SNARE vesicles in the absence or presence of αS and was monitored by a fluorescence increase upon lipid mixing.
αS inhibits SNARE-mediated vesicle fusion in a concentration dependent manner (Fig 1A). Inhibition is observed at αS:accessible lipid ratios of less than 1:500. The extent of inhibition increases with increasing αS and saturation of the effect occurs at a αS:accessible lipid ratio of ~1:20. Fusion is inhibited a maximum of ~50% relative to the control. Effects of similar magnitude have been observed for established regulators of SNARE-mediated fusion (14, 15). αS does not appear to stall the fusion process at hemifusion (Fig S1).
Figure 1.
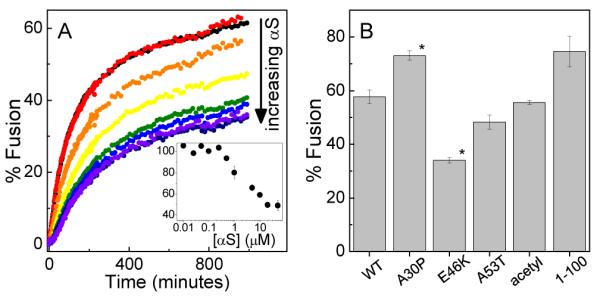
αS inhibits vesicle fusion. A. SNARE-mediated vesicle fusion as a function of increasing αS concentration (800 μM lipid). Inset. Quantification of the extent of inhibition normalized to fusion in the absence of αS. B. Comparison of αS variants (5 μM αS, 800 μM lipid). N=3, error is s.e.m., *P<0.01 compared to WT by Student’s T-test.
To gain further insight into this phenomenon, we exploited previous work in our lab which quantified differences in membrane affinity between αS variants associated with familial forms of PD (2). Each of the variants inhibits SNARE-mediated fusion, although to differing extents relative to wild-type (WT): E46K>WT, A30P<WT, and A53T≈WT(Fig 1B). Importantly, the extent to which each of these variants inhibits fusion correlates with its affinity for the lipid bilayer and thus with the amount of αS associated with the vesicles. To illustrate, E46K binds more tightly to membranes and also inhibits more strongly relative to WT, while A30P binds membranes more weakly and inhibits fusion less effectively than WT (2). αS from mammalian sources is acetylated on its N-terminus (16); this modification does not interfere with its ability to inhibit fusion (Fig 1B). αS truncated at residue 100 is also capable of inhibiting fusion (Fig 1B), although to a lesser extent than predicted based on its binding affinity (3). Although the C-terminus does not interact directly with the lipid bilayer, this supports a role for it in inhibiting fusion, perhaps through electrostatic repulsion. It may also reflect a more general ability of amphipathic α-helical membrane-binding proteins to alter lipid bilayer properties and affect processes such as fusion and tubulation. Lastly, of note is a very recent study where oligomeric αS was reported to inhibit SNARE-mediated vesicle fusion (17). Although monomer αS was not found to have the same effect in this study, this is likely due to the low content of anionic lipids used in the vesicles (see Supporting Information).
The results of the fusion assays indicate a correlation between extent of inhibition and amount of αS bound to the vesicles, strongly supported by our previous work with pure lipid vesicles (2). However, the presence of the t- and v-SNARE proteins could be expected to alter αS partitioning to SNARE vesicles, as would binding of αS to either of the SNARE proteins (18). To directly assess the impact of t- and v-SNARE proteins, we measured binding of αS to vesicles containing t- and v-SNAREs. αS was labeled at residue 33 with NBD, an environment sensitive fluorophore (αS-NBD). The intensity of the αS-NBD fluorescence changes upon insertion into the lipid bilayer, reporting on binding (Fig S2).
αS binds with lower affinity to vesicles containing either v- or t-SNARE proteins as compared with pure lipid vesicles (Fig 2A). The results are quantified in Table S1. Using a floatation assay as an orthogonal approach, we screened a broader range of lipid compositions and found a similar result for all compositions tested (Fig S3). There are several mechanisms by which the presence of SNARE proteins could impact binding of αS to vesicles. The simplest explanation is that the SNARE proteins occupy lipid binding sites that would otherwise be accessible to αS, thereby decreasing protein binding. Electrostatics are important for αS binding to pure lipid vesicles (2, 19), and the presence of the SNARE proteins, particularly the highly acidic t-SNARE, will alter the effective charge of the vesicles. However, given that αS binds with higher affinity to more negatively charged lipid vesicles and has also been observed to interact with a variety of negatively charged macromolecules (20-22), we might expect an increase in binding t-SNARE vesicles, rather than the observed decrease. Lastly, direct binding of αS to VAMP2, mediated by the C-terminus of αS and the N-terminus of VAMP2, both of which should be accessible in the membrane-associated forms of the proteins (23, 24), has been proposed to enhance assembly of the SNARE fusion complex (18). Our expectation is that binding of αS to VAMP2 would result in a net decrease in the measured Kd for v-SNARE vesicles relative to lipid-only vesicles, whereas we observe nearly a three-fold increase in Kd (Table S1).
Figure 2.
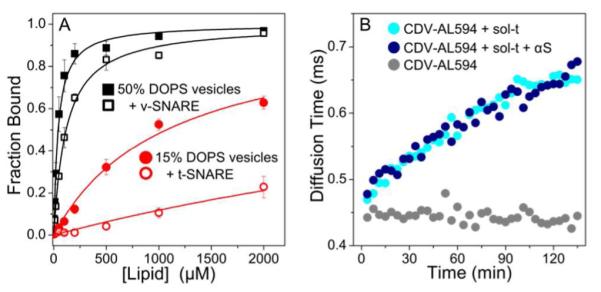
αS does not bind to SNARE proteins. A. αS (500 nM) binds more weakly to SNARE vesicles (open) than to comparable pure lipid vesicles (closed). N=3, error is s.e.m. B. An increase in diffusion time is observed by FCS upon mixing of CDV-AL594 (gray) with sol-t (cyan) reflecting soluble SNARE complex formation. An excess of αS (blue) does not affect the extent or rate of complex formation.
The use of soluble t- and v-SNARE constructs allowed us to examine their interactions with αS in the absence of the lipid bilayer. In particular, we considered the possibility that the excess of lipid relative to protein found in our model system, as compared to actual synaptic vesicles (25), interfere with or mask evidence of a direct interaction between αS and either of the SNARE proteins. Soluble SNARE constructs were created by removing the transmembrane anchors, resulting in the cytoplasmic domain of VAMP2 (CDV: residues 1-95) and soluble t-SNARE (sol-t: syntaxin residues 1-265 and full-length SNAP-25). SNARE complex formation of these soluble constructs was observed by monitoring an increase in diffusion time of Alexa 594 labeled CDV (CDV-AL594) upon binding to unlabeled sol-t using fluorescence correlation spectroscopy (FCS). Incorporation of CDV-AL594 into the larger complex results in a more slowly diffusing fluorescent species due to the increased hydrodynamic size (Fig 2B). The addition of an excess of αS does not alter either the kinetics or the extent of complex formation (Fig 2B). Interaction of αS with the SNARE complex was assessed directly by repeating the experiment using Alexa 488 labeled αS (αS-AL488) and unlabeled CDV and sol-t (Fig S4). No change in the diffusion time of αS was observed on the timescale of SNARE complex formation, indicating it is not associated with the SNARE complexes. Lastly, complex formation was examined by using size exclusion chromatography to separate SNARE complexes from the free constituent proteins in CDV/sol-t samples in the absence and presence of αS (26, 27). αS was observed to elute as free monomer and the extent of complex formation was independent of αS (Fig S5). This finding is consistent with our FCS measurements and indicates that αS does not appear to have a direct role in chaperoning SNARE complex formation. With these measurements, however, we cannot exclude the possibility that interactions with αS could be mediated by any of the numerous proteins, such as synaptotagmin and complexin, that regulate SNARE complex formation in vivo.
Our results strongly support a model where αS inhibits SNARE-mediated vesicle fusion through its interactions with lipid membranes. These findings are broadly consistent with the body of in vitro and in vivo work that suggests that the function of αS may derive from its ability to alter lipid bilayer properties, rendering it less fusogenic. The most direct support for this model comes from the observation that αS inhibits calcium-mediated, SNARE-independent vesicle fusion (28)(Fig S6), suggesting that decreased membrane fusability may be independent of the fusion mechanism. αS has been observed to anneal defects in bilayers, resulting in a more uniform membrane with a higher energy barrier to fusion (29). Both experiment and simulations have shown that αS binding thins lipid bilayers and increases their rigidity (3), an effect which increases the energy required to form the highly curved fusion intermediates (30). More generally, αS is capable of inducing curvature and tubulation in bilayers composed of a range of lipids (3-5). Induction of curvature and tubulation of lipid bilayers is also a feature of many BAR domain proteins which have well-studied roles in remodeling membranes in various endocytic and exoctytic pathways. This last feature is particularly intriguing in light of a recent study which found upregulation of the BAR domain proteins endophilin A1 and endophilin B2 in mice deficient in α, β, and γ synuclein (5). The endophilins are implicated specifically in sensing/induction of membrane curvature (31, 32), indicating there may be a compensation for the loss of these functions in the absence of all the synucleins (Fig S7).
It is of interest to consider the consequences of our model in interpreting αS functional studies in a variety of model systems. Increased dopamine release in αS knock-out mice and reduced dopamine release in mice overexpressing αS can both be explained by αS functioning to inhibit fusion (10-13). In cultured mammalian cells and in yeast, overexpression of αS disrupts ER to Golgi transport (26, 33), a process highly dependent on vesicle fusion. Overexpression of αS in C. elegans resulted in increased fragmentation of mitochondria, due to decreased fusion between mitochondria (28). Moreover, downregulation of αS was associated with mitochondrial elongation, reflecting increased fusion. Finally, our observation that αS does not enhance SNARE complex formation is consistent with a study which observed a normal abundance of SNARE complexes in mice lacking all three members of the synuclein family (13). Our results here help in the mechanistic understanding of altered membrane fusion events observed in vivo and strongly implicates a role for αS in membrane remodeling at the synaptic terminal.
Supplementary Material
ACKNOWLEDGMENT
We thank James Rothman for providing SNARE plasmids, Jeff Coleman and Li Shi for helpful discussions.
Funding Sources This research was supported by grants from the Ellison Medical Foundation (to E.R.) and the NIH Training Grant 5T32GM007223 (to D.C.D.).
Footnotes
Supporting Information Figures S1-S7 and detailed experimental methods. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes The authors declare no competing financial interests.
REFERENCES
- 1.Jakes R, Spillantini MG, Goedert M. FEBS Lett. 1994;345:27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- 2.Middleton ER, Rhoades E. Biophys J. 2010;99:2279–2288. doi: 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braun AR, Sevcsik E, Chin P, Rhoades E, Tristram-Nagle S, Sachs JN. J Am Chem Soc. 2012;134:2613–2620. doi: 10.1021/ja208316h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Varkey J, Isas JM, Mizuno N, Jensen MB, Bhatia VK, Jao CC, Petrlova J, Voss JC, Stamou DG, Steven AC, Langen R. J Biol Chem. 2010;285:32486–32493. doi: 10.1074/jbc.M110.139576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Westphal CH, Chandra SS. J Biol Chem. 2013;288:1829–1840. doi: 10.1074/jbc.M112.418871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Auluck PK, Caraveo G, Lindquist S. Annu Rev Cell Dev Biol. 2010;26:211–233. doi: 10.1146/annurev.cellbio.042308.113313. [DOI] [PubMed] [Google Scholar]
- 7.Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 8.Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 9.Sutton RB, Fasshauer D, Jahn R, Brunger AT. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 10.Lundblad M, Decressac M, Mattsson B, Bjorklund A. Proc Natl Acad Sci U S A. 2012;109:3213–3219. doi: 10.1073/pnas.1200575109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, Savalle M, Nemani V, Chaudhry FA, Edwards RH, Stefanis L, Sulzer D. J Neurosci. 2006;26:11915–11922. doi: 10.1523/JNEUROSCI.3821-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anwar S, Peters O, Millership S, Ninkina N, Doig N, Connor-Robson N, Threlfell S, Kooner G, Deacon RM, Bannerman DM, Bolam JP, Chandra SS, Cragg SJ, Wade-Martins R, Buchman VL. J Neurosci. 2011;31:7264–7274. doi: 10.1523/JNEUROSCI.6194-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schaub JR, Lu X, Doneske B, Shin YK, McNew JA. Nat Struct Mol Biol. 2006;13:748–750. doi: 10.1038/nsmb1124. [DOI] [PubMed] [Google Scholar]
- 15.Malsam J, Parisotto D, Bharat TA, Scheutzow A, Krause JM, Briggs JA, Sollner TH. EMBO J. 2012;31:3270–3281. doi: 10.1038/emboj.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim PS, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, Chilcote TJ. J Biol Chem. 2006;281:29739–29752. doi: 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- 17.Choi BK, Choi MG, Kim JY, Yang Y, Lai Y, Kweon DH, Lee NK, Shin YK. Proc Natl Acad Sci U S A. 2013;110:4087–4092. doi: 10.1073/pnas.1218424110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rhoades E, Ramlall TF, Webb WW, Eliezer D. Biophys J. 2006;90:4692–4700. doi: 10.1529/biophysj.105.079251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cohlberg JA, Li J, Uversky VN, Fink AL. Biochemistry. 2002;41:1502–1511. doi: 10.1021/bi011711s. [DOI] [PubMed] [Google Scholar]
- 21.Alim MA, Hossain MS, Arima K, Takeda K, Izumiyama Y, Nakamura M, Kaji H, Shinoda T, Hisanaga S, Ueda K. J Biol Chem. 2002;277:2112–2117. doi: 10.1074/jbc.M102981200. [DOI] [PubMed] [Google Scholar]
- 22.Payton JE, Perrin RJ, Clayton DF, George JM. Brain Res Mol Brain Res. 2001;95:138–145. doi: 10.1016/s0169-328x(01)00257-1. [DOI] [PubMed] [Google Scholar]
- 23.Eliezer D, Kutluay E, Bussell R, Jr., Browne G. J Mol Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 24.Williams D, Vicogne J, Zaitseva I, McLaughlin S, Pessin JE. Mol Biol Cell. 2009;20:4910–4919. doi: 10.1091/mbc.E09-04-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, Urlaub H, Schenck S, Brugger B, Ringler P, Muller SA, Rammner B, Grater F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmuller H, Heuser J, Wieland F, Jahn R. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 26.Thayanidhi N, Helm JR, Nycz DC, Bentley M, Liang Y, Hay JC. Mol Biol Cell. 2010;21:1850–1863. doi: 10.1091/mbc.E09-09-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fasshauer D, Otto H, Eliason WK, Jahn R, Brunger AT. J Biol Chem. 1997;272:28036–28041. doi: 10.1074/jbc.272.44.28036. [DOI] [PubMed] [Google Scholar]
- 28.Kamp F, Exner N, Lutz AK, Wender N, Hegermann J, Brunner B, Nuscher B, Bartels T, Giese A, Beyer K, Eimer S, Winklhofer KF, Haass C. EMBO J. 2010;29:3571–3589. doi: 10.1038/emboj.2010.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kamp F, Beyer K. J Biol Chem. 2006;281:9251–9259. doi: 10.1074/jbc.M512292200. [DOI] [PubMed] [Google Scholar]
- 30.Aeffner S, Reusch T, Weinhausen B, Salditt T. Proc Natl Acad Sci U S A. 2012;109:E1609–1618. doi: 10.1073/pnas.1119442109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farsad K, Ringstad N, Takei K, Floyd SR, Rose K, De Camilli P. J Cell Biol. 2001;155:193–200. doi: 10.1083/jcb.200107075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gallop JL, Jao CC, Kent HM, Butler PJ, Evans PR, Langen R, McMahon HT. EMBO J. 2006;25:2898–2910. doi: 10.1038/sj.emboj.7601174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.