

Abstract
Although the late phase of ischemic preconditioning is known to be mediated by increased inducible nitric oxide synthase (iNOS) activity, controversy persists regarding the role of iNOS in ischemia/reperfusion (I/R) injury and, specifically, whether this protein is protective or detrimental. We hypothesized that iNOS is protective in myocytes but detrimental in inflammatory cells. To test this hypothesis, we created chimeric mice with iNOS-deficient peripheral blood cells by transplanting iNOS knockout (KO) bone marrow in wild-type (WT) mice after lethal irradiation. 2 months later, the mice underwent a 30-min coronary occlusion followed by 24 h of reperfusion. In WT naïve mice (iNOS+/+ naïve; group I, n = 17), infarct size was 56.9 ± 2.8% of the risk region. In iNOS KO naïve mice with whole-body iNOS deletion (iNOS−/− naïve; group II, n = 10), infarct size was comparable to group I (53.4 ± 3.5%). When irradiated WT mice received marrow from WT mice (iNOS+/+ chimera; group III, n = 10), infarct size was slightly reduced versus group I (44.3 ± 3.2%), indicating that irradiation and/or transplantation slightly decrease I/R injury. However, when WT mice received marrow from iNOS KO mice (iNOS−/− chimera; group IV, n = 14), infarct size was profoundly reduced (22.8 ± 2.1%, P <0.05 vs. group III), indicating that selective deletion of iNOS from peripheral blood cells (with no change in myocardial iNOS content) induces protection against myocardial infarction. Together with our previous work showing the cardioprotective actions of NO donors, iNOS gene therapy, and cardiac-specific overexpression of iNOS, these data support a complex, dual role of iNOS in myocardial infarction (i.e., protective in myocytes but deleterious in blood cells). To our knowledge, this is the first study to identify a critical role of iNOS in peripheral blood cells as a mediator of myocardial I/R injury. The results support heretofore unknown differential actions of iNOS depending on cell source and have important translational implications.
Keywords: Inducible nitric oxide synthase, Myocardial infarction, Infarct size, Chimeric mice
Introduction
Despite the fact that prompt restoration of perfusion (with thrombolysis and/or interventional strategies) has dramatically decreased the early morbidity and mortality from myocardial infarction (MI), to date no infarct-sparing intervention is clinically available [7]. Nitric oxide (NO) is known to participate not only in the normal functions of the heart but also in the pathophysiology of cardiac ischemic injury [5]. Under physiological conditions, constitutive production of NO by NO synthase (NOS) in endothelial cells (eNOS) is thought to be cytoprotective [4]. These salubrious actions of eNOS differ from the primarily pro-inflammatory and cytotoxic actions that have been ascribed to NO derived from the inducible isoform of NO (iNOS) [5]. Whether the opposing actions of these two sources of NO relate to the differing amounts and/or cellular sources of the gaseous monoxide remains unclear.
We have previously reported that NO plays a critical role in mediating cardioprotection during the late phase of ischemic preconditioning (PC) [8–10, 16, 17, 44, 49, 52, 53]. Accordingly, increased generation of NO from eNOS on day 1 triggers multiple signaling pathways [5, 6]. These lead to the upregulation of a number of proteins, including iNOS, which in turn mediate the cardioprotective effects of late PC on day 2 [5, 6]. The obligatory role of iNOS is also supported by the observation that targeted deletion of the iNOS gene abrogates late PC induced by a variety of stimuli, including ischemia, adenosine A1 agonists, opioid δ1 agonists, endotoxin derivatives, and exercise, suggesting that iNOS is the final common effector of cardioprotection [5, 6, 16]. Furthermore, it has been demonstrated that NO contributes to myocardial hibernation by preserving regional contractile function [2, 23]. In contrast to these findings, NO is not involved in the protection against infarction by early/classical PC in pigs in vivo [42]. Although these data collectively support a key role of iNOS-derived NO in mitigating ischemic injury, the role of this protein remains controversial. Considerable evidence strongly suggests that the cellular origin of NOS is one of the major factors that determine whether the NO producing enzyme exerts a beneficial or detrimental effect in models of tissue injury [27]. Despite the fact that iNOS is found in a variety of cells, including circulating blood cells of myeloid origin (e.g., leukocytes) and resident cells (e.g., endothelial cells), it remains unclear as to whether the protective effect of iNOS deficiency in ischemia/ reperfusion (I/R) injury reflects the involvement of iNOS associated with circulating blood cells, resident cells, or both. These issues are important for developing therapeutically viable anti-ischemic interventions and for understanding the mechanisms underlying the cardioprotective effects of iNOS.
Accordingly, the goal of the present study was to elucidate the cell-specificity of the effects of iNOS on acute myocardial I/R injury. To this end, we determined whether selective iNOS deletion in peripheral blood cells modulates the extent of cell death in the mouse heart in vivo.
Materials and methods
This study was performed in accordance with the Guide for the Care and Use of Laboratory Animals (Department of Health and Human Services, Publication No. [NIH] 86–23) and with the guidelines of the Animal Care and Use Committee of the University of Louisville, School of Medicine (Louisville, KY, USA).
Experimental protocol
Wild-type (WT) naïve (iNOS+/+ naïve) and iNOS knockout (KO) naïve (iNOS−/− naïve) male mice (age 14–22 weeks), both on the same genetic C57Bl/6 background, were obtained from the Jackson Laboratories (Bar Harbor, Maine) and housed under specific pathogen-free conditions. Reciprocal bone marrow transplant chimeras were generated using iNOS+/+ and iNOS−/− mice. All mice underwent a 30-min coronary occlusion followed by 24 h of reperfusion and were assigned as follows: group I (WT naïve mice; iNOS+/+ naïve), group II (iNOS KO naïve mice; iNOS−/− naïve), group III (WT chimeric mice; iNOS+/+ chimera), and group IV (iNOS KO chimeric mice; iNOS−/− chimera). At the end of reperfusion, mice were euthanized for the analysis of morphometric and pathologic parameters.
Animal preparation
The preparation of chimeric mice was performed as described previously [28, 29]. In brief, recipient mice were total body irradiated (950 cGy), and their bone marrow was reconstituted by intravenous infusion (via a tail vein injection) of 5 million bone marrow cells (4–6 h after irradiation) harvested from donor mice. Two chimeric groups were generated in which (1) non-irradiated WT bone marrow was transferred into iNOS+/+ mice (iNOS+/+ chimera); and (2) iNOS−/− bone marrow was transferred into WT mice (iNOS−/− chimera). 2 months after receiving bone marrow cells, the mice were anesthetized with sodium pentobarbital (60 mg/kg i.p.), intubated, and ventilated with room air supplemented with oxygen at a rate of 105 strokes/min and with a tidal volume of 0.27 ± 0.05 ml using a mouse ventilator (MiniVent 845). These respiratory settings were found to result in optimal values of arterial pH (7.42 ± 0.03), PO2 (146 ± 2 mmHg), and PCO2 (33.7 ± 2.6 mmHg). Body temperature was carefully monitored with a rectal probe and maintained between 36.8 and 37.2°C throughout the experiment by using a heating pad and heat lamps (Table 1). To prevent hypotension, clinical grade Dextran 40 was administered i.v. at 40 ml/ kg, divided in three equal bolus doses. With the aid of a dissecting microscope and a microcoagulator, the chest was opened through a midline sternotomy. An 8-0 nylon suture was passed with a tapered needle under the left anterior descending (LAD) coronary artery 2–3 mm from the tip of the left auricle, and a non-traumatic balloon occluder was applied on the artery. Coronary occlusion was induced by inflating the balloon occluder for 30 min. Successful performance of coronary occlusion and reperfusion was verified by visual inspection (i.e., by noting the development of a pale color in the distal myocardium on inflation of the balloon and the return of a bright red color due to hyperemia after deflation) and by observing S-T segment elevation and widening of the QRS on the ECG during ischemia and their resolution after reperfusion). After the coronary occlusion/reperfusion, the chest was closed in layers, and a small catheter was left in the thorax for 10–20 min to evacuate air and fluids.
Table 1.
Rectal temperature and heart rate in iNOS+/+ naïve, iNOS−/− naïve, iNOS+/+ chimeric and iNOS−/− chimeric mice before coronary occlusion and during I/R
| Group | Pre-occlusion | Occlusion
|
Reperfusion
|
||||
|---|---|---|---|---|---|---|---|
| 5 min | 15 min | 30 min | 5 min | 15 min | 30 min | ||
| Rectal temperature (°C) | |||||||
| Group I (iNOS+/+ naïve mice, n = 17) | 37.0 ± 0.1 | 37.1 ± 0.1 | 37.0 ± 0.1 | 37.1 ± 0.1 | 37.0 ± 0.1 | 37.1 ± 0.1 | 37.1 ± 0.1 |
| Group II (iNOS−/− naïve mice, n = 10) | 36.9 ± 0.1 | 37.2 ± 0.1 | 37.1 ± 0.1 | 37.2 ± 0.1 | 36.9 ± 0.1 | 37.0 ± 0.0 | 37.0 ± 0.1 |
| Group III (iNOS+/+ chimeric mice, n = 10) | 37.0 ± 0.1 | 37.1 ± 0.1 | 37.1 ± 0.0 | 37.0 ± 0.1 | 37.1 ± 0.1 | 37.0 ± 0.1 | 37.0 ± 0.1 |
| Group IV (iNOS−/− chimeric mice, n = 14) | 37.0 ± 0.1 | 37.2 ± 0.0 | 37.0 ± 0.1 | 37.1 ± 0.1 | 37.0 ± 0.1 | 37.0 ± 0.1 | 37.0 ± 0.0 |
| Heart rate (beats/min) | |||||||
| Group I (iNOS+/+ naïve mice, n = 17) | 521 ± 20 | 542 ± 17 | 538 ± 16 | 549 ± 18 | 549 ± 19 | 551 ± 19 | 560 ± 24 |
| Group II (iNOS−/− naïve mice, n = 10) | 503 ± 17 | 598 ± 17 | 592 ± 22 | 503 ± 24 | 502 ± 20 | 516 ± 19 | 491 ± 29 |
| Group III (iNOS+/+ chimeric mice, n = 10) | 558 ± 17 | 569 ± 13 | 551 ± 13 | 525 ± 15 | 539 ± 18 | 516 ± 20 | 523 ± 27 |
| Group IV (iNOS−/− chimeric mice, n = 14) | 565 ± 15 | 580 ± 16 | 570 ± 15 | 554 ± 12 | 562 ± 13 | 567 ± 16 | 538 ± 18 |
Data are mean ± SEM. Measurements of rectal temperature and heart rate were taken before the 30-min coronary occlusion (pre-occlusion), at 5, 15, and 30 min into the occlusion, and at 5, 15, and 30 min after reperfusion. Rectal temperature was continuously monitored and carefully controlled throughout the experiment, as detailed in the text
Postmortem tissue analysis
At the end of the study, the thorax was opened; the heart was excised and perfused with Krebs–Henseleit solution through an aortic cannula. To delineate infarcted from viable myocardium, the heart was perfused with 1% triphenyltetrazolium chloride (TTC) in phosphate buffer [16, 18]. To identify the occluded/reperfused bed, the coronary artery was tied at the site of the previous occlusion and the aortic root was perfused with 10% phthalo blue dye [16, 18]. As a result of this procedure, the region at risk was identified by the absence of blue dye, whereas the rest of the left ventricle was stained dark blue. The left ventricle was cut into 5–7 transverse slices, which were fixed in 10% neutral buffered formaldehyde, weighed, and photographed under a microscope. The corresponding areas were measured by computerized videoplanimetry and from these measurements infarct size was calculated as a percentage of the region at risk.
Statistical analysis
Data are reported as mean ± SEM. All data were analyzed with one-way ANOVA for normally distributed data or Kruskal–Wallis one-way ANOVA on ranks for data that are not normally distributed, as appropriate, followed by unpaired Student’s t Tests with the Bonferroni correction. A P value less than 0.05 was considered statistically significant. All statistical analyses were performed using the SPSS software (version 8, SPSS Inc., Chicago, IL).
Results
Exclusions
A total of 61 mice were enrolled in this study. Of these, seven mice died during the surgical procedure and three were excluded from the study due to failure of the coronary occluder, leaving a total of 17, 10, 10, and 14 mice in iNOS+/+ naïve, iNOS−/− naïve, iNOS+/+ chimeric, and iNOS−/− chimeric groups, respectively.
General characteristics
The general characteristics (heart rate and rectal temperature) taken before the 30-min coronary occlusion (pre-occlusion), at 5, 15, and 30 min into the occlusion, and at 5, 15, and 30-min after reperfusion in iNOS+/+ naïve, iNOS−/− naïve, iNOS+/+ chimeric, and iNOS−/− chimeric groups are shown in Table 1. Heart rate, a fundamental physiological parameter that may impact infarct size [18, 30], was similar in all the groups. Within the same group, heart rate did not differ significantly at any time-point before and during the 30-min occlusion or the ensuing reperfusion. By experimental design, rectal temperature remained within a narrow, physiologic range (36.8–37.2°C) in all groups. Also, there was no significant difference in the body weight between the groups (Table 2).
Table 2.
General characteristics and pathologic parameters in iNOS+/+ naïve, iNOS−/− naïve, iNOS+/+ chimeric, and iNOS−/− chimeric mice at the end of follow-up period
| Group | Age (wk) | Body wt (g) | Heart wt (mg) | Heart wt/ body wt | LV wt (mg) | Risk region wt (mg) | Infarct wt (mg) | Risk region (% of LV) | Infarct (% of risk region) | Infarct (% of LV) |
|---|---|---|---|---|---|---|---|---|---|---|
| Group I (iNOS+/+ naïve mice, n = 17) | 14 ± 1 | 30.4 ± 0.9 | 154 ± 7 | 0.50 ± 0.01 | 115 ± 6 | 38 ± 3 | 23 ± 3 | 33.8 ± 2.0 | 56.9 ± 2.8 | 19.7 ± 1.8 |
| Group II (iNOS−/− naïve mice, n = 10) | 17 ± 1 | 30.8 ± 2.4 | 167 ± 15 | 0.54 ± 0.02 | 121 ± 9 | 44 ± 2 | 23 ± 2 | 38.0 ± 2.8 | 53.4 ± 3.5 | 20.3 ± 2.2 |
| Group III (iNOS+/+ chimeric mice, n = 10) | 17 ± 2 | 27.8 ± 0.7 | 139 ± 7 | 0.50 ± 0.03 | 104 ± 5 | 36 ± 2 | 16 ± 2a,b | 34.9 ± 1.1 | 44.3 ± 3.2a,b | 15.6 ± 1.5a,b |
| Group IV (iNOS−/− chimeric mice, n = 14) | 22 ± 0 | 30.5 ± 0.7 | 139 ± 7 | 0.51 ± 0.01 | 118 ± 3 | 37 ± 2 | 8 ± 1a,b,c | 31.3 ± 2.1 | 22.8 ± 2.1a,b,c | 7.0 ± 0.8a,b,c |
Data are mean ± SEM
LV left ventricle
P <0.001 versus group I
P <0.001 versus group II
P <0.001 versus group III
Myocardial area at risk and infarct size
The size of the region at risk did not differ in iNOS+/+ naïve and iNOS−/− naïve groups (38 ± 3 mg vs. 44 ± 2 mg; 33.8 ± 2.0% vs. 38.0 ± 2.8%, respectively, Fig. 1; Table 2); as shown in Table 2, the weight of the risk region was slightly (but not significantly) smaller in iNOS+/+ chimeras and iNOS−/− chimeras compared with their naïve groups (36 ± 2 and 37 ± 2 mg, 34.9 ± 1.1% and 31.3 ± 2.1%, respectively). To determine whether iNOS deletion from peripheral blood cells produces a protected cardiac phenotype, infarct size was measured in iNOS+/+ naïve, iNOS−/− naïve, iNOS+/+ chimeric, and iNOS−/− chimeric mice after subjecting them to a 30-min coronary occlusion and 24 h of reperfusion. In iNOS−/− naïve mice, infarct size did not differ from that measured in iNOS+/+ naïve mice (53.4 ± 3.5% of the risk region, n = 10, vs. 56.9 ± 2.8%, n = 17; P <0.05; Fig. 1; Table 2), indicating that whole-body deletion of iNOS did not affect the extent of cell death. In contrast, infarct size in iNOS−/− chimeric mice was reduced by an average of 60, 57 and 49% versus iNOS+/+ naïve, iNOS−/− naïve, and iNOS+/+ chimeric mice, respectively, indicating significant protection conferred by iNOS deletion from peripheral blood cells (Fig. 1; Table 2). In iNOS+/+ chimeric mice, infarct size was also significantly smaller than in the iNOS+/+ naïve group (44.3 ± 3.2% of the risk region, n = 10, versus 56.9 ± 2.8%, n = 17; P <0.05; Fig. 1; Table 2), demonstrating that whole-body irradiation and/or bone marrow cell transplantation, in themselves, slightly decreased the extent of cell death in the chimeric mouse heart.
Fig. 1.
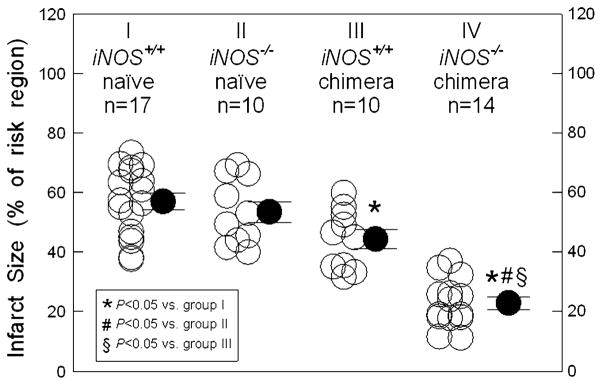
iNOS deletion from peripheral blood cells limits infarct size after I/R in vivo. WT naïve mice (iNOS+/+ naïve; group I), iNOS KO naïve mice (iNOS−/− naïve; group II), iNOS+/+ chimeras (group III), and iNOS−/− chimeras (group IV) were subjected to a 30-min coronary occlusion followed by 24 h of reperfusion. Infarct size was measured by TTC staining and expressed as a percent of the risk region. * P <0.05 versus group I, #P <0.05 versus group II, and §P <0.05 versus group III
Representative examples of the infarctions observed in different groups are shown in Fig. 2. Large, confluent areas of infarction spanning most of the thickness of the left ventricular wall, with thin rims of viable subendocardial tissue, were characteristic of the iNOS+/+ naïve and iNOS−/− naïve mice. In contrast, in the iNOS+/+ chimeric and iNOS−/− chimeric groups, only small, sporadic areas of cell death were noted (Fig. 2; Table 2).
Fig. 2.

Representative heart slices from WT naïve mice (iNOS+/+ naïve; group I, Panel A), iNOS KO naïve mice (iNOS−/− naïve; group II, Panel B), iNOS+/+ chimeric mice (group III, Panel C), and iNOS−/− chimeric mice (group IV, Panel D) after a 30-min coronary occlusion followed by 24 h of reperfusion. The slices shown here were obtained from the apex to the base. The region at risk and the infarct were identified by postmortem perfusion with TTC and phthalo blue dye, as described in “Materials and methods”. The non-ischemic portion of the left ventricular was stained dark blue, and viable tissue within the region at risk was stained bright red, whereas infarcted tissue was light yellow/white. The left ventricular endocardial surface was stained dark blue with phthalo blue to facilitate identification of the endocardial border of the slice. The hearts in groups I and II exhibited large, confluent areas of infarction. In contrast, the hearts in groups III and IV exhibited small patchy areas of infarction, indicating a profound cardioprotective effect
Discussion
The major finding of this study is that selective deletion of iNOS from peripheral blood cells (with no change in myocardial iNOS content) reduces infarct size, thus supporting the concept that iNOS derived from peripheral blood cells is detrimental during myocardial I/R. This is the first study to specifically examine the relative role of iNOS in different cell populations (i.e., myocardial vs. peripheral blood cells) during MI. Together with our previous work showing the cardioprotective actions of NO donors, iNOS gene therapy, and cardiac-specific overexpression of iNOS, these data support a complex, dual role of iNOS in MI (i.e., protective in myocytes but deleterious in blood cells). Many previous studies have addressed the role of iNOS in MI [10, 12, 20, 26, 32, 35, 38, 47]. However, to our knowledge, this is the first study to identify a role of iNOS in peripheral blood cells as a mediator of myocardial I/R injury. The results reveal heretofore unknown differential actions of iNOS and offer new insights into the mechanism of NO-mediated protection; in addition, they have significant clinical implications for developing long-term prophylactic anti-ischemic interventions.
Previous studies indicate that generation of NO by iNOS appears to be a final common pathway that mediates the late phase of ischemic PC, pharmacological PC, and PC induced by physical stimuli. NO has been shown to play an obligatory role in the delayed cardioprotection elicited by NO donors, adenosine A1 receptor agonists, opioid δ1 receptor agonists, and endotoxin derivatives [5, 6, 16, 17, 51]. The role of iNOS was further supported by studies using a recombinant adeno-associated viral vector encoding the human iNOS gene (rAAV/iNOS) [32–34, 50], which showed that iNOS gene therapy confers a powerful cardioprotective phenotype by limiting infarct size at 3 days after gene therapy [34], an effect that was sustained in the long-term (2 months and 1 year after gene therapy) without inflammation or adverse functional consequences [32, 33]. Additional studies demonstrated that cyclooxygenase-2 and heme oxygenase-1 are obligatory downstream effectors of iNOS-dependent cardioprotection and that iNOS imparts its protective effects, at least in part, by recruiting nuclear factor-κB, leading to cyclooxygenase-2 and heme oxygenase-1 upregulation [30, 31]. These observations were corroborated by the finding that cardiac myocyte-specific transgenic overexpression of iNOS [20] protects the myocardium against I/R injury by preventing mitochondrial permeability transition [22, 55]. Collectively, these data support a key role of myocyte iNOS-derived NO in mitigating ischemic injury.
The reason for the divergent effect of myocyte iNOS versus peripheral blood iNOS remains unclear. It is conceivable that after an inflammatory stimulus, such as I/R injury, the bone marrow-derived inflammatory cell population [comprising both mononuclear (e.g., lymphocytes and macrophages) and polymorphonuclear (e.g., neutrophils and eosinophils) cells] expresses high levels of iNOS and produces large amounts of NO for a sustained period of time [4, 37]. This excessive amount of NO may exert pro-oxidant effects by various mechanisms, such as activation of soluble guanylate cyclase, nitrosylation of protein thiols, and formation of nitrosyl complexes with metal ions [1, 3, 24, 40, 45, 46]. Importantly, since inflammatory cells are known to produce large quantities of superoxide anion, the interaction of NO with superoxide will lead to the formation of the highly toxic peroxynitrite anion, which causes tissue injury [3, 25, 43]. In contrast, since myocytes probably express lower levels of iNOS and produce smaller amounts of superoxide, the amount of NO produced by these cells is likely to be smaller and to be protective rather than toxic. It has been hypothesized that NO, at lower concentrations, protects myocytes from myocardial ischemia by several mechanisms: (1) decrease in apoptosis by inactivation of caspases [54], (2) reduction in generation of reactive oxygen species (ROS) by modulation of the mitochondrial electron transfer [36], and (3) activation of mitochondrial ATP-sensitive potassium channels [48], which triggers early ischemic PC by generating free radicals [14, 15, 41].
On the basis of these present data and of our previous data showing that iNOS upregulation in myocytes is protective [16, 34, 55], we propose that during MI, iNOS plays a diametrically opposite role in myocytes and in peripheral blood cells: protective in the former and detrimental in the latter. In normal naïve myocardium, iNOS is almost completely absent in myocytes [16]; in this setting, iNOS deletion has no effect on infarct size, as shown by the present data and previous studies [16]. In preconditioned myocardium (or in the setting of iNOS transgenesis or gene transfer), iNOS is upregulated in myocytes but not in inflammatory cells, conferring protection [34, 55]; hence, iNOS deletion or inhibition is detrimental. In both settings, however, inflammatory cell iNOS is detrimental, which explains why in the present study iNOS deletion in peripheral blood cells was beneficial.
Limitations of the current study include the lack of direct evidence that the deletion of iNOS in peripheral blood cells leads to a reduction in total NO bioavailability. As the storage pool (filled by NOS-dependent and NOS-independent sources) is critically involved in NO availability during I/R, it remains to be determined whether the bio-available NO storage pool in blood or tissues is reduced during I/R [21, 39]. Since our results demonstrate that iNOS chimeras are viable without any overt phenotypic abnormality, and that iNOS deletion in the peripheral blood cells induces profound cardioprotection by decreasing infarct size, a logical next step will be to determine the number of blood cells that are effectively iNOS−/− after irradiation/transplantation and to test the impact of iNOS activity in the peripheral blood cells on the total bioavailability of NO during I/R. Based on our previous experience with donor chimerism [28, 29], over 90% of the blood cells are effectively donor cells (in recipient mice) after irradiation. In the current study, as both our donor and recipient mice were created on the same C57BL/6 background, we were not able to evaluate the level of donor chimerism using flow cytometry techniques.
In conclusion, our study shows, for the first time, that selective deletion of iNOS in peripheral blood cells dramatically mitigates the extent of myocardial I/R injury. Together with our previous findings that myocyte iNOS is protective [16, 32–34, 55], these data reveal a novel paradigm regarding the role of iNOS; specifically they support the concept that the function of this enzyme is more complex than heretofore thought, and that during MI, iNOS can be beneficial or detrimental depending on the cell type in which it is expressed. This new paradigm may reconcile the apparent discrepancy among previous studies, some of which have concluded that iNOS is beneficial [10, 11, 20, 32, 38, 56], whereas others have found it to be detrimental [12, 13, 19, 26, 35, 47]. These differences may depend on the level of inflammatory cell activation and infiltration into the infarct, which in turn may be determined by model specific factors. The new paradigm that we propose challenges the conventional wisdom that iNOS is deleterious during I/R and instead supports the concept that iNOS, when expressed in cardiac myocytes, is a profoundly protective protein. Further investigation of the concept of cell-source specific effects of iNOS in MI is needed to better characterize the effects of iNOS from individual cells.
Acknowledgments
This study was supported in part by NIH grants R01 HL55757, HL-70897, HL-76794, and P01HL78825.
Footnotes
Conflict of interest The authors declare that they have no conflicts of interest.
Contributor Information
Yiru Guo, Institute of Molecular Cardiology, University of Louisville, 550 S Jackson Street ACB Bldg, 3rd Floor, Louisville, KY 40202, USA.
Santosh K. Sanganalmath, Institute of Molecular Cardiology, University of Louisville, 550 S Jackson Street ACB Bldg, 3rd Floor, Louisville, KY 40202, USA
Wenjian Wu, Institute of Molecular Cardiology, University of Louisville, 550 S Jackson Street ACB Bldg, 3rd Floor, Louisville, KY 40202, USA.
Xiaoping Zhu, Institute of Molecular Cardiology, University of Louisville, 550 S Jackson Street ACB Bldg, 3rd Floor, Louisville, KY 40202, USA.
Yiming Huang, Institute for Cellular Therapeutics, University of Louisville, Louisville, KY, USA.
Wei Tan, Institute of Molecular Cardiology, University of Louisville, 550 S Jackson Street ACB Bldg, 3rd Floor, Louisville, KY 40202, USA.
Suzanne T. Ildstad, Institute for Cellular Therapeutics, University of Louisville, Louisville, KY, USA
Qianhong Li, Institute of Molecular Cardiology, University of Louisville, 550 S Jackson Street ACB Bldg, 3rd Floor, Louisville, KY 40202, USA.
Roberto Bolli, Email: rbolli@louisville.edu, Institute of Molecular Cardiology, University of Louisville, 550 S Jackson Street ACB Bldg, 3rd Floor, Louisville, KY 40202, USA.
References
- 1.Abu-Soud HM, Hazen SL. Nitric oxide modulates the catalytic activity of myeloperoxidase. J Biol Chem. 2000;275:5425–5430. doi: 10.1074/jbc.275.8.5425. [DOI] [PubMed] [Google Scholar]
- 2.Baker CS, Dutka DP, Pagano D, Rimoldi O, Pitt M, Hall RJ, Polak JM, Bonser RS, Camici PG. Immunocytochemical evidence for inducible nitric oxide synthase and cyclooxygenase-2 expression with nitrotyrosine formation in human hibernating myocardium. Basic Res Cardiol. 2002;97:409–415. doi: 10.1007/s0039502 00050. [DOI] [PubMed] [Google Scholar]
- 3.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and super-oxide. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2:907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 5.Bolli R. Cardioprotective function of inducible nitric oxide synthase and role of nitric oxide in myocardial ischemia and preconditioning: an overview of a decade of research. J Mol Cell Cardiol. 2001;33:1897–1918. doi: 10.1006/jmcc.2001.1462. [DOI] [PubMed] [Google Scholar]
- 6.Bolli R. The late phase of preconditioning. Circ Res. 2000;87:972–983. doi: 10.1161/01.res.87.11.972. [DOI] [PubMed] [Google Scholar]
- 7.Bolli R, Becker L, Gross G, Mentzer R, Jr, Balshaw D, Lathrop DA. Myocardial protection at a crossroads: the need for translation into clinical therapy. Circ Res. 2004;95:125–134. doi: 10.1161/01.RES.0000137171.97172.d7. [DOI] [PubMed] [Google Scholar]
- 8.Bolli R, Bhatti ZA, Tang XL, Qiu Y, Zhang Q, Guo Y, Jadoon AK. Evidence that late preconditioning against myocardial stunning in conscious rabbits is triggered by the generation of nitric oxide. Circ Res. 1997;81:42–52. doi: 10.1161/01.res.81.1.42. [DOI] [PubMed] [Google Scholar]
- 9.Bolli R, Dawn B, Tang XL, Qiu Y, Ping P, Xuan YT, Jones WK, Takano H, Guo Y, Zhang J. The nitric oxide hypothesis of late preconditioning. Basic Res Cardiol. 1998;93:325–338. doi: 10.1007/s003950050101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bolli R, Manchikalapudi S, Tang XL, Takano H, Qiu Y, Guo Y, Zhang Q, Jadoon AK. The protective effect of late preconditioning against myocardial stunning in conscious rabbits is mediated by nitric oxide synthase. Evidence that nitric oxide acts both as a trigger and as a mediator of the late phase of ischemic preconditioning. Circ Res. 1997;81:1094–1107. doi: 10.1161/01.res.81.6.1094. [DOI] [PubMed] [Google Scholar]
- 11.Das A, Xi L, Kukreja RC. Phosphodiesterase-5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis. Essential role of nitric oxide signaling. J Biol Chem. 2005;280:12944–12955. doi: 10.1074/jbc.M404706200. [DOI] [PubMed] [Google Scholar]
- 12.Feng Q, Lu X, Jones DL, Shen J, Arnold JM. Increased inducible nitric oxide synthase expression contributes to myocardial dysfunction and higher mortality after myocardial infarction in mice. Circulation. 2001;104:700–704. doi: 10.1161/hc3201.092284. [DOI] [PubMed] [Google Scholar]
- 13.Gealekman O, Abassi Z, Rubinstein I, Winaver J, Binah O. Role of myocardial inducible nitric oxide synthase in contractile dysfunction and beta-adrenergic hyporesponsiveness in rats with experimental volume-overload heart failure. Circulation. 2002;105:236–243. doi: 10.1161/hc0202.102015. [DOI] [PubMed] [Google Scholar]
- 14.Gross GJ, Fryer RM. Mitochondrial K(ATP) channels: triggers or distal effectors of ischemic or pharmacological preconditioning? Circ Res. 2000;87:431–433. doi: 10.1161/01.res.87.6.431. [DOI] [PubMed] [Google Scholar]
- 15.Grover GJ, Garlid KD. ATP-Sensitive potassium channels: a review of their cardioprotective pharmacology. J Mol Cell Cardiol. 2000;32:677–695. doi: 10.1006/jmcc.2000.1111. [DOI] [PubMed] [Google Scholar]
- 16.Guo Y, Jones WK, Xuan YT, Tang XL, Bao W, Wu WJ, Han H, Laubach VE, Ping P, Yang Z, Qiu Y, Bolli R. The late phase of ischemic preconditioning is abrogated by targeted disruption of the inducible NO synthase gene. Proc Natl Acad Sci USA. 1999;96:11507–11512. doi: 10.1073/pnas.96.20.11507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo Y, Stein AB, Wu WJ, Zhu X, Tan W, Li Q, Bolli R. Late preconditioning induced by NO donors, adenosine A1 receptor agonists, and delta1-opioid receptor agonists is mediated by iNOS. Am J Physiol Heart Circ Physiol. 2005;289:H2251–H2257. doi: 10.1152/ajpheart.00341.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo Y, Wu WJ, Qiu Y, Tang XL, Yang Z, Bolli R. Demonstration of an early and a late phase of ischemic preconditioning in mice. Am J Physiol. 1998;275:H1375–H1387. doi: 10.1152/ajpheart.1998.275.4.H1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hataishi R, Rodrigues AC, Morgan JG, Ichinose F, Derumeaux G, Bloch KD, Picard MH, Scherrer-Crosbie M. Nitric oxide synthase 2 and pressure-overload-induced left ventricular remodelling in mice. Exp Physiol. 2006;91:633–639. doi: 10.1113/ expphysiol.2005.033068. [DOI] [PubMed] [Google Scholar]
- 20.Heger J, Godecke A, Flogel U, Merx MW, Molojavyi A, Kuhn-Velten WN, Schrader J. Cardiac-specific overexpression of inducible nitric oxide synthase does not result in severe cardiac dysfunction. Circ Res. 2002;90:93–99. doi: 10.1161/hh0102.102757. [DOI] [PubMed] [Google Scholar]
- 21.Hendgen-Cotta UB, Merx MW, Shiva S, Schmitz J, Becher S, Klare JP, Steinhoff HJ, Goedecke A, Schrader J, Gladwin MT, Kelm M, Rassaf T. Nitrite reductase activity of myoglobin regulates respiration and cellular viability in myocardial ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2008;105:10256–10261. doi: 10.1073/pnas.0801336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heusch G, Boengler K, Schulz R. Cardioprotection: nitric oxide, protein kinases, and mitochondria. Circulation. 2008;118:1915–1919. doi: 10.1161/CIRCULATIONAHA.108.805242. [DOI] [PubMed] [Google Scholar]
- 23.Heusch G, Post H, Michel MC, Kelm M, Schulz R. Endogenous nitric oxide and myocardial adaptation to ischemia. Circ Res. 2000;87:146–152. doi: 10.1161/01.res.87.2.146. [DOI] [PubMed] [Google Scholar]
- 24.Ignarro LJ. Signal transduction mechanisms involving nitric oxide. Biochem Pharmacol. 1991;41:485–490. doi: 10.1016/0006-2952(91)90618-f. [DOI] [PubMed] [Google Scholar]
- 25.Ischiropoulos H, Zhu L, Beckman JS. Peroxynitrite formation from macrophage-derived nitric oxide. Arch Biochem Biophys. 1992;298:446–451. doi: 10.1016/0003-9861(92)90433-w. [DOI] [PubMed] [Google Scholar]
- 26.Jones SP, Greer JJ, Ware PD, Yang J, Walsh K, Lefer DJ. Deficiency of iNOS does not attenuate severe congestive heart failure in mice. Am J Physiol Heart Circ Physiol. 2005;288:H365–H370. doi: 10.1152/ajpheart.00245.2004. [DOI] [PubMed] [Google Scholar]
- 27.Kolios G, Valatas V, Ward SG. Nitric oxide in inflammatory bowel disease: a universal messenger in an unsolved puzzle. Immunology. 2004;113:427–437. doi: 10.1111/j.1365-2567.2004. 01984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Colson YL, Ildstad ST. Mixed allogeneic chimerism achieved by lethal and nonlethal conditioning approaches induces donor-specific tolerance to simultaneous islet allografts. Transplantation. 1995;60:523–529. doi: 10.1097/00007890-199509270-00001. [DOI] [PubMed] [Google Scholar]
- 29.Li H, Kaufman CL, Ildstad ST. Allogeneic chimerism induces donor-specific tolerance to simultaneous islet allografts in nonobese diabetic mice. Surgery. 1995;118:192–197. doi: 10.1016/s0039-6060(05)80323-x. (discussion 197–198) [DOI] [PubMed] [Google Scholar]
- 30.Li Q, Guo Y, Ou Q, Cui C, Wu WJ, Tan W, Zhu X, Lanceta LB, Sanganalmath SK, Dawn B, Shinmura K, Rokosh GD, Wang S, Bolli R. Gene transfer of inducible nitric oxide synthase affords cardioprotection by upregulating heme oxygenase-1 via a nuclear factor-{kappa}B-dependent pathway. Circulation. 2009;120:1222–1230. doi: 10.1161/CIRCULATIONAHA.108.778688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Q, Guo Y, Tan W, Ou Q, Wu WJ, Sturza D, Dawn B, Hunt G, Cui C, Bolli R. Cardioprotection afforded by inducible nitric oxide synthase gene therapy is mediated by cyclooxygenase-2 via a nuclear factor-kappaB dependent pathway. Circulation. 2007;116:1577–1584. doi: 10.1161/CIRCULATIONAHA.107.689810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Q, Guo Y, Tan W, Stein AB, Dawn B, Wu WJ, Zhu X, Lu X, Xu X, Siddiqui T, Tiwari S, Bolli R. Gene therapy with iNOS provides long-term protection against myocardial infarction without adverse functional consequences. Am J Physiol Heart Circ Physiol. 2006;290:H584–H589. doi: 10.1152/ajpheart.00855.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Q, Guo Y, Wu WJ, Ou Q, Zhu X, Tan W, Yuan F, Chen N, Dawn B, Luo L, O’Brien E, Bolli R. Gene transfer as a strategy to achieve permanent cardioprotection I: rAAV-mediated gene therapy with inducible nitric oxide synthase limits infarct size 1 year later without adverse functional consequences. Basic Res Cardiol. 2011;106:1355–1366. doi: 10.1007/s00395-011-0207-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Q, Guo Y, Xuan YT, Lowenstein CJ, Stevenson SC, Prabhu SD, Wu WJ, Zhu Y, Bolli R. Gene therapy with inducible nitric oxide synthase protects against myocardial infarction via a cyclooxygenase-2-dependent mechanism. Circ Res. 2003;92:741–748. doi: 10.1161/01.RES.0000065441.72685.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu YH, Carretero OA, Cingolani OH, Liao TD, Sun Y, Xu J, Li LY, Pagano PJ, Yang JJ, Yang XP. Role of inducible nitric oxide synthase in cardiac function and remodeling in mice with heart failure due to myocardial infarction. Am J Physiol Heart Circ Physiol. 2005;289:H2616–H2623. doi: 10.1152/ajpheart.00546.2005. [DOI] [PubMed] [Google Scholar]
- 36.Loke KE, McConnell PI, Tuzman JM, Shesely EG, Smith CJ, Stackpole CJ, Thompson CI, Kaley G, Wolin MS, Hintze TH. Endogenous endothelial nitric oxide synthase-derived nitric oxide is a physiological regulator of myocardial oxygen consumption. Circ Res. 1999;84:840–845. doi: 10.1161/01.res.84.7.840. [DOI] [PubMed] [Google Scholar]
- 37.MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–350. doi: 10.1146/ annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 38.Marfella R, Di Filippo C, Esposito K, Nappo F, Piegari E, Cuzzocrea S, Berrino L, Rossi F, Giugliano D, D’Amico M. Absence of inducible nitric oxide synthase reduces myocardial damage during ischemia reperfusion in streptozotocin-induced hyperglycemic mice. Diabetes. 2004;53:454–462. doi: 10.2337/diabetes.53.2.454. [DOI] [PubMed] [Google Scholar]
- 39.Martin C, Schulz R, Post H, Boengler K, Kelm M, Kleinbongard P, Gres P, Skyschally A, Konietzka I, Heusch G. Microdialysis-based analysis of interstitial NO in situ: NO synthase-independent NO formation during myocardial ischemia. Cardiovasc Res. 2007;74:46–55. doi: 10.1016/j.cardiores.2006.12.020. [DOI] [PubMed] [Google Scholar]
- 40.Mohr S, Hallak H, de Boitte A, Lapetina EG, Brune B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem. 1999;274:9427–9430. doi: 10.1074/jbc.274.14.9427. [DOI] [PubMed] [Google Scholar]
- 41.Pain T, Yang XM, Critz SD, Yue Y, Nakano A, Liu GS, Heusch G, Cohen MV, Downey JM. Opening of mitochondrial K(ATP) channels triggers the preconditioned state by generating free radicals. Circ Res. 2000;87:460–466. doi: 10.1161/01.res.87.6.460. [DOI] [PubMed] [Google Scholar]
- 42.Post H, Schulz R, Behrends M, Gres P, Umschlag C, Heusch G. No involvement of endogenous nitric oxide in classical ischemic preconditioning in swine. J Mol Cell Cardiol. 2000;32:725–733. doi: 10.1006/jmcc.2000.1117. [DOI] [PubMed] [Google Scholar]
- 43.Pryor WA, Squadrito GL. The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. Am J Physiol. 1995;268:L699–L722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- 44.Qiu Y, Rizvi A, Tang XL, Manchikalapudi S, Takano H, Jadoon AK, Wu WJ, Bolli R. Nitric oxide triggers late preconditioning against myocardial infarction in conscious rabbits. Am J Physiol. 1997;273:H2931–H2936. doi: 10.1152/ajpheart.1997.273.6.H2931. [DOI] [PubMed] [Google Scholar]
- 45.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of super-oxide and nitric oxide. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 46.Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, Kirk M, Freeman BA. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidized lipid derivatives. J Biol Chem. 1994;269:26066–26075. [PubMed] [Google Scholar]
- 47.Sam F, Sawyer DB, Xie Z, Chang DL, Ngoy S, Brenner DA, Siwik DA, Singh K, Apstein CS, Colucci WS. Mice lacking inducible nitric oxide synthase have improved left ventricular contractile function and reduced apoptotic cell death late after myocardial infarction. Circ Res. 2001;89:351–356. doi: 10.1161/hh1601.094993. [DOI] [PubMed] [Google Scholar]
- 48.Sasaki N, Sato T, Ohler A, O’Rourke B, Marban E. Activation of mitochondrial ATP-dependent potassium channels by nitric oxide. Circulation. 2000;101:439–445. doi: 10.1161/01.cir.101.4.439. [DOI] [PubMed] [Google Scholar]
- 49.Shinmura K, Xuan YT, Tang XL, Kodani E, Han H, Zhu Y, Bolli R. Inducible nitric oxide synthase modulates cyclooxygenase-2 activity in the heart of conscious rabbits during the late phase of ischemic preconditioning. Circ Res. 2002;90:602–608. doi: 10.1161/01.res.0000012202.52809.40. [DOI] [PubMed] [Google Scholar]
- 50.Szelid Z, Pokreisz P, Liu X, Vermeersch P, Marsboom G, Gillijns H, Pellens M, Verbeken E, Van de Werf F, Collen D, Janssens SP. Cardioselective nitric oxide synthase 3 gene transfer protects against myocardial reperfusion injury. Basic Res Cardiol. 2010;105:169–179. doi: 10.1007/s00395-009-0077-4. [DOI] [PubMed] [Google Scholar]
- 51.Takano H, Bolli R, Black RG, Jr, Kodani E, Tang XL, Yang Z, Bhattacharya S, Auchampach JA. A(1) or A(3) adenosine receptors induce late preconditioning against infarction in conscious rabbits by different mechanisms. Circ Res. 2001;88:520–528. doi: 10.1161/01.res.88.5.520. [DOI] [PubMed] [Google Scholar]
- 52.Takano H, Manchikalapudi S, Tang XL, Qiu Y, Rizvi A, Jadoon AK, Zhang Q, Bolli R. Nitric oxide synthase is the mediator of late preconditioning against myocardial infarction in conscious rabbits. Circulation. 1998;98:441–449. doi: 10.1161/01.cir.98.5.441. [DOI] [PubMed] [Google Scholar]
- 53.Takano H, Tang XL, Qiu Y, Guo Y, French BA, Bolli R. Nitric oxide donors induce late preconditioning against myocardial stunning and infarction in conscious rabbits via an antioxidant-sensitive mechanism. Circ Res. 1998;83:73–84. doi: 10.1161/01.res.83.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vegh A, Papp JG, Szekeres L, Parratt JR. Prevention by an inhibitor of the L-arginine-nitric oxide pathway of the antiarrhythmic effects of bradykinin in anaesthetized dogs. Br J Pharmacol. 1993;110:18–19. doi: 10.1111/j.1476-5381.1993.tb13764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.West MB, Rokosh G, Obal D, Velayutham M, Xuan YT, Hill BG, Keith RJ, Schrader J, Guo Y, Conklin DJ, Prabhu SD, Zweier JL, Bolli R, Bhatnagar A. Cardiac myocyte-specific expression of inducible nitric oxide synthase protects against ischemia/reperfusion injury by preventing mitochondrial permeability transition. Circulation. 2008;118:1970–1978. doi: 10.1161/CIRCULATIONAHA. 108.791533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ziolo MT, Maier LS, Piacentino V, 3rd, Bossuyt J, Houser SR, Bers DM. Myocyte nitric oxide synthase 2 contributes to blunted beta-adrenergic response in failing human hearts by decreasing Ca2+ transients. Circulation. 2004;109:1886–1891. doi: 10.1161/01.CIR.0000124231.98250.A8. [DOI] [PubMed] [Google Scholar]