

New software has been developed for automating the experimental and data-processing stages of fragment-based drug discovery at a macromolecular crystallography beamline. A new workflow-automation framework orchestrates beamline-control and data-analysis software while organizing results from multiple samples.
Keywords: AutoDrug, fragment-based drug discovery, workflow automation
Abstract
AutoDrug is software based upon the scientific workflow paradigm that integrates the Stanford Synchrotron Radiation Lightsource macromolecular crystallography beamlines and third-party processing software to automate the crystallography steps of the fragment-based drug-discovery process. AutoDrug screens a cassette of fragment-soaked crystals, selects crystals for data collection based on screening results and user-specified criteria and determines optimal data-collection strategies. It then collects and processes diffraction data, performs molecular replacement using provided models and detects electron density that is likely to arise from bound fragments. All processes are fully automated, i.e. are performed without user interaction or supervision. Samples can be screened in groups corresponding to particular proteins, crystal forms and/or soaking conditions. A single AutoDrug run is only limited by the capacity of the sample-storage dewar at the beamline: currently 288 samples. AutoDrug was developed in conjunction with RestFlow, a new scientific workflow-automation framework. RestFlow simplifies the design of AutoDrug by managing the flow of data and the organization of results and by orchestrating the execution of computational pipeline steps. It also simplifies the execution and interaction of third-party programs and the beamline-control system. Modeling AutoDrug as a scientific workflow enables multiple variants that meet the requirements of different user groups to be developed and supported. A workflow tailored to mimic the crystallography stages comprising the drug-discovery pipeline of CoCrystal Discovery Inc. has been deployed and successfully demonstrated. This workflow was run once on the same 96 samples that the group had examined manually and the workflow cycled successfully through all of the samples, collected data from the same samples that were selected manually and located the same peaks of unmodeled density in the resulting difference Fourier maps.
1. Introduction
A key step in drug design is the discovery of drug leads, which are low-molecular-weight compounds with some affinity for a protein drug target from which higher affinity compounds can be derived. Fragment-based design is an approach that employs macromolecular crystallography to detect compounds that bind to target proteins, often with affinities lower than those detected by conventional assays. Potential drug fragments are soaked into crystals of the target protein, usually in cocktail mixtures of five to ten compounds (Rees et al., 2004 ▶; Sharff & Jhoti, 2003 ▶; Nienaber et al., 2000 ▶). With the development of high-throughput crystallographic methods, it is feasible to screen large numbers of cocktail mixtures and to identify bound compounds that may lead to new drug leads in a systematic and efficient manner. Although the crystallographic steps in the process of identifying these potential drug leads are straightforward and are traditionally performed manually at a synchrotron-radiation beamline, the procedures are time-consuming and labor-intensive, even when robotic sample-handling systems and predefined data-processing scripts are employed. The need to organize and evaluate the large number of diffraction images, reduced data sets and structure-solution results for drug screening further complicates the effort. Consequently, the number of fragment screens attempted may be limited by the overall workload. End-to-end automation of these workflows thus promises to yield significant value to researchers and organizations developing new drugs.
Complete automation of fragment-based drug-design experiments faces a number of technical challenges that complicate any effort to achieve automation that spans the experimental and computational operations at a macromolecular crystallography beamline. Firstly, the sample-handling hardware system must be sufficiently robust and of high enough capacity to support hundreds of samples without failure or manual intervention. AutoDrug employs the Stanford Automated Mounting (SAM) system (Cohen et al., 2002 ▶), a robotic system for transferring frozen samples between cryogenically cooled cassettes and the goniometer. SAM systems are deployed at all Stanford Synchrotron Radiation Lightsource (SSRL) macromolecular crystallography beamlines and have mounted over a half million crystal samples to date.
A second challenge is posed by diverse programming interfaces. The interfaces to beamline-control systems and data-collection software packages, which are typically network-based, differ significantly from the interfaces to data-processing and structure-determination software packages, which are frequently command-line based. These differences in software architecture can complicate the implementation of a single application that controls both types of software. At SSRL, the Blu-Ice/DCS beamline-control software is the interface between the user and the instrumentation for sample screening and data collection (McPhillips et al., 2002 ▶). We have previously automated numerous sample-oriented processes within this system, including automatic loop and sample centering, fluorescence-energy scans and multi-wavelength diffraction data collection. Web-Ice and Crystal Analysis Server systems assist users performing auto-indexing and strategy analysis (González et al., 2008 ▶), but do not provide automatic feedback to ongoing experimental operations at the beamline. Similarly, the Xsolve system developed by the Joint Center for Structural Genomics takes as input complete diffraction data sets, yields MAD-phased electron-density maps and automatically builds molecular models, but does not interact with the beamline instrumentation (van den Bedem et al., 2011 ▶). To facilitate the integration of the experimental and analysis stages of crystallographic experiments, we have developed a scientific workflow-automation framework, RestFlow (http://www.restflow.org/). A scientific workflow defines a series of computational tasks necessary to carry out a scientific process, such as data collection or data analysis (Taylor et al., 2007 ▶). Brockhauser et al. (2012 ▶) recently applied the scientific workflow concept to automate several individual macromolecular crystallography tasks (namely, sample characterization, strategy determination and sample grid scanning). Because RestFlow supports workflow steps that employ diverse programming languages, which in turn support a broad range of programming interfaces, AutoDrug workflows can readily include all beamline-control and data-analysis tasks typically associated with macromolecular crystallography experiments.
A third challenge of end-to-end automation at a crystallography beamline is the large number of data and result files that must be stored, organized and passed between steps in the workflow. AutoDrug addresses this problem by employing features in RestFlow for organizing the data produced by a workflow in a directory structure similar to those that scientists use when performing experiments and processing data manually. RestFlow also facilitates the generation of flexible reports from ongoing and completed experiments for debugging, auditing and summarizing experimental results.
AutoDrug thus expands the capabilities of the existing automation technology at SSRL (Soltis et al., 2008 ▶) to meet the demanding requirements of high-throughput drug discovery. AutoDrug automatically carries out the tedious and sometimes difficult process of screening many hundreds to perhaps thousands of samples and analyzing numerous electron-density maps. Unlike other automated systems such as Xsolve (van den Bedem et al., 2011 ▶), the SPACE and PERON systems (Sugahara et al., 2008 ▶) and the library-screening system reported by Mooij et al. (2006 ▶), AutoDrug runs unassisted and unsupervised at the beamline.
2. AutoDrug workflows
Fragment-based drug discovery generally begins with soaking crystals of the target protein in solutions that contain small chemical compounds representing potential drug fragments and freezing them in liquid nitrogen. The compounds that bind to the protein are detected by collecting X-ray diffraction data and applying molecular replacement to determine the molecular structure of the protein. Excess electron density in difference maps between the native protein and the protein soaked with chemicals often indicates the presence of a bound compound.
AutoDrug runs beamline-control software and third-party data-processing programs to screen, collect X-ray data, process data and to determine the structure and any excess electron density that may be present. AutoDrug workflows are initiated from the command line and require the following input files: (i) a standard SSRL Excel spreadsheet that contains information about the samples mounted in cassettes or uni-pucks installed at the beamline, (ii) an AutoDrug-specific input file describing the experimental parameters for screening, data collection, processing etc. and sample-acceptance criteria for determining which samples should be used for data collection and (iii) a Protein Data Bank file of the target protein structure for molecular replacement. All files created during a particular run are stored in a single directory tree and organized according to path templates defined in the workflow. AutoDrug provides customizable reports providing real-time and post-run feedback on outcomes and statistics. Post-run reports can also be used to facilitate comparisons across multiple AutoDrug runs.
Fig. 1 ▶ illustrates a simplified AutoDrug workflow for the steps of X-ray diffraction screening, data collection, data processing, structure solution and electron-density analysis.
Figure 1.
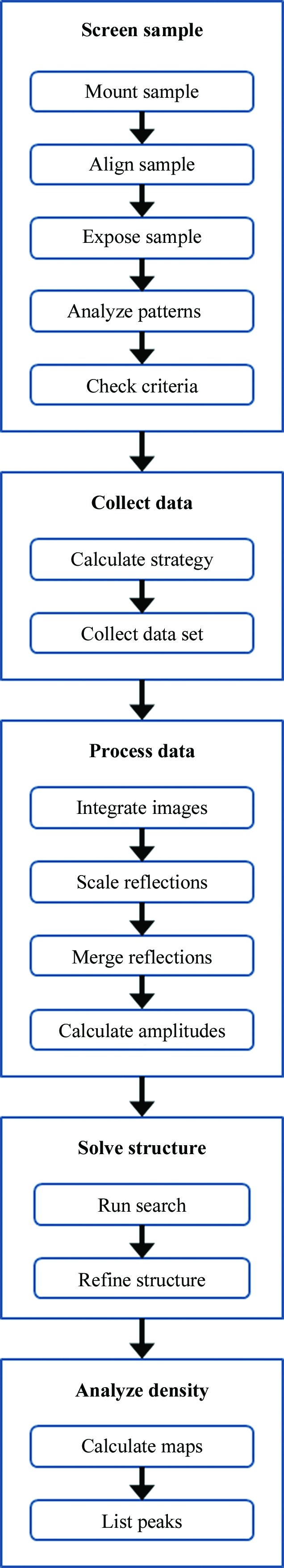
A simplified generic AutoDrug workflow. The entire workflow is completely automated at the SSRL beamlines and runs without user interaction or supervision.
2.1. Screen sample
AutoDrug uses procedures previously implemented in the SSRL Blu-Ice/DCS control system (McPhillips et al., 2002 ▶) and Web-Ice (González et al., 2008 ▶) to screen samples and to collect data. AutoDrug uses the SAM robot (Cohen et al., 2002 ▶) to individually mount cooled crystal samples onto the beamline diffractometer and then uses a loop-centering algorithm (Miller et al., 2004 ▶) to align the crystal sample with the X-ray beam. Next, AutoDrug collects two diffraction images 90° apart and analyzes them. AutoDrug runs LABELIT (Sauter et al., 2004 ▶) to determine the unit-cell, diffraction-limit and spot-mosaicity parameters. AutoDrug uses these parameters to check against user-specified thresholds and other criteria to decide which samples should be queued for data collection.
2.2. Collect data
For each sample that passes the specified criteria, AutoDrug collects a complete X-ray diffraction data set using data-collection parameters that are based on strategy programs such as ipmosflm (Leslie & Powell, 2007 ▶) or BEST (Bourenkov & Popov, 2006 ▶). AutoDrug will override these parameters if the user specifies them in the input file.
2.3. Process data
Once a complete data set has been collected, AutoDrug uses XDS (Kabsch, 2010 ▶) to integrate the Bragg reflections, POINTLESS (Winn et al., 2011 ▶) for analysis and sorting and SCALA (Evans, 2006 ▶) to scale and merge the reflections. AutoDrug uses TRUNCATE to calculate amplitudes and RFREE to select a free reflection set for refinement validation (Winn et al., 2011 ▶).
2.4. Solve structure
Current AutoDrug workflows require that a suitable structure model already exists and that structure solution by molecular replacement is straightforward. Although the existing model could be directly refined against new data in many cases, molecular replacement is used to cope with pronounced non-isomorphism or conflicting indexing schemes. AutoDrug carries out molecular replacement using MOLREP (Vagin & Teplyakov, 2010 ▶) and structure refinement using REFMAC (Murshudov et al., 2011 ▶).
2.5. Analyze density
Once the structure has been solved and refined, AutoDrug calculates an electron-density difference Fourier map and then identifies and tabulates areas of excess electron density using FFT (Eyck, 1973 ▶). Although current versions of AutoDrug do not attempt to model the excess density with the compounds that were used in the soak, this step could be implemented using programs such as X-LIGAND (Oldfield, 2001 ▶) or LigandFit (Terwilliger et al., 2007 ▶).
3. AutoDrug customization
While all versions of AutoDrug have the basic steps described above, the steps can be expanded, interleaved or carried out in a different order depending on the requirements of the users. In one version, for example, AutoDrug runs data collection and the subsequent data-processing steps concurrently when possible, thereby maximizing the efficiency of the workflow. Other variations include screening samples in groups rather than individually. In this case, crystals soaked in the same cocktail mixtures can be compared against each other to determine the best diffracting sample among the group for data collection. In another workflow, AutoDrug screens for space-group symmetry when the samples crystallize in more than one space group.
AutoDrug workflows have also been designed to improve scientific experiments. In a fragment-based drug-discovery study involving HIV protease (Perryman et al., 2010 ▶) as the target protein, diffraction-based rastering (Song et al., 2007 ▶) was implemented in the AutoDrug workflow to automatically align samples to the X-ray beam where the highest quality diffraction could be obtained. Replacing automated loop alignment with diffraction-based rastering increased the number of samples that met the data-collection criteria when the sample and loop size were mismatched. Once a workflow has been written, it is straightforward to test new sample criteria and modify routines that could potentially improve the efficiency and productivity of the workflow.
4. AutoDrug criteria
AutoDrug software developers work closely with experimenters to accurately model their decision-making process. In practice, user groups may use soft thresholds and/or sliding limits. Because AutoDrug requires the specification of fixed criteria, it yields very consistent results. Examples of criteria that are used in current AutoDrug workflows include a comparison of the unit-cell parameters and crystal quality parameters that are determined during the screening process. In one case, the unit-cell lengths are required to be within 1% of the standard values. In another case, the diffraction resolution and crystal mosaicity have to be within a given range. Versions of AutoDrug also compare an overall score as calculated in Web-Ice (González et al., 2008 ▶), which incorporates into a single value the diffraction resolution, mosaicity and the r.m.s.d. of the observed and predicted Bragg spots.
5. Demonstration of AutoDrug
A customized AutoDrug workflow was commissioned in collaboration with CoCrystal Discovery (CoCD) Inc. The workflow closely mimicked the steps that CoCD researchers had been carrying out on SSRL beamlines as part of their drug-discovery pipeline. The workflow was tested on 96 crystals containing samples that had previously been manually screened and analyzed for bound fragments by CoCD researchers. Samples with the same cocktail soaks were grouped together, with approximately three per group. The groups were identified in a column in the SSRL Excel spreadsheet. Since this workflow was designed for crystals with little variation in shape and size, the data-collection parameters such as the resolution at the edge of the detector, the oscillation per image and the exposure time were specified in the AutoDrug-specific input file for each sample rather than calculated. Once the cassette containing the samples had been loaded into the robot dewar and the spreadsheet had been uploaded, a script invoking AutoDrug was run from the command line with the location of the sample spreadsheet and user input file as arguments. Each sample in a group was mounted by the robot, centered in the beam using loop centering, exposed to X-rays 90° apart and returned to the cassette. The resulting diffraction images were analyzed and a WebIce-based score was calculated. When all of the crystals in a group had been screened, the scores were compared for those samples meeting the basic criteria specified in the input file (unit-cell parameters within 1%, mosaicity less than 1° and a maximum diffraction resolution better than 2 Å). The crystal with the highest score in a group was remounted, re-centered and re-indexed and a full data set was then collected. If no sample in the group met the minimum criteria no data collection was carried out and the next group of samples was examined. When data collection was finished for each sample, the data were processed and scaled and the structure was solved based on the PDB structure. After a few rounds of refinement, a weighted electron-density difference Fourier map was calculated and peaks in the map greater than 5σ were listed.
AutoDrug successfully screened all 96 samples and selected the same 16 crystals that the CoCD researchers had selected for data collection. Seven data sets yielded density above 5σ in the target regions of the structure, fully matching the results of the prior experiments carried out by CoCD researchers. However, AutoDrug managed the entire process in a single run without interruption or manual intervention.
6. Specifying workflows with RestFlow
AutoDrug is implemented using RestFlow, a new application-development framework for creating automated scientific workflows (http://www.restflow.org/). RestFlow emphasizes the flow of data between tasks, while leaving the implementation details of each task open to the developer. A functional RestFlow application is architecturally similar to the scientist’s understanding of the process under development (Fig. 2 ▶). This similarity has empowered scientists in our group to write workflows for end users using RestFlow and to easily verify the work of software developers.
Figure 2.

Diagram of an AutoDrug workflow for data processing. The workflow reflects the exact steps that a crystallographer takes when executing programs for data processing. The subworkflows RunXds, RunPointless, RunScala, RunTruncate and RunFreeR run the programs XDS, POINTLESS, SCALA, TRUNCATE and FREERFLAG for data processing. The subworkflow RunXtriage runs xtriage, a program that analyzes the data. The diagram is understood by both software programmers and scientists, and simplifies communication during workflow development. The diagram was generated by RestFlow based on the workflow specification and was rendered using GraphViz (http://www.graphviz.org).
This dataflow-oriented approach to modeling and executing scientific workflows in RestFlow is similar to the approaches taken by Kepler (Ludäscher et al., 2006 ▶), DAWB (Brockhauser et al., 2012 ▶), Taverna (Oinn et al., 2006 ▶), Triana (Taylor et al., 2007 ▶), VisTrails (Bavoil et al., 2005 ▶) and other general-purpose scientific workflow systems. RestFlow is fundamentally distinguished from these systems by its use of uniform resource identifier (URI) template expressions (described in detail below) to declare the paths of data flow, to describe how the data produced by a run of a workflow should be organized and retained, and to uniquely name resulting directories and output files using metadata.
The design of a RestFlow application involves splitting the scientific workflow into computational and experimental steps to be carried out by software components termed actors. These components are analogous to the actors of Kepler and DAWB and of the Ptolemy II system (Eker et al., 2003 ▶), on which both Kepler and DAWB are based. However, in RestFlow each actor is encapsulated by a generic component, called a workflow node, which manages the flow of data into and out of the actor. A node invokes the actor it manages when data required by the actor are received on the inflow(s) of the node; the node subsequently publishes the data produced by the actor on the node’s outflow(s). A workflow comprises one or more nodes with interconnected inflows and outflows (Fig. 3 ▶). Nodes can also encapsulate and invoke entire subworkflows. A multi-step workflow such as that required for fragment-based drug discovery will typically consist of many nodes.
Figure 3.

Conceptual view of a simple workflow implemented with RestFlow. A workflow comprises one or more nodes connected by data inflows and outflows. Each node encapsulates either an actor or another workflow. Each data flow is associated with a universal resource identifier (URI) template that specifies where data passing across the data flow are persisted (i.e. saved as a file).
In RestFlow, all outflows are named uniquely and provide locations for results to be stored. The unique name takes the form of a URI template (http://code.google.com/p/uri-templates/), which has the familiar appearance of a directory path with integrated variable names (Fig. 4 ▶ a). Variables in the URI template are expanded at runtime using the latest values of data passing through the inflows or outflows (Fig. 4 ▶ b), such that each data item produced on an outflow is also named uniquely. In this way, RestFlow dynamically creates directory paths and filenames based on metadata from the experiment, such as sample name, X-ray energy used, LABELIT output etc.
Figure 4.

The role of URI templates in the management of data in a simplified RestFlow program for crystallographic data collection. Rounded boxes represent nodes carrying out tasks at the beamline and the expressions in the square boxes are URI templates that specify routes of data flow. (a) Variables are specified in the URI templates (shown in bold curly braces: {variable}). (b) The variables are expanded at run time using current values of data and metadata passing through the node to yield unique identifiers and destinations on the file system.
RestFlow actors currently can be implemented in Python, Perl, Bash, Tcl, Groovy and Java. Support for multiple languages allows the developer to select the most appropriate language for a particular task. For example, procedures written in Tcl can control the beamline using the Blu-Ice event-driven API, while procedures written in Groovy can expand text templates to yield input files for third-party crystallographic applications. The node transfers inflow data to variables of the selected language before execution and exports output variable values back to the outflows at the end of the task (see Fig. 3 ▶). This allows data values and references to data files to easily flow from actor to actor even when the actors are implemented in different programming languages. For actors that require access to a file system, the node automatically creates unique scratch directories in which to run external programs. RestFlow copies necessary data files into these scratch directories and copies generated files from the scratch directory to the destinations indicated by the node’s outflows.
At runtime, RestFlow directs each node to execute its actor (or sub-workflow) repeatedly until inflow data are no longer available. This data-driven behavior is similar to that of many dataflow systems, but is distinct from the behavior of task-dependency-based workflow systems that execute each task node just once per workflow run1. The data-driven approach is compatible with multiple scheduling schemes, and RestFlow, like Kepler and Ptolemy II, delegates the scheduling of actor invocations to an explicit component, the director, associated with each workflow, so that the scheduling of actor invocations can be chosen separately for each workflow and subworkflow. RestFlow currently provides two data-driven directors supporting single-threaded execution of workflows in which only one actor is invoked at a time and one that implements a multi-threaded scheduling scheme that invokes actors concurrently where possible. The latter can yield the maximum parallelism compatible with the topology of the workflow and the availability of data. Each subworkflow is assigned its own director component independently of the choice of director for the containing workflow. Directors in RestFlow also are responsible for determining how data flowing between nodes is buffered; the data-driven directors currently provided by RestFlow implement unbounded first-in-first-out (FIFO) buffers on all node inflows so that slower downstream nodes do not block the further execution of faster upstream nodes. All of the data-driven directors in RestFlow support workflow cycles (e.g. for/while loops and feedback loops), conditional control flow constructs (e.g. if/then) and exception handling. The latter allows a workflow to shut down and/or change the flow of the data through the workflow following an unexpected error.
RestFlow applications are specified using plain-text Spring (Spring Framework; http://static.springsource.org/spring/docs/3.0.x/spring-framework-reference/html/) configuration files written in readable YAML (YAML Ain’t Markup Language. http://www.yaml.org/) format. At runtime, Spring loads the files and assembles an object-oriented Java application. As such, RestFlow applications may mix workflow technology, such as actors, nodes, inflows and outflows, with traditional object-oriented techniques and pre-existing libraries. This combination of programming paradigms provides a powerful and flexible framework for implementing complex automated scientific workflows.
RestFlow is one of the first scientific workflow-automation systems that implements a very simple actor interface that shields actor developers from the details of dataflow programming through a separate node entity. RestFlow nodes thus resemble the frames introduced in Bowers et al. (2006 ▶) and Ngu et al. (2008 ▶) to separate dataflow and control-flow concerns. In RestFlow, this division of labor between general-purpose workflow nodes responsible for managing the flow of data into and out of actors on the one hand and task-specific actors on the other enables a number of innovations that further simplify workflow development. The simplicity of the actor API not only makes it extremely easy to develop new actors as needed, but also makes it relatively easy to add to RestFlow support for additional actor programming languages. Scientists consequently can use the scripting languages that they are already familiar with. Because nodes can be configured to persist outflowing data to files automatically, workflows need not include explicit file-reading and file-writing actors, and the organization of data produced by a workflow is immediately apparent to (and easily changed by) a scientist reading the workflow specification. Finally, the use of URI template expressions in the routing, naming and storing of data enables RestFlow to effectively organize data associated with numerous subprojects, samples and experiments during and following workflow runs.
7. Summary
AutoDrug is an automated scientific workflow for fragment-based drug discovery. The workflow specified by CoCrystal Discovery, from sample screening and selection through electron-density map analysis, was successfully automated and carried out by AutoDrug. This demonstrates the potential of AutoDrug to perform experiments involving large numbers of crystals unassisted and unattended, thus expanding the search space and expediting the discovery of drug leads. We plan to automate other workflows at our beamlines using the underlying application framework RestFlow, including data-reduction tasks carried out by users and alignment procedures carried out by staff. Future workflows will automate experiments that require complex sample-screening and data-collection strategies, such as projects that require large numbers of crystals in order to collect a complete data set (e.g. radiation-sensitive microcrystals). RestFlow and several generic AutoDrug workflows (http://smb.slac.stanford.edu/autodrug) are licensed as free and open-source software and are available to the general community upon request.
Acknowledgments
The project described was supported by Grant Nos. P41 RR001209 and P01 GM083658-01 from the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the US Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program and the National Institute of General Medical Sciences.
Footnotes
The data-driven approach of RestFlow also is distinct from event-driven models of computation such as that implemented in Evans and Sutherland function networks (Yamaguchi et al., 1985 ▶). In the latter, data used by the functions in a network are never consumed but are only replaced by newly arriving data; each function re-computes its outputs each time data arrive on any one of its inputs. This contrasts with the behavior of RestFlow, in which all inputs to an actor are consumed each time an actor is invoked; new data must be received on all inflows before a node will invoke its actor again.
References
- Bavoil, L., Callahan, S., Crossno, P., Freire, J., Scheidegger, C., Silva, C. & Vo, H. (2005). Visualization, 2005. VIS 05. IEEE, pp. 135–142. doi:10.1109/VISUAL.2005.1532788.
- Bedem, H. van den, Wolf, G., Xu, Q. & Deacon, A. M. (2011). Acta Cryst. D67, 368–375. [DOI] [PMC free article] [PubMed]
- Bourenkov, G. P. & Popov, A. N. (2006). Acta Cryst. D62, 58–64. [DOI] [PubMed]
- Bowers, S., Ludaescher, B., Ngu, A. H. & Critchlow, T. (2006). Workshop on Workflow and Data Flow for Scientific Applications (SciFlow)
- Brockhauser, S., Svensson, O., Bowler, M. W., Nanao, M., Gordon, E., Leal, R. M. F., Popov, A., Gerring, M., McCarthy, A. A. & Gotz, A. (2012). Acta Cryst. D68, 975–984. [DOI] [PMC free article] [PubMed]
- Cohen, A. E., Ellis, P. J., Miller, M. D., Deacon, A. M. & Phizackerley, R. P. (2002). J. Appl. Cryst. 35, 720–726. [DOI] [PMC free article] [PubMed]
- Eker, J., Janneck, J., Lee, E., Liu, J., Liu, X., Ludvig, J., Neuendorffer, S., Sachs, S. & Xiong, Y. (2003). Proc. IEEE, 91, 127–144.
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- González, A., Moorhead, P., McPhillips, S. E., Song, J., Sharp, K., Taylor, J. R., Adams, P. D., Sauter, N. K. & Soltis, S. M. (2008). J. Appl. Cryst. 41, 176–184.
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Leslie, A. G. W. & Powell, H. R. (2007). Evolving Methods for Macromolecular Crystallography, edited by R. J. Read & J. L. Sussman, pp. 41–51. Dordrecht: Springer.
- Ludäscher, B., Altintas, I., Berkley, C., Higgins, D., Jaeger, E., Jones, M., Lee, E. A., Tao, J. & Zhao, Y. (2006). Concurrency Comput. Pract. Exper. 18, 1039–1065.
- McPhillips, T. M., McPhillips, S. E., Chiu, H.-J., Cohen, A. E., Deacon, A. M., Ellis, P. J., Garman, E., Gonzalez, A., Sauter, N. K., Phizackerley, R. P., Soltis, S. M. & Kuhn, P. (2002). J. Synchrotron Rad. 9, 401–406. [DOI] [PubMed]
- Miller, M. D., Brinen, L. S., Cohen, A., Deacon, A. M., Ellis, P., McPhillips, S. E., McPhillips, T. M., Phizackerley, R. P., Soltis, S. M., van den Bedem, H., Wolf, G., Xu, Q. & Zhang, Z. (2004). AIP Conf. Proc. 705, 1233.
- Mooij, W. T., Hartshorn, M. J., Tickle, I. J., Sharff, A. J., Verdonk, M. L. & Jhoti, H. (2006). ChemMedChem, 1, 827–838. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Ngu, A. H. H., Bowers, S., Haasch, N., McPhillips, T. M. & Critchlow, T. (2008). Scientific and Statistical Database Management, edited by B. Ludäscher & N. Mamoulis, pp. 566–572. Berlin: Springer.
- Nienaber, V. L., Richardson, P. L., Klighofer, V., Bouska, J. J., Giranda, V. L. & Greer, J. (2000). Nature Biotechnol. 18, 1105–1108. [DOI] [PubMed]
- Oinn, T. M. et al. (2006). Concurrency Comput. Pract. Exper. 18, 1067–1100.
- Oldfield, T. J. (2001). Acta Cryst. D57, 696–705. [DOI] [PubMed]
- Perryman, A. L., Zhang, Q., Soutter, H. H., Rosenfeld, R., McRee, D. E., Olson, A. J., Elder, J. E. & Stout, C. D. (2010). Chem. Biol. Drug Des. 75, 257–268. [DOI] [PMC free article] [PubMed]
- Rees, D. C., Congreve, M., Murray, C. W. & Carr, R. (2004). Nature Rev. Drug Discov. 3, 660–672. [DOI] [PubMed]
- Sauter, N. K., Grosse-Kunstleve, R. W. & Adams, P. D. (2004). J. Appl. Cryst. 37, 399–409. [DOI] [PMC free article] [PubMed]
- Sharff, A. & Jhoti, H. (2003). Curr. Opin. Chem. Biol. 7, 340–345. [DOI] [PubMed]
- Soltis, S. M. et al. (2008). Acta Cryst. D64, 1210–1221. [DOI] [PMC free article] [PubMed]
- Song, J., Mathew, D., Jacob, S. A., Corbett, L., Moorhead, P. & Soltis, S. M. (2007). J. Synchrotron Rad. 14, 191–195. [DOI] [PubMed]
- Sugahara, M. et al. (2008). J. Struct. Funct. Genomics, 9, 21–28. [DOI] [PubMed]
- Taylor, I. J., Deelman, E., Gannon, D. B. & Shields, M. (2007). Editors. Workflows for e-Science. New York: Springer.
- Ten Eyck, L. F. (1973). Acta Cryst. A29, 183–191.
- Terwilliger, T. C., Adams, P. D., Moriarty, N. W. & Cohn, J. D. (2007). Acta Cryst. D63, 101–107. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yamaguchi, K., Inamoto, N. & Kunii, T. L. (1985). IEEE Comput. Graph. Appl. 5(3), 48–60.