

Crystal structures of the cytoplasmic domain of FlhB from S. typhimurium and A. aeolicus were solved at 2.45 and 2.55 Å resolution, respectively. The deletion of a short loop in the cytoplasmic domain of Salmonella FlhB completely abolishes secretion by the type III secretion system. A molecular-dynamics simulation shows that the deletion of the loop affects the flexibility of a linker between the transmembrane and cytoplasmic domains of FlhB.
Keywords: FlhB, bacterial flagellum, type III secretion system, Salmonella, Aquifex
Abstract
The membrane protein FlhB is a highly conserved component of the flagellar secretion system. It is composed of an N-terminal transmembrane domain and a C-terminal cytoplasmic domain (FlhBC). Here, the crystal structures of FlhBC from Salmonella typhimurium and Aquifex aeolicus are described at 2.45 and 2.55 Å resolution, respectively. These flagellar FlhBC structures are similar to those of paralogues from the needle type III secretion system, with the major difference being in a linker that connects the transmembrane and cytoplasmic domains of FlhB. It was found that deletion of a short flexible loop in a globular part of Salmonella FlhBC leads to complete inhibition of secretion by the flagellar secretion system. Molecular-dynamics calculations demonstrate that the linker region is the most flexible part of FlhBC and that the deletion of the loop reduces this flexibility. These results are in good agreement with previous studies showing the importance of the linker in the function of FlhB and provide new insight into the relationship between the different parts of the FlhBC molecule.
1. Introduction
The bacterial flagellum is a large complex structure that is used as a motility organelle by many bacteria. It consists of three main substructures: the basal body, the hook and the filament. Most of the flagellar proteins are localized outside the cell and are translocated across the cell membrane by the flagellum-specific secretion apparatus, which is evolutionarily and structurally related to the virulence type III secretion system (Aizawa, 2001 ▶; Blocker et al., 2003 ▶). Protein export by the flagellar type III secretion system is highly regulated. The secretion system first exports rod/hook-type proteins until the hook reaches an appropriate length. The secretion system then switches substrate specificity from hook-type export to filament-type export (Minamino & Macnab, 1999 ▶; Hirano et al., 2003 ▶). In Salmonella, the substrate-specificity switch is controlled by two proteins: FliK and FlhB (Kutsukake et al., 1994 ▶; Hirano et al., 1994 ▶; Williams et al., 1996 ▶).
FlhB is an essential membrane protein of the flagellar type III secretion system. It consists of two domains: a hydrophobic N-terminal part (FlhBTM) that is predicted to contain four transmembrane helices and a C-terminal cytoplasmic domain (FlhBC) (Minamino et al., 1994 ▶). The two domains are connected by a flexible linker. This linker is a highly conserved part of the FlhB protein and is essential for the type III secretion system (TTSS). Deletions or point mutations in the linker region completely abolish or significantly reduce secretion (Fraser et al., 2003 ▶; Zarivach et al., 2008 ▶). The wild-type cytoplasmic domain of Salmonella FlhB undergoes autocatalytic cleavage between amino-acid residues Asn269 and Pro270 within a highly conserved NPTH sequence (Minamino & Macnab, 2000a ▶). This autocleavage is essential for the switching process (Fraser et al., 2003 ▶; Ferris et al., 2005 ▶). Mutation of Asn269 to Ala prevents cleavage and locks the export apparatus in the hook-type specificity state.
FlhBC has been shown to interact with several soluble components of the TTSS: FliH, FliI, FliJ (Minamino & Macnab, 2000b ▶), the cytoplasmic part of the membrane protein FlhA (Zhu et al., 2002 ▶) and the hook-length control protein FliK (Minamino et al., 2004 ▶; Morris et al., 2010 ▶). Interaction of FlhB with FliK has been suggested to be important for the substrate-specificity switching process (Ferris & Minamino, 2006 ▶). Cells with a deleted fliK gene produce an abnormally long hook, termed a ‘polyhook’, without any filament attached (Hirano et al., 1994 ▶).
Several structures of the cytoplasmic domain of FlhB paralogues from the needle TTSS have been published (Zarivach et al., 2008 ▶; Deane et al., 2008 ▶; Wiesand et al., 2009 ▶; Lountos et al., 2009 ▶). However, no structural information is available for FlhB from the flagellar secretion system. Here, we describe crystal structures of the cytoplasmic domain of flagellar FlhB from two organisms: Salmonella typhimurium and Aquifex aeolicus.
2. Materials and methods
2.1. Structure determination
Details of the purification, crystallization and data collection of Salmonella and Aquifex FlhBc have been described previously (Meshcheryakov et al., 2011 ▶; Meshcheryakov & Samatey, 2011 ▶). Both structures were solved by multiwavelength anomalous diffraction (MAD) using the program SHELXD (Sheldrick, 2008 ▶). Initial protein models were built automatically with Buccaneer (Cowtan, 2006 ▶) from the CCP4 package (Winn et al., 2011 ▶). The models were refined through an iterative combination of refinement with REFMAC5 (Murshudov et al., 2011 ▶) and manual model building in Coot (Emsley et al., 2010 ▶). In the case of Salmonella FlhBC, TLS refinement was performed in the final stages with two TLS groups per FlhBC molecule (residues 229–269 and 270–353; Painter & Merritt, 2006 ▶). Structural figures were produced using PyMOL (http://www.pymol.org).
2.2. DNA manipulation and motility assay
Mutations of S. typhimurium flhB carried by the plasmid pMM26 (Minamino & Macnab, 2000a ▶) were performed as described previously (Wang & Malcolm, 1999 ▶). For the motility assay, freshly transformed Salmonella cells were directly inoculated as colonies into soft tryptone agar containing 0.35%(w/v) agar and incubated at 303 K.
2.3. Preparation of the whole-cell and culture-supernatant fractions and immunoblotting
Salmonella MKM50 (ΔflhB strain) cells (Fraser et al., 2003 ▶) carrying an appropriate plasmid were incubated at 310 K in LB medium containing 100 µg ml−1 ampicillin until the optical density OD600 reached 1.4–1.5. Aliquots of culture containing a constant amount of cells were centrifuged. Cell pellets were suspended in an equal volume of SDS loading buffer. Proteins in the culture supernatant were precipitated using 10% trichloroacetic acid and were suspended in SDS loading buffer. After SDS–PAGE, proteins were detected with anti-FlgE and anti-FliC antibodies using a WesternBreeze chromogenic immunodetection kit (Invitrogen).
2.4. Molecular-dynamics simulations
Molecular-dynamics (MD) simulations were performed using the SCUBA (Simulation Codes for hUge Biomolecular Assembly) program package (Ishida et al., 2006 ▶). The AMBER ff99SB force field (Hornak et al., 2006 ▶) was used for the protein. The simulated systems were solvated with SPC/E water molecules (Berendsen et al., 1987 ▶) with 100 mM KCl in the periodic boundary separated by at least 12 Å from the FlhBC molecule in the initial stage. After energy minimization and 0.27 ns MD simulation to adjust the temperature and pressure of the system to 300 K and 101 kPa with positional restraints, 40 ns MD simulation was performed without restraints in the canonical ensemble. The last 20 ns trajectory was used for the analysis. A shifted-force cutoff of real-space nonbonded energy was made at 12 Å and the particle-particle-particle-mesh (PPPM) method (Deserno & Holm, 1998 ▶) was employed for electrostatic energy calculation in Fourier space. Integration of the equation of motion was carried out using the multi-time-step method XO-RESPA (Martyna et al., 1996 ▶) in the canonical ensemble. Integrations of fast (bond and angle), medium (torsion and real-space nonbonded) and slow (Fourier-space nonbonded) energy terms were performed every 0.5, 1.0 and 2.0 fs, respectively.
2.5. Accession numbers
Atomic coordinates and structure factors have been deposited in the PDB with accession codes 3b0z and 3b1s for Salmonella and Aquifex FlhBC, respectively. The structures reported here are explained in interactive three dimensions at http://proteopedia.org/w/Samatey.
3. Results and discussion
3.1. Flagellar FlhBC structure description
The Salmonella FlhBC (SalFlhBC) and Aquifex FlhBC (AquFlhBC) structures were solved by multiwavelength anomalous diffraction (MAD) using selenomethionine derivatives (Meshcheryakov et al., 2011 ▶; Meshcheryakov & Samatey, 2011 ▶; Table 1 ▶).
Table 1. X-ray data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell. MAD data-collection statistics for Salmonella FlhBC have been published in Meshcheryakov Samatey (2011 ▶).
| Salmonella FlhBc | Aquifex FlhBc | ||||
|---|---|---|---|---|---|
| Native | Native | SeMet derivative | |||
| Peak | Inflection | Remote | |||
| Data collection | |||||
| Space group | P42212 | C2 | C2 | ||
| Unit-cell parameters (, ) | a = b = 49.1, c = 143.1, = = = 90 | a = 114.6, b = 33.8, c = 122.4, = = 90, = 107.8 | a = 113.4, b = 33.6, c = 122.2, = = 90, = 107.9 | ||
| Molecules in asymmetric unit | 1 | 3 | 3 | ||
| Wavelength () | 0.9 | 0.9 | 0.9791 | 0.97936 | 0.99508 |
| Resolution () | 40.452.45 (2.582.45) | 47.762.55 (2.692.55) | 503.00 (3.163.00) | 503.00 (3.163.00) | 503.00 (3.163.00) |
| R merge † | 0.075 (0.380) | 0.056 (0.386) | 0.094 (0.452) | 0.069 (0.407) | 0.064 (0.368) |
| I/(I) | 16.2 (5.7) | 12.5 (3.4) | 7.6 (2.4) | 9.9 (3.0) | 10.5 (3.2) |
| Completeness (%) | 98.8 (100) | 99.3 (100) | 100 (100) | 100 (100) | 100 (100) |
| Multiplicity | 7.7 (7.9) | 3.7 (3.8) | 3.6 (3.7) | 3.7 (3.7) | 3.7 (3.7) |
| Refinement | |||||
| Resolution () | 28.072.45 | 29.752.55 | |||
| R work/R free (%) | 23.1/24.7 | 24.1/26.2 | |||
| No. of atoms | |||||
| Protein | 992 | 2707 | |||
| Ligand/ion | 4 | 0 | |||
| Water | 20 | 48 | |||
| Wilson plot B factor (2) | 79.3 | 83.7 | |||
| Average B factor (2) | |||||
| Protein | 78.8 | 73.6 | |||
| Ligand/ion | 77.2 | N/A | |||
| Water | 39.3 | 61.2 | |||
| R.m.s. deviations | |||||
| Bond lengths () | 0.021 | 0.019 | |||
| Bond angles () | 2.090 | 1.844 | |||
| Ramachandran plot (%) | |||||
| Most favoured | 97.5 | 99.7 | |||
| Additionally allowed | 2.5 | 0.3 | |||
| Disallowed | 0 | 0 | |||
R
merge = 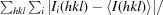
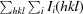 , where Ii(hkl) is the intensity of the ith measurement of reflection hkl and I(hkl) is the mean value of Ii(hkl) for all i measurements.
, where Ii(hkl) is the intensity of the ith measurement of reflection hkl and I(hkl) is the mean value of Ii(hkl) for all i measurements.
The SalFlhBC and AquFlhBC crystals belonged to different space groups: P42212 and C2, respectively. In the case of the AquFlhBC crystal there are three protein molecules in the asymmetric unit. The three molecules in the asymmetric unit are very similar, with r.m.s.d.s on pairwise superposition in the range 0.40–0.76 Å. Each molecule consists of two polypeptide chains resulting from proteolytic cleavage after Asn263. In all molecules no electron density was observed for residues 213–231 at the N-terminus; depending on the molecule, two to six residues at the C-terminus were disordered.
In the case of SalFlhBC the final model comprises residues 229–353 out of 219–383 in the crystallized protein, with a cleavage after Asn269. No electron density was observed for residues 219–228 and 354–383. The model of Salmonella FlhBC includes two Zn2+ ions and two Na+ ions (Fig. 1 ▶ a). All of these atoms mediate intermolecular interactions in the crystal lattice. Zn2+ was added to the crystallization solution and was necessary to obtain well diffracting crystals. Analysis of the crystallographic packing shows that one of the zinc ions coordinates three glutamate residues from three symmetry-related SalFhBC molecules: Glu230, Glu258 and Glu307 (Fig. 1 ▶ b). This interaction fixes N-terminal helix α1, which is one of the most flexible parts of Salmonella FlhBC (see below), between two symmetrical molecules.
Figure 1.

Molecular packing in the crystal of Salmonella FlhBC. (a) The asymmetric molecule and symmetry-related molecules are displayed in green and grey, respectively. Sodium ions are represented as magenta spheres and zinc ions are shown as blue spheres. (b) Rotated enlarged view of the zinc-binding site (black box in Fig. 1 ▶ a). The F o −F c electron-density map is displayed in grey at a contour level of 5σ and was calculated without a Zn atom.
Both the Salmonella and Aquifex FlhBC structures show very similar folds, with an r.m.s.d. of 1.03 Å for 102 Cα atoms (Fig. 2 ▶ a). Flagellar FlhBC consists of a globular domain composed of a four-stranded β-sheet surrounded by four α-helices. The globular domain is preceded by a long N-terminal α-helix (α1) that connects the cytoplasmic globular part of FlhB to the transmembrane domain. The α1 helix engages in a crystal contact in both the Salmonella and Aquifex FlhBC crystals, which may affect its orientation relative to the globular domain. However, these crystal contacts differ. In the SalFlhBC crystal α1 primarily contacts α1 and α2 of adjacent molecules, while in the AquFlhBC crystal α1 primarily contacts α4 and the cleavage site between β1 and β2.
Figure 2.

Structure of flagellar FlhBc. (a) Ribbon representation of the crystal structures of Salmonella and Aquifex FlhBC. (b) Electrostatic potential mapped onto the surface of Salmonella FlhBC. Electrostatics were calculated using the APBS software (Baker et al., 2001 ▶) and plotted at ±5kT e−1. (c) Evolutionarily conserved residues of FlhBC. The figure was prepared with ConSurf (http://consurf.tau.ac.il/; Ashkenazy et al., 2010 ▶). Residues are coloured according to the conservation in amino-acid sequences of 200 different FlhB proteins. Arrows mark the position of the autocleavage site between β1 and β2.
The major difference between SalFlhBC and AquFlhBC is in the N-terminal region. In the model of SalFlhBC helix α1 is longer and has a kink at the highly conserved residue Gly236. However, a longer helix with a kink is not excluded in AquFlhBC, in which a highly conserved Gly230 occurs just two residues into the disordered segment 213–231 which is present in the crystallized protein but is absent in the model. Although the kink may arise from the crystal packing, our data show potential flexibility of the linker around this conserved glycine residue. The importance of such flexibility has previously been shown for EscU, an FlhB paralogue from the needle TTSS. Mutation of Gly229 (which corresponds to Gly236 of SalFlhB) to the less flexible proline in EscU completely abolished secretion (Zarivach et al., 2008 ▶).
The conserved NPTH autocleavage site is exposed on a surface between strands β1 and β2. Both Salmonella and Aquifex FlhBC show different conformations of the PTH region that suggest its flexibility. This is very different from the needle paralogues. In all known paralogue structures the PTH region has the same orientation, which is stabilized by contacts with surrounding residues (Zarivach et al., 2008 ▶; Deane et al., 2008 ▶; Wiesand et al., 2009 ▶; Lountos et al., 2009 ▶). It is difficult to say for the moment whether the greater flexibility of the PTH site in flagellar FlhBC has any functional meaning. In SalFlhBC, the PTH region, together with adjacent residues in the globular domain and the C-terminal part of the linker α-helix, forms a positively charged cleft (Fig. 2 ▶ b). A similar positive cleft is also present in AquFlhBC. Such a cleft might be a potential recognition site for proteins secreted by the flagellar secretion system. The autocleavage of FlhB has been suggested to create an interaction site for the other components of the type III secretion system (Zarivach et al., 2008 ▶; Deane et al., 2008 ▶). In particular, there is a model that describes the binding of FliK to the cleaved NPTH loop of FlhB (Mizuno et al., 2011 ▶). However, the linker helix, which is one of the most conserved parts of the FlhB protein (Fig. 2 ▶ c), could also participate in the recognition of secreted proteins, since deletions or point mutations in this region of FlhB completely block secretion (Fraser et al., 2003 ▶; Zarivach et al., 2008 ▶).
3.2. Comparison with needle paralogue structures
Despite low sequence identity (Fig. 3 ▶ a), the overall structure of flagellar FlhBC is very similar to the structures of paralogues from the needle secretion system: EscUC, SpaSC, YscUC and Spa40C (Fig. 3 ▶ b). The obvious difference between these proteins is the linker region between the N-terminal transmembrane domain and the globular cytoplasmic domain. All of the proteins show a large difference in the conformation of their N-terminal parts, indicating flexibility of this region of the molecule. In our structures no electron density was observed for residues 219–228 of SalFlhBC and residues 213–231 of AquFlhBC, which is consistent with flexibility of this part of FlhBC. However, the remainder of the residues of the linker form a well defined α-helix, which in the case of SalFlhBC is kinked at position Gly236. In contrast to the needle paralogues, it might be a general property of flagellar FlhB to have a more stable linker helix.
Figure 3.


Comparison of flagellar FlhBC and its paralogues from the needle type III secretion system. (a) Multiple sequence alignment of FlhBC from S. typhimurium (SalFlhBC; Swiss-Prot accession No. P40727) with FlhBC from A. aeolicus (AquFlhBC; O67813), EscUC from E. coli (Q7DB59), YscUC from Yersinia pestis (P69986), SpaSC from S. typhimurium (P40702) and Spa40C from Shigella flexneri (Q6XVW1). Identical residues are boxed in red; similar residues are coloured red. Alignment was performed with ClustalW (Larkin et al., 2007 ▶; Goujon et al., 2010 ▶). (b) Stereoviews of the superposition of Salmonella FlhBC (blue; PDB entry 3b0z; this work), Aquifex FlhBC (red; PDB entry 3b1s; this work), EscUC (orange; PDB entry 3bzo; Zarivach et al., 2008 ▶), YscUC (grey; PDB entry 2jli; Lountos et al., 2009 ▶), SpaSC (green; PDB entry 3c01; Zarivach et al., 2008 ▶) and Spa40C (purple; PDB entry 2vt1; Deane et al., 2008 ▶).
The proteins of the FlhB family exhibit a significant variation in length, mainly because of differences at the C-terminus. For instance, Salmonella FlhB is longer than the Aquifex protein by 33 amino acids. However, these additional residues (residues 354–383) were not visible in the electron-density map, suggesting that they are unfolded. This region in SalFlhB is rich in proline residues, making it unlikely to form any stable structure. The function of the elongated C-terminal part of FlhB is not known, but it is dispensable for motility (Kutsukake et al., 1994 ▶). It apparently participates in the regulation of secretion because C-terminal truncation of Salmonella FlhB can partially suppress the ΔfliK phenotype (Kutsukake et al., 1994 ▶; Williams et al., 1996 ▶). However, it is unlikely to directly interact with FliK since the truncation has almost no effect in a wild-type fliK background (Williams et al., 1996 ▶).
3.3. Effect of mutations of residues 281–285 of Salmonella FlhB on TTSS function
The two strands β2 and β3 are connected by a long flexible loop. This loop is not conserved within the FlhB family, although it is flanked by highly conserved residues: Tyr279 and Pro287 (in Salmonella numbering). The length of the loop, which is longer than necessary just to connect two β-strands, made us think that it might be of functional importance. To investigate this hypothesis, we created three mutants of Salmonella FlhB. In the first mutant the loop residues 281–285 were deleted (Fig. 4 ▶ a). In the second and third mutants residues 281–285 were substituted by Ala or Pro residues, respectively. We then carried out swarming assays on soft agar plates to investigate whether the Salmonella cells containing mutated FlhB were still motile. We found that the deletion of the loop completely abolished motility (Fig. 4 ▶ b). At the same time, substitution by Ala residues had no effect on motility and Pro substitution decreased motility. To check whether these changes in motility were because of changes in export activity, we analyzed secretion of the hook protein FlgE and the filament protein FliC by the flagellar secretion system containing mutated FlhB (Fig. 4 ▶ c). We found that motility is correlated to the secretion of FlgE and FliC. In the case in which the loop (281–285) of FlhB was deleted neither FlgE nor FliC were secreted, whereas proline substitution reduced the secretion of both proteins. No difference in secretion was observed between the wild-type FlhB and the Ala substitution.
Figure 4.

Effect of mutations of the ENKMS281–285 region of Salmonella FlhB on protein function. (a) Ribbon representation of Salmonella FlhBC; the region is shown in black. (b) The ability of FlhB variants with a modified ENKMS region to complement the ΔflhB Salmonella strain MKM50. Motility assays were carried out on semi-solid agar plates at 303 K for 5 h. FlhB products were 1, none, empty vector; 2, wild-type FlhB; 3, FlhB Δ(281–285); 4, FlhB AAAAA281–285; 5, FlhB PPPPP281–285. (c) Immunoblotting using anti-FlgE and anti-FliC antibodies on the whole-cell (Cell) and culture-supernatant (Sup) fractions from Salmonella strain MKM50 producing different FlhB variants.
3.4. Molecular-dynamics (MD) simulations
To further investigate the effect of mutation of the loop on the FlhBC molecule, we performed MD simulations of the wild-type SalFlhBC and the Δ(281–285), AAAAA281–285 and PPPPP281–285 mutants. During the MD simulations, we observed that the globular domain is relatively rigid in all of the cases, while the N-terminal α-helix of the wild-type FlhBC is very flexible and becomes less flexible in the mutants (Figs. 5 ▶ b–5e). In addition to the kink around Gly236–Pro238 (Fig. 2 ▶ a), a significant kink was observed near Met256 during the MD simulations. To characterize the flexibility of the N-terminal α-helix, we defined a distance D, angles θ12, θ23, θ34 and θ14 and torsion angles χ3 and χ5 (see explanations in Fig. 5 ▶ a and Table 2 ▶). A notable structural difference is demonstrated by torsion angle χ3, which determines the direction of the V2 region of the N-terminal α-helix relative to the globular domain. The χ3 value is positive for wild-type FlhBC and the AAAAA281–285 mutant, but is negative for the Δ(281–285) and PPPPP281–285 mutants, which is consistent with the structural differences shown in Figs. 5 ▶(b)–5(e). Since the latter mutations reduce Salmonella motility, this structural change might have some functional effects. Another notable difference is observed as a reduction in the θ12, θ23, θ14, χ3 and χ5 fluctuations in the PPPPP281–285 mutant, indicating significant structural change and loss of flexibility of the N-terminal α-helix.
Figure 5.

Flexibility of the N-terminal α-helix of Salmonella FlhBC observed in MD simulations. (a) Key residues and vectors used for MD analysis in Table 2 ▶. The vectors connecting residues 229–238, 238–256, 256–264, 265–269 and 238–264 are defined as V1–V5, respectively. (b–e) Structural variations of the N-terminal α-helix during MD simulations when the globular domain is superimposed for (b) the wild-type FlhBC, (c) FlhBC Δ(281–285), (d) FlhBC AAAAA281–285 and (e) FlhBC PPPPP281–285. For each case, six snapshots were chosen based on the maximum and minimum values of D, θ14 and χ5 (see Table 2 ▶ for their definitions).
Table 2. Structure and fluctuation differences between wild-type Salmonella FlhBC and its mutants during MD simulation shown by the key distance and angles defined by vectors V15 shown in Fig. 4 ▶(a).
D, length of V5. 12, 23, 34 and 14, angles defined between V1 and V2, V2 and V3, V3 and V4, and V1 and V4, respectively. 3 and 5, torsion angles defined by the sets of three vectors V2V3V4 (torsion around V3) and V1V5V4 (around V5), respectively. Averages and standard deviations over the last 20ns of MD are shown, with negative values shown in bold.
| Protein | D () | 12 () | 23 () | 34 () | 14 () | 3 () | 5 () |
|---|---|---|---|---|---|---|---|
| Salmonella FlhBC | |||||||
| Wild type | 37.2 3.9 | 96.0 21.9 | 53.8 19.4 | 105.5 2.2 | 77.1 38.3 | 83.8 23.5 | 103.8 44.8 |
| (281285) | 36.9 0.7 | 133.2 11.7 | 64.1 5.2 | 108.2 1.7 | 34.0 9.5 | 23.1 7.9 | 168.3 21.0 |
| AAAAA281285 | 38.1 1.5 | 113.3 34.6 | 45.7 6.1 | 113.2 1.8 | 47.9 35.4 | 30.3 11.6 | 175.5 43.6 |
| PPPPP281285 | 36.6 3.1 | 69.6 37.1 | 51.4 12.2 | 112.9 2.4 | 59.2 26.9 | 77.3 18.9 | 165.4 43.1 |
4. Conclusions
The flagellar secretion system is closely related to the needle type III secretion system utilized by a number of virulent bacteria for the secretion of toxins into the host-cell cytoplasm. The structure of flagellar FlhBC described in this paper once again confirms this relationship. Basically, the structure is similar to already known structures of needle paralogues. The differences (such as the orientation of the conserved PTH sequence or the longer linker helix) could be peculiarities of the flagellar protein. The functional implications of such differences, if any, are a subject for further investigation.
The most important of our findings is that flexibility of the large nonconserved loop in the globular domain of FlhBC is necessary for function of the whole secretion system. Deletion of this loop or its mutation to less flexible proline residues makes FlhBC more rigid and thus abolishes or significantly reduces secretion. Taking into account the similarity between the flagellar and needle proteins, this loop could be a promising target for the creation of novel drugs against pathogenic bacteria.
Supplementary Material
PDB reference: Salmonella FlhBC, 3b0z
PDB reference: Aquifex FlhBC, 3b1s
Acknowledgments
We thank Y.-H. Yoon and I. Meshcheryakova for help with the experiments. We thank the staff at beamlines BL44XU and BL41XU at the SPring-8 synchrotron, Harima, Japan for support in obtaining beam time and assistance with X-ray data collection. We are grateful to the School of Frontier Biosciences, Osaka University for the Salmonella strains. We thank E. Martz at the University of Massachusetts, Amherst, USA for making the Proteopedia web pages and the videos. This work was supported by direct funding provided by OIST. Computations were performed using the computers at OIST, IMCB/University of Tokyo, the Research Center for Computational Science/Okazaki Research Facilities/National Institute of Natural Science and the Institute for Solid State Physics/University of Tokyo.
References
- Aizawa, S.-I. (2001). FEMS Microbiol. Lett. 202, 157–164. [DOI] [PubMed]
- Ashkenazy, H., Erez, E., Martz, E., Pupko, T. & Ben-Tal, N. (2010). Nucleic Acids Res. 38, W529–W533. [DOI] [PMC free article] [PubMed]
- Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. (2001). Proc. Natl Acad. Sci. USA, 98, 10037–10041. [DOI] [PMC free article] [PubMed]
- Berendsen, H. J. C., Grigera, J. R. & Straatsma, T. P. (1987). J. Phys. Chem. 91, 6269–6271.
- Blocker, A., Komoriya, K. & Aizawa, S. (2003). Proc. Natl Acad. Sci. USA, 100, 3027–3030. [DOI] [PMC free article] [PubMed]
- Cowtan, K. (2006). Acta Cryst. D62, 1002–1011. [DOI] [PubMed]
- Deane, J. E., Graham, S. C., Mitchell, E. P., Flot, D., Johnson, S. & Lea, S. M. (2008). Mol. Microbiol. 69, 267–276. [DOI] [PMC free article] [PubMed]
- Deserno, M. & Holm, C. (1998). J. Chem. Phys. 109, 7678.
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Ferris, H. U., Furukawa, Y., Minamino, T., Kroetz, M. B., Kihara, M., Namba, K. & Macnab, R. M. (2005). J. Biol. Chem. 280, 41236–41242. [DOI] [PubMed]
- Ferris, H. U. & Minamino, T. (2006). Trends Microbiol. 14, 519–526. [DOI] [PubMed]
- Fraser, G. M., Hirano, T., Ferris, H. U., Devgan, L. L., Kihara, M. & Macnab, R. M. (2003). Mol. Microbiol. 48, 1043–1057. [DOI] [PubMed]
- Goujon, M., McWilliam, H., Li, W., Valentin, F., Squizzato, S., Paern, J. & Lopez, R. (2010). Nucleic Acids Res. 38, W695–W699. [DOI] [PMC free article] [PubMed]
- Hirano, T., Minamino, T., Namba, K. & Macnab, R. M. (2003). J. Bacteriol. 185, 2485–2492. [DOI] [PMC free article] [PubMed]
- Hirano, T., Yamaguchi, S., Oosawa, K. & Aizawa, S. (1994). J. Bacteriol. 176, 5439–5449. [DOI] [PMC free article] [PubMed]
- Hornak, V., Abel, R., Okur, A., Strockbine, B., Roitberg, A. & Simmerling, C. (2006). Proteins, 65, 712–725. [DOI] [PMC free article] [PubMed]
- Ishida, H., Higuchi, M., Yonetani, Y., Kano, T., Joti, Y., Kitao, A. & Go, N. (2006). Annual Report of the Earth Simulator Center, pp. 237–239. Yokohama: Earth Simulator Center, Japan Agency for Marine-Earth Science and Technology.
- Kutsukake, K., Minamino, T. & Yokoseki, T. (1994). J. Bacteriol. 176, 7625–7629. [DOI] [PMC free article] [PubMed]
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J. & Higgins, D. G. (2007). Bioinformatics, 23, 2947–2948. [DOI] [PubMed]
- Lountos, G. T., Austin, B. P., Nallamsetty, S. & Waugh, D. S. (2009). Protein Sci. 18, 467–474. [DOI] [PMC free article] [PubMed]
- Martyna, G. J., Tuckerman, M. E., Tobias, D. J. & Klein, M. L. (1996). Mol. Phys. 87, 1117–1157.
- Meshcheryakov, V. A. & Samatey, F. A. (2011). Acta Cryst. F67, 808–811. [DOI] [PMC free article] [PubMed]
- Meshcheryakov, V. A., Yoon, Y.-H. & Samatey, F. A. (2011). Acta Cryst. F67, 280–282. [DOI] [PMC free article] [PubMed]
- Minamino, T., Iino, T. & Kutuskake, K. (1994). J. Bacteriol. 176, 7630–7637. [DOI] [PMC free article] [PubMed]
- Minamino, T. & Macnab, R. M. (1999). J. Bacteriol. 181, 1388–1394. [DOI] [PMC free article] [PubMed]
- Minamino, T. & Macnab, R. M. (2000a). J. Bacteriol. 182, 4906–4914. [DOI] [PMC free article] [PubMed]
- Minamino, T. & Macnab, R. M. (2000b). Mol. Microbiol. 35, 1052–1064. [DOI] [PubMed]
- Minamino, T., Saijo-Hamano, Y., Furukawa, Y., González-Pedrajo, B., Macnab, R. M. & Namba, K. (2004). J. Mol. Biol. 341, 491–502. [DOI] [PubMed]
- Mizuno, S., Amida, H., Kobayashi, N., Aizawa, S. & Tate, S. (2011). J. Mol. Biol. 409, 558–573. [DOI] [PubMed]
- Morris, D. P., Roush, E. D., Thompson, J. W., Moseley, M. A., Murphy, J. W. & McMurry, J. L. (2010). Biochemistry, 49, 6386–6393. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Painter, J. & Merritt, E. A. (2006). Acta Cryst. D62, 439–450. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Wang, W. & Malcolm, B. A. (1999). Biotechniques, 26, 680–682. [DOI] [PubMed]
- Wiesand, U., Sorg, I., Amstutz, M., Wagner, S., van den Heuvel, J., Lührs, T., Cornelis, G. R. & Heinz, D. W. (2009). J. Mol. Biol. 385, 854–866. [DOI] [PubMed]
- Williams, A. W., Yamaguchi, S., Togashi, F., Aizawa, S.-I., Kawagishi, I. & Macnab, R. M. (1996). J. Bacteriol. 178, 2960–2970. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Zarivach, R., Deng, W., Vuckovic, M., Felise, H. B., Nguyen, H. V., Miller, S. I., Finlay, B. B. & Strynadka, N. C. (2008). Nature (London), 453, 124–127. [DOI] [PubMed]
- Zhu, K., González-Pedrajo, B. & Macnab, R. M. (2002). Biochemistry, 41, 9516–9524. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Salmonella FlhBC, 3b0z
PDB reference: Aquifex FlhBC, 3b1s