

The potential of second-harmonic generation (SHG) microscopy for automated crystal centering to guide synchrotron X-ray diffraction of protein crystals has been explored.
Keywords: second-harmonic generation microscopy, crystal centering, imaging
Abstract
The potential of second-harmonic generation (SHG) microscopy for automated crystal centering to guide synchrotron X-ray diffraction of protein crystals was explored. These studies included (i) comparison of microcrystal positions in cryoloops as determined by SHG imaging and by X-ray diffraction rastering and (ii) X-ray structure determinations of selected proteins to investigate the potential for laser-induced damage from SHG imaging. In studies using β2 adrenergic receptor membrane-protein crystals prepared in lipidic mesophase, the crystal locations identified by SHG images obtained in transmission mode were found to correlate well with the crystal locations identified by raster scanning using an X-ray minibeam. SHG imaging was found to provide about 2 µm spatial resolution and shorter image-acquisition times. The general insensitivity of SHG images to optical scatter enabled the reliable identification of microcrystals within opaque cryocooled lipidic mesophases that were not identified by conventional bright-field imaging. The potential impact of extended exposure of protein crystals to five times a typical imaging dose from an ultrafast laser source was also assessed. Measurements of myoglobin and thaumatin crystals resulted in no statistically significant differences between structures obtained from diffraction data acquired from exposed and unexposed regions of single crystals. Practical constraints for integrating SHG imaging into an active beamline for routine automated crystal centering are discussed.
1. Introduction
X-ray diffraction of protein crystals using synchrotron radiation has become an indispensable tool for high-resolution protein structure determination. However, the high cost of the construction and maintenance of synchrotron sources results in a corresponding high value being placed on beam time. Methods that can improve the throughput and reliability of automated data collection at synchrotron sources can significantly improve the overall pace of protein structure determination. For these reasons, robotic sample handling and mounting under cryogenic conditions are now routine. Nevertheless, the reliable centering of small crystals within small-diameter X-ray beams is an ongoing challenge as technological improvements continue to provide capabilities for studying smaller crystals with smaller beams and as the increased use of turbid crystallization matrices, such as cryo-cooled lipidic mesophases, provides additional crystal-visualization difficulties (Sanishvili et al., 2008 ▶; Fischetti et al., 2009 ▶; Cherezov et al., 2009 ▶).
Numerous approaches have been developed to confront the problem of reliable automated crystal centering. Algorithms to identify the loop based on bright-field image analysis and to position the center of the loop in the center of the X-ray beam are routine and are reasonably reliable when the crystal size is comparable to that of the loop (Lavault et al., 2006 ▶; Pothineni et al., 2006 ▶). However, such approaches often fail in reliable centering of microcrystals and/or crystals prepared within turbid matrices. UV-excited fluorescence (UVF) has also been used to complement bright-field imaging, with some degree of success (Asanov et al., 2001 ▶; Bourgeois et al., 2002 ▶; Chavas et al., 2011 ▶; Vernede et al., 2006 ▶). However, crystals are routinely encountered for which the contrast is insufficient for reliable centering. Neither bright-field imaging nor UVF is applicable to highly scattering samples, including crystals extracted from lipidic mesophase. In addition, extended UV exposure is also known to cause damage, including the breakage of disulfide bonds (Nanao & Ravelli, 2006 ▶), and possible perturbation of aromatic residues (Sgarbossa et al., 2003 ▶). Recent work with two-photon excited UV fluorescence has been shown to limit some, but not all, of these effects (Madden et al., 2011 ▶).
More recently, automated raster scanning of loops across a small-diameter X-ray minibeam has been used for crystal centering based on identifying sample locations that provide significant X-ray diffraction or X-ray fluorescence (Cherezov et al., 2009 ▶; Aishima et al., 2010 ▶; Hilgart et al., 2011 ▶; Stepanov et al., 2011 ▶). The first approach has the distinct advantage of using diffraction efficiency itself as a probe for good diffraction quality on a length scale commensurate with the X-ray beam diameter. However, the time required for completing a raster scan can vary from a few minutes up to about 60 min per sample depending on the number of grid points, the diffracting power of the crystals and the detector readout speed (Cherezov et al., 2009 ▶). X-ray fluorescence rastering is faster, but requires a fluorescing element in the crystal. Recent advents in fast-readout detectors and shutterless data collection can also increase the speed substantially (Aishima et al., 2010 ▶), but do not limit the X-ray exposure necessary to judge adequate diffraction. Although in principle fast X-ray array detectors can enable single-pixel integration times as low as 2 ms (Aishima et al., 2010 ▶), in practice longer integration times are often used to increase signal levels, to allow for computer processing time and/or to allow for sample translation. The fastest X-ray raster images that have been reported to date were acquired with exposures of 0.2 s per pixel (Aishima et al., 2010 ▶), and typical X-ray raster times with CCD detectors are of the order of 2 s per pixel (Cherezov et al., 2009 ▶). All of these methods expose the sample to a significant X-ray dose before a high-resolution diffraction data set can be collected, which can lead to additional X-ray damage, with the effects generally being greater for small or weakly diffracting crystals by nature of the longer integration times required or lowered attenuations (Garman & Nave, 2009 ▶; Holton & Frankel, 2010 ▶; Meents et al., 2010 ▶; Beitlich et al., 2007 ▶).
In the present work, crystal imaging by SHG was initially evaluated as a potentially viable complement to bright-field imaging and an alternative to initial coarse X-ray rastering. SHG imaging has recently been shown to be a high-contrast technique for imaging non-centrosymmetric inorganic crystals (Hellwart & Christen, 1974 ▶; Gannaway & Sheppard, 1978 ▶; Gauderon et al., 1998 ▶), organic crystals (Shen et al., 2001 ▶; Wanapun et al., 2010 ▶; Zheng et al., 2011 ▶), biological fibers (Brown et al., 2003 ▶; Cox et al., 2003 ▶; Stoller et al., 2003 ▶; Zoumi et al., 2002 ▶; Campagnola & Loew, 2003 ▶) and, more recently, protein crystals (Kissick et al., 2010 ▶; Gualtieri et al., 2011 ▶; Wampler et al., 2008 ▶; Haupert & Simpson, 2011 ▶; Padayatti et al., 2012 ▶; Fromme & Spence, 2011 ▶; Cherezov, 2011 ▶; Höfer et al., 2011 ▶; Caffrey, 2011 ▶; Luft et al., 2011 ▶). In crystallization wells, SHG can improve hit rates for crystal detection significantly over a combined suite of bright-field, UVF, birefringence and trace-labeled fluorescence imaging, routinely detecting crystals that are 5–10 µm in size (Kissick et al., 2010 ▶). SHG imaging of bacteriorhodopsin purple membrane protein crystals, grown in live cells, has shown the detection of crystals <1 µm2 and one monolayer thick (Gualtieri et al., 2011 ▶). In this study, the ability of SHG imaging to pre-screen cryogenically preserved membrane-protein crystals within lipidic mesophase was assessed. As with all beam-scanning nonlinear optical measurements, SHG has the distinct advantage of enabling high-resolution imaging within highly scattering media (Kissick et al., 2010 ▶; Ying et al., 2000 ▶; Yoo et al., 1991 ▶) such as lipidic mesophases. Scattering of the incident light may reduce the overall intensity surviving to the focal plane, but only the focal point of the remaining unscattered beam contains sufficient intensity to enable efficient SHG. Assessment of the impact of laser exposure on the protein structures generated by XRD was also performed using laser fluences significantly greater than those required for routine imaging. These collective results provide a basis for assessing the strengths and limitations of SHG imaging for automated protein crystal centering to aid in synchrotron X-ray diffraction.
2. Methods
2.1. SHG imaging measurements
The instrument used for this work has been described previously (Kissick et al., 2010 ▶) and is similar to those used for other applications (Hellwart & Christen, 1974 ▶; Gannaway & Sheppard, 1978 ▶; Gauderon et al., 1998 ▶; Shen et al., 2001 ▶; Zheng et al., 2011 ▶; Brown et al., 2003 ▶; Cox et al., 2003 ▶; Stoller et al., 2003 ▶; Zoumi et al., 2002 ▶; Campagnola & Loew, 2003 ▶). In brief, a femtosecond infrared (IR) laser is focused onto a sample. High-intensity electromagnetic fields probe nonresonant anharmonicity in chiral molecules, causing frequency doubling and the generation of coherent visible light or SHG. The SHG light is separated from the IR light with dichroic mirrors and filters and detected via photomultiplier tubes (PMTs) in both the forward (transmission) and backward (epi) directions. The coherent nature of this signal ensures that the signal propagates either coparallel (i.e. forwards) or antiparallel (i.e. backwards) to the fundamental beam. It also gives rise to crystalline selectivity, as isotropic systems (i.e. liquids and amorphous aggregates) lead to complete destructive interference of the SHG signal. Similarly, coherent SHG does not arise in centrosymmetric crystals (e.g. those of most salts). Two-photon excited fluorescence (TPEF) probes resonant absorption, vibrational relaxation and subsequent emission of frequency-shifted light by a molecule as in conventional fluorescence, except that the excitation energy is achieved by the simultaneous absorption of two photons. TPEF is an incoherent process and is thus not directional or intrinsically crystal-selective. This signal is excited with the same source as the SHG signal and is detected simultaneously with an additional set of dichroic mirror, filters and PMTs. Both of these signals utilize tight focusing to achieve the high local electromagnetic fields necessary for efficient nonlinear optical interactions. As in confocal microscopy, the tight focal spot is moved rapidly around the sample throughout the sample plane and an image is produced from temporal reconstruction of this movement. Adaptations to enable imaging of looped protein crystals under cryogenic conditions have included the design of a custom mounting apparatus capable of XYZ positioning along with rotation about the loop axis. An Oxford Cryosystems cryostream was maintained at 100 K for all reported measurements. The ∼16 mm working distances of a matched pair of 10× objectives (Nikon) were sufficient to allow reliable imaging with minimal perturbation to or from the cryostream. A MaiTai laser (Spectra-Physics) was used for all of the nonlinear optical images, with a fundamental wavelength of 1.0 µm and 150 mW average power (80 MHz, <150 fs pulses). Images were obtained using an Electro-Optic Products resonant scanner operating at 8 kHz. Photon counts from approximately 1000 line scans of the resonant mirror were pooled to generate each line of the image, with the slow-scan motion on the orthogonal axis controlled using a galvanometer (Electro-Optic Products) to generate images. All software for scan-mirror control and data acquisition was written in-house.
2.2. Characterization of the focal volume
Dilute solutions of 200 nm barium titanate nanocrystals (US Research Nanomaterials Inc.) in polyethylene glycol molecular weight 2050 were produced by serial dilution at 333 K. Samples were mounted in CryoLoops (Hampton Research) and allowed to solidify at room temperature. Loops were imaged at 100 nm resolution and the average full width at half maximum of the SHG peaks in the image minus the size of the particles was used as the beam width. The depth of field was characterized by full width at half maximum in depth of 2 µm resolution images stepped along the beam in 5 µm intervals.
2.3. LCP experiments
Crystals of human β2 adrenergic receptor fused to lysozyme from T4 phage (β2AR-T4L) in lipidic cubic phase were prepared as described in Hanson et al. (2008 ▶), mounted in MiTeGen loops and cryocooled in liquid nitrogen.
2.4. Protein-damage studies
Horse-heart myoglobin was obtained from Sigma–Aldrich (≥90%) and crystals were grown according to Maurus et al. (1998 ▶). A crystal of ∼330 × 65 × 15 µm was soaked in a cryosolvent consisting of 20% glycerol, 68% ammonium sulfate, 20 mM Tris pH 7.5, 5 mM EDTA and mounted in a CryoLoop. Thaumatin from Thaumatococcus daniellii was obtained from Sigma–Aldrich. Tetragonal crystals were grown by the hanging-drop vapor-diffusion method using a drop consisting of equal parts 30 mg ml−1 aqueous protein solution and a reservoir solution consisting of 0.95 M potassium sodium tartrate with 5% glycerol. Thaumatin crystals were transferred to a cryoprotectant solution with an increased glycerol concentration of 30% prior to flash-cooling in liquid nitrogen.
For both proteins, a cryocooled crystal was mounted on a goniometer at Purdue University in a cryostream held at 100 K and one half of the crystal was exposed to SHG imaging conditions using a 1.0 µm laser and scanning at an average laser power of 500 mW, which yielded a saturating SHG signal. For all crystals used in the damage assessments, approximately half of the protein crystal was exposed to laser fluences that were approximately five times more intense than those typically used for SHG imaging (e.g. 500 mW compared with 100 mW), as shown in Fig. 2. Given the nonlinear dependence of multi-photon absorption, a fivefold increase in intensity corresponds to a 25-fold increase in two-photon absorption and a 125-fold increase in three-photon absorption. The absorption of the fundamental frequency was negligible. The absorbed dose of the doubled light at 500 nm by heme in the crystal was also negligible, as the power at 500 nm was <1 fW. The crystals were maintained under cryoconditions and X-ray diffraction data were subsequently collected using a 20 µm mini-beam collimator on GM/CA-CAT beamline 23ID-B (Fischetti et al., 2009 ▶) to isolate laser-exposed and unexposed regions of the crystals. Data were processed and scaled with HKL-2000 (Otwinowski & Minor, 1997 ▶) and were phased and refined with CNSsolve v.1.2 (Brünger et al., 1998 ▶; Brunger, 2007 ▶) using PDB entries 3lr7 (Yi et al., 2010 ▶) and 2vi3 (Asherie et al., 2009 ▶) as the initial phasing models for myoglobin and thaumatin, respectively. Data-collection statistics are shown in Table 1 ▶ and the structure-refinement statistics are shown in Table 2 ▶. The structures were modeled in Coot (Emsley et al., 2010 ▶). The atomic coordinates and structure factors have been deposited with PDB codes 4dc5, 4dc6, 4dc7 and 4dc8.
Table 1. Data-collection statistics.
Values in parentheses are for the highest shell.
| Myoglobin | Thaumatin | |||
|---|---|---|---|---|
| No laser | Plus laser | No laser | Plus laser | |
| Space group | P21 | P21 | P41212 | P41212 |
| Unit-cell parameters (Å, °) | a = 35.36, b = 28.79, c = 63.06, β = 106.0 | a = 35.35, b = 28.79, c = 63.04, β = 106.1 | a = 57.91, b = 57.91, c = 150.32 | a = 57.93, b = 57.93, c = 150.35 |
| Resolution (Å) | 50.00–1.50 (1.55–1.50) | 50.00–1.50 (1.55–1.50) | 50.00–1.48 (1.52–1.48) | 50.00–1.48 (1.52–1.48) |
| No. of reflections | 92663 | 97119 | 930164 | 888486 |
| No. of unique reflections | 19833 | 19856 | 43152 | 43518 |
| Completeness (%) | 97.3 (90.1) | 97.7 (91.4) | 98.9 (97.9) | 98.9 (95.4) |
| 〈I/σ(I)〉 | 21.7 (4.7) | 21.7 (5.1) | 40.9 (9.6) | 44.3 (8.3) |
| R merge (%) | 6.7 (29.9) | 6.8 (29.9) | 6.3 (30.0) | 6.1 (32.8) |
| Mosaicity range (°) | 0.31–0.44 | 0.27–0.44 | 0.14–0.27 | 0.12–0.18 |
Table 2. Refinement statistics.
| Myoglobin | Thaumatin | |||
|---|---|---|---|---|
| No laser | Plus laser | No laser | Plus laser | |
| R cryst (%) | 18.3 | 18.1 | 17.5 | 17.8 |
| R free (%) | 21.8 | 21.3 | 19.2 | 19.4 |
| No. of nonsolvent atoms | 1246 | 1246 | 1572 | 1572 |
| No. of solvent atoms | 129 | 129 | 252 | 252 |
| Mean B (Å2) | 16.1 | 16.3 | 13.3 | 13.2 |
| Ramachandran plot, residues in (%) | ||||
| Allowed regions | 98.6 | 98.0 | 98.5 | 98.5 |
| Additional regions | 1.4 | 2.0 | 1.5 | 1.5 |
| Disallowed regions | 0.0 | 0.0 | 0.0 | 0.0 |
| R.m.s.d. bonds (Å) | 0.005 | 0.005 | 0.01 | 0.01 |
| R.m.s.d. angles (°) | 1.0 | 1.0 | 1.4 | 1.4 |
3. Results and discussion
3.1. Focal volume studies
The beam width was determined to be 1.6 ± 0.2 µm and the depth of field was 25 ± 5 µm. The theoretical diffraction-limited focal volume in air for this system is 1.4 × 22.5 µm. The small difference in the focal volume shows that the cryostream and the relatively turbid solid PEG sample do not have a large effect on imaging resolution.
3.2. Crystal location studies
A comparison of SHG and X-ray raster-scanned imaging is shown in Fig. 1 ▶ for protein crystals of human β2 adrenergic receptor fused to lysozyme from T4 phage (β2AR-T4L) in MiTeGen loops at cryogenic temperatures. Using a 10 µm minibeam, X-ray raster-scanned images were acquired over a time course of 10–15 min with a flux of 1.1 × 1010 photons s−1 to produce raster grids containing approximately 100 cells with 10 × 10 µm per cell, exposing crystals to ∼1 MGy of the estimated ∼10 MGy dose limit reported previously (Cherezov et al., 2009 ▶). (A 5 µm minibeam is also a routine option for raster scanning, but the time needed for rastering an equivalent sample area is increased by roughly fourfold.) The SHG images were acquired in about 45 s to produce 22 000 pixel images with 2 × 2 µm per pixel. Fig. 1 ▶ includes bright-field images, a false-color composite image of epi SHG, transmitted SHG and TPEF, and an image indicating the number of Bragg reflections in each pixel overlaid on the loop image acquired at the beamline. Six loops are shown, five of which contained protein crystals and one control that did not. The areas that produce strong SHG in transmission correlate well with areas of relatively strong protein diffraction. While in this study the full loop was imaged with both SHG and XRD raster imaging so that correlations could be shown, an operational crystal-centering algorithm would be different. A crystal could be located and its position recorded either relative to the loop center for nylon loops or relative to the fiducial marks on the polyimide loops. In order to solve a structure from the crystal, it will need to be located in all three dimensions so it would remain in the X-ray beam as the crystal is rotated, which would require additional SHG images to be obtained with the loop rotated 90°. The precision with which SHG imaging can locate crystals and the size of the crystals will determine whether XRD data can be collected centering based solely on the SHG data or whether a small-area XRD raster will still be required.
Figure 1.

Comparison of bright-field (top row) X-ray diffraction raster scanning for crystal centering (center row) and SONICC (bottom row) for four representative β2AR-T4L crystals within lipidic mesophase and a control with no protein crystal (rightmost images). In the top row, blue regions correspond to locations of protein-like diffraction overlaid on bright-field images. The bottom row represents composite images: red, transmitted SHG; blue, epi-detected SHG; green, two-photon excited fluorescence. Red regions correlate well with locations of protein crystals of size appropriate for diffraction analysis. Scale bar = 50 µm.
A nonzero TPEF background Stokes-shifted from the doubled frequency was observed for both of the loops and several of the proteins investigated. In general, luminescence varied significantly for crystals of different proteins, with some generating fairly strong visible-light TPEF when probed by an 800 nm source and others having only weak fluorescence. In unfavorable conditions, the TPEF was strong enough to interfere with SHG detection. Indeed, TPEF may also serve as a potential contrast mechanism for crystal detection, but without the crystal selectivity of SHG (both the crystals and the polyamide loops produced TPEF). In general, the luminescence from both the proteins and the loops was substantially suppressed by using wavelengths of >1 µm for excitation.
While scanning at incident wavelengths of >1 µm, neither MiTeGen nor Hampton Research nylon loops were noticeably affected by exposure to the laser. However, shorter wavelengths centered on 800 nm produced significant physical damage to the MiTeGen loops under normal imaging conditions (∼100 mW). Several explanations of this susceptibility to laser-induced damage were considered. Heating from one-photon absorption is not likely, as the loops are optically transparent in the visible and near-infrared. Contributions from high-order vibrational overtones that typically dominate near-IR absorption spectra would be expected to be reduced at 800 nm compared with 1 µm. Consequently, the most likely mechanism for damage to the MiTeGen loops was local heating from efficient two-photon absorption. Two-photon absorption of 800 nm and 1 µm light corresponds to equivalent one-photon excitation at 400 nm (purple) and 500 nm (green), respectively. The yellow color of the Mitogen loops is consistent with the presence of electronic states capable of absorption in the blue region of the spectrum, which may explain the marked decrease in susceptibility to damage at longer wavelengths.
Based on analysis of Fig. 1 ▶ and others, the locations of protein diffraction correlate well with SHG, particularly when detected in transmission. Fig. 1 ▶(c) shows areas where the intensities of SHG in the epi and transmission directions are comparable. Several explanations were considered, including crystalline lipid, fibrous contaminants and protein nanocrystals too small to produce detectable diffraction from the highly attenuated beam used during rastering. These points are tentatively attributed to areas containing protein crystals or other ordered material too small to detect by X-ray diffraction under the conditions used in this experiment (e.g. <200 nm). The coherent nature of SHG can result in marked differences in efficiency in the transmitted versus epi detected signals depending on the nature of the SHG-active source. For large samples (larger than a few micrometres), SHG is expected to be significantly stronger in the transmission direction owing to the difference in coherence lengths for these two directions. In brief, SHG of samples longer than the depth of field scales inversely with the sum of the fundamental and SHG refractive indices in the epi direction and the difference in the refractive indices in the transmission direction. Based on the measured coherence lengths for collagen, the forward coherence length for proteins can be expected to be ∼7 µm, while the backward coherence length is ∼100 nm (Lacomb et al., 2008 ▶). Consequently, crystals larger than about 100–200 nm will generally produce stronger SHG in transmission, while crystals less than about 150 nm in thickness will produce comparable SHG in both the forward and backward directions, where the difference in crystalline order would scale in both directions equally. Combining SHG measurements in both the epi and transmission directions provides unique additional information that is inaccessible from measurements of only one direction, although at the expense of additional instrumental complexity.
Although these results are encouraging, significant effort is still required before SHG imaging can be used in practice for high-throughput crystal centering to guide synchrotron XRD. Firstly, these initial studies were performed in a two-step process in which the loops were pre-screened by SHG imaging at Purdue and then characterized by XRD at the APS. Any mismatch in positioning between the two systems would introduce errors in the reliability of locating a crystal for diffraction. Routinely achieving 1–2 µm absolute accuracy in crystal position may be challenging in such a two-step approach. An alternative option would be to perform SHG measurements directly on samples positioned with a goniometer at a beamline. However, the engineering challenges associated with the design and construction of such a system are not trivial to overcome. The instrument used to acquire the images described here occupied a four foot by ten foot laser table. Significant reductions in size, particularly immediately adjacent to the sample, would be necessary for practical implementation given the severe space constraints in most robotic-enabled synchrotron diffraction apparatuses. The benefit of data collection in transmission also represents a nontrivial practical constraint. Efforts are under way to develop these capabilities; if these engineering challenges can be successfully addressed for either design (pre-screening or in situ), then SHG and TPEF may potentially help to address a major throughput bottleneck in protein structure determination by synchrotron XRD.
3.3. Damage studies
Measurements designed to assess the impact of extended ultrafast laser exposure on crystal and diffraction quality are shown in Fig. 2 ▶. The crystals selected for the damage analysis were generally known to diffract to high resolution in order to assess the presence of subtle structural changes within the proteins. Myoglobin was selected as a potentially challenging protein by nature of the color center of the heme group, which is capable of significant two-photon absorption, and thaumatin was chosen for the presence of disulfide bonds. The study sought to determine whether evidence of laser-induced damage could be detected in protein structures and electron-density maps by collecting data where one part of a crystal was exposed to an excessive imaging dose by SHG and the other part of the crystal was unexposed. For both the myoglobin and thaumatin protein crystals the unit cells and mosaicities showed no significant deviations between laser-exposed and unexposed data sets. For both laser-exposed and unexposed myoglobin, σA-weighted 2|F o| − |F c| maps were well defined for all residues and the hemes and their water ligands, with the exception of some amino-acid side chains where the density was relatively poor or partially lacking (Ile21, Asp44, Lys47, His48, Lys50, Glu54, Lys62, Leu89, Lys96 and Lys98). Thr95 side chains were modeled as two conformers in both structures. A water molecule was modeled as a ligand of the heme iron in each case. For both the ‘no laser’ structure and the ‘plus laser’ structure, 114 waters and three sulfate ions were added as solvent (all but seven waters were modeled in the same positions). The two protein structures superpose with insignificant deviations. No systematic differences in their electron-density maps were observed, as illustrated in Fig. 3 ▶, which shows conformations and electron density for heme as well as for representative Trp and Tyr residues. Similarly, for thaumatin the structures refined against diffraction data obtained from laser-exposed and unexposed regions of the crystal are essentially identical. Only minor differences are observed for the side chains of residues Asn46, Tyr57, Arg76, Arg79 and Lys189, all of which have poorly defined or incomplete electron density and are interpreted as being poorly ordered in both structures. Good electron density is observed for all eight disulfide linkages and shows no significant differences between laser-exposed and unexposed data sets (Fig. 4 ▶).
Figure 2.
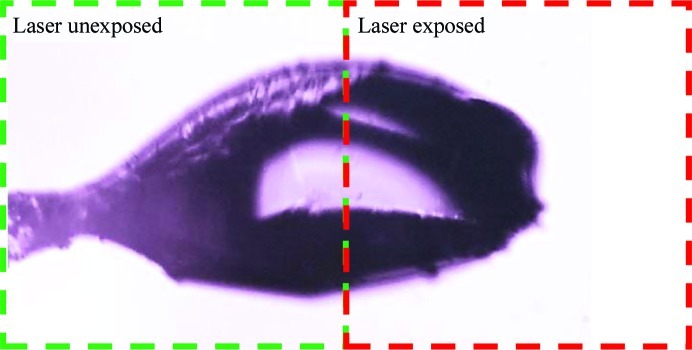
Representative bright-field image illustrating the approach taken for assessment of laser-induced structural perturbations (shown here for thaumatin). In each study, two structures were solved from exposed and unexposed regions of each single crystal.
Figure 3.

Myoglobin. (a) Heme (iron, brown; oxygen, red; nitrogen, blue), no laser (green molecule, cyan map); (b) heme, plus laser (cyan molecule, magenta map); (c) heme, no laser; (d) heme, plus laser; (e) Trp7, no laser; (f) Trp7, plus laser; (g) Tyr146, no laser; (h) Tyr146, plus laser. Figures were made with PyMOL (DeLano, 2002 ▶).
Figure 4.

Thaumatin disulfide Cys134–Cys145: (a) no laser, (b) plus laser.
Collectively, these results suggest that the laser fluences required for SHG imaging are well below the threshold required to induce readily detectable crystal damage under cryogenic conditions as assessed by synchrotron XRD. It should be noted that these studies were limited to measurements on crystals previously prepared and maintained under cryogenic conditions; therefore, inferences regarding damage to crystals still contained within the mother liquor at room temperature should be made cautiously.
Although little has been reported previously on the tolerance of protein crystals to optical exposure to ∼1000 nm light, this wavelength falls within the near-infrared biological transparency window (Weissleder, 2001 ▶; Frangioni, 2003 ▶), sandwiched by visible light scattering and absorption at the blue end, and by vibrational overtone absorption at the red end. Unlike UV or X-rays, the energy of IR photons is too low to break covalent bonds. Although little is known about the two-photon absorption cross-sections of purified proteins to near-IR incident light, proteins that lack obvious color centers at the doubled frequencies can also be reasonably expected to exhibit negligible two-photon absorption. Three-photon and four-photon absorption could supply sufficient energy to induce covalent-bond breaking in principle. However, in practice the efficiency of multi-photon excitation diminishes significantly with increasing order, i.e. the number of photons involved. Given the relatively low numerical aperture used in these studies to ensure a long working distance, it is reasonable to assume that three and four-photon processes do not contribute to sample damage. These expectations are consistent with the results of the damage studies, in which extended optical exposure produced a negligible impact on the resulting solved structures, even in the case of myoglobin, which contains a strong visible-wavelength color center.
In the absence of direct damage through one-photon or two-photon absorption, indirect damage from local heating arising from weak one-photon or two-photon absorption was also considered. High vibrational overtones and combination bands can result in weak absorption of 1 µm radiation. However, fast beam scanning with a resonant scanner (in this case 8 kHz) suppresses the rapid localized heating that can potentially arise with tightly focused beams. Energy deposition can also arise from nonradiative relaxation following multi-photon absorption. In the conditions under which these data were acquired (e.g. using a long working-distance, low numerical aperture objective) three-photon absorption is negligible, such that heat deposition to the sample was likely to be dominated by two-photon absorption. Conservatively assuming about ten multi-photon absorption events per detected SHG photon, the SHG flux of <103 photons per second corresponds to an equivalent of only a few femtowatts of deposited visible-light energy. For uncolored proteins lacking chromophores capable of absorption of visible wavelengths, the rate of energy deposition can reasonably be expected to be substantially lower. The absence of detectable perturbation in structures generated from myoglobin crystals was particularly encouraging given the presence of a strong color center in the protein that is capable of two-photon absorption. The absence of detectable structural changes indicates that heat can be transferred rapidly enough to maintain cryogenic temperatures even in proteins that are capable of undergoing significant two-photon absorption.
4. Conclusion
Here, the first steps are described towards the reliable automated positioning of protein crystals based on SHG imaging prior to synchrotron X-ray diffraction, driven by the growing desire for high-throughput automated diffraction data collection. As X-ray beam diameters continue to decrease, the size of the protein crystals amenable to diffraction analysis also decreases, challenging the reliability of existing routine methods used for automated centering. In this study, SHG microscopy was explored for the production of high-contrast crystal-specific images that may be more amenable to image-analysis algorithms targeting automated positioning. A good correlation was observed between the protein-crystal locations identified by SHG imaging and by X-ray diffraction. The degree of perturbation of SHG on the diffraction quality was assessed by comparison of structures produced from exposed and unexposed portions of single crystals. Crystal structures determined from proteins that absorb strongly in the visible (i.e. that are most likely to be susceptible to laser-induced perturbations) and for proteins that contain disulfide bonds showed no detectable evidence of damage, even under laser powers that were much greater than those typically used for SHG imaging. Conversely, even carefully planned X-ray raster strategies for samples that are not especially prone to X-ray damage can introduce crystals to ∼10% of the radiation-dose limit. SHG microscopy enabled relatively high spatial resolution (2 × 2 µm with exposure times of 2 ms per pixel) compared with X-ray raster scanning (typically 10 × 10 µm at 2 s per pixel) (Cherezov et al., 2009 ▶). Furthermore, previous studies by SHG microscopy demonstrating the detection of protein crystals <1 µm2 and one molecular layer thick (Gualtieri et al., 2011 ▶) and the routine detection of crystals 5–10 µm in size (Kissick et al., 2010 ▶) suggest that a further lowering of the detection limits may be expected under favorable conditions. However, on these size scales the uncertainties in absolute positioning are likely to be dominated by the subtle differences in sample positioning between the two instrument platforms (SHG versus synchrotron XRD), suggesting the potential need to integrate the two systems.
Supplementary Material
PDB reference: thaumatin, 4dc5
PDB reference: 4dc6
PDB reference: myoglobin, 4dc7
PDB reference: 4dc8
Acknowledgments
Financial interest statement: two of the authors (GJS and DJK) are co-inventors of instrumentation designed for protein crystal detection using nonlinear optical imaging (US Patent 8030077). This technology has been licensed by Formulatrix for well-plate crystallization screening. DJK and GJS gratefully acknowledge support from NIH Grant No. R01GM103401 from the National Center for Research Resources. VC acknowledges support from the NIH Common Fund’s Structural Biology program (P50 GM073197). Results shown in this report are derived from work performed at Argonne National Laboratory at the Advanced Photon Source. GM/CA@APS has been funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Sciences (Y1-GM-1104). IMCA-CAT is supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman–Woodward Medical Research Institute. The Structural Biology Center at Argonne is operated by UChicago Argonne LLC for the US Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
References
- Aishima, J., Owen, R. L., Axford, D., Shepherd, E., Winter, G., Levik, K., Gibbons, P., Ashton, A. & Evans, G. (2010). Acta Cryst. D66, 1032–1035. [DOI] [PMC free article] [PubMed]
- Asanov, A. N., McDonald, H. M., Oldham, P. B., Jedrzejas, M. J. & Wilson, W. (2001). J. Cryst. Growth, 232, 603–609.
- Asherie, N., Jakoncic, J., Ginsberg, C., Greenbaum, A., Stojanoff, V., Hrnjez, B. J., Blass, S. & Berger, J. (2009). Cryst. Growth Des. 9, 4189–4198.
- Beitlich, T., Kühnel, K., Schulze-Briese, C., Shoeman, R. L. & Schlichting, I. (2007). J. Synchrotron Rad. 14, 11–23. [DOI] [PubMed]
- Bourgeois, D., Vernede, X., Adam, V., Fioravanti, E. & Ursby, T. (2002). J. Appl. Cryst. 35, 319–326.
- Brown, E., McKee, T., diTomaso, E., Pluen, A., Seed, B., Boucher, Y. & Jain, R. K. (2003). Nature Med. 9, 796–800. [DOI] [PubMed]
- Brunger, A. T. (2007). Nature Protoc. 2, 2728–2733. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Caffrey, M. (2011). Biochem. Soc. Trans. 39, 725–732. [DOI] [PMC free article] [PubMed]
- Campagnola, P. J. & Loew, L. M. (2003). Nature Biotechnol. 21, 1356–1360. [DOI] [PubMed]
- Chavas, L. M. G., Yamada, Y., Hiraki, M., Igarashi, N., Matsugaki, N. & Wakatsuki, S. (2011). J. Synchrotron Rad. 18, 11–15. [DOI] [PMC free article] [PubMed]
- Cherezov, V. (2011). Curr. Opin. Struct. Biol. 21, 559–566. [DOI] [PMC free article] [PubMed]
- Cherezov, V., Hanson, M. A., Griffith, M. T., Hilgart, M. C., Sanishvili, R., Nagarajan, V., Stepanov, S., Fischetti, R. F., Kuhn, P. & Stevens, R. C. (2009). J. R. Soc. Interface, 6, S587–S597. [DOI] [PMC free article] [PubMed]
- Cox, G., Kable, E., Jones, A., Fraser, I., Manconi, F. & Gorrell, M. D. (2003). J. Struct. Biol. 141, 53–62. [DOI] [PubMed]
- DeLano, W. L. (2002). PyMOL http://www.pymol.org.
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Fischetti, R. F., Xu, S., Yoder, D. W., Becker, M., Nagarajan, V., Sanishvili, R., Hilgart, M. C., Stepanov, S., Makarov, O. & Smith, J. L. (2009). J. Synchrotron Rad. 16, 217–225. [DOI] [PMC free article] [PubMed]
- Frangioni, J. V. (2003). Curr. Opin. Chem. Biol. 7, 626–634. [DOI] [PubMed]
- Fromme, P. & Spence, J. C. (2011). Curr. Opin. Struct. Biol. 21, 509–516. [DOI] [PMC free article] [PubMed]
- Gannaway, J. N. & Sheppard, C. J. R. (1978). Opt. Quantum Electron. 10, 435–439.
- Garman, E. F. & Nave, C. (2009). J. Synchrotron Rad. 16, 129–132.
- Gauderon, R., Lukins, P. B. & Sheppard, C. J. (1998). Opt. Lett. 23, 1209–1211. [DOI] [PubMed]
- Gualtieri, E. J., Guo, F., Kissick, D. J., Jose, J., Kuhn, R. J., Jiang, W. & Simpson, G. J. (2011). Biophys. J. 100, 207–214. [DOI] [PMC free article] [PubMed]
- Hanson, M. A., Cherezov, V., Griffith, M. T., Roth, C. B., Jaakola, V.-P., Chien, E. Y. T., Velasquez, J., Kuhn, P. & Stevens, R. C. (2008). Structure, 16, 897–905. [DOI] [PMC free article] [PubMed]
- Haupert, L. M. & Simpson, G. J. (2011). Methods, 55, 379–386. [DOI] [PMC free article] [PubMed]
- Hellwart, R. & Christen, P. (1974). Opt. Commun. 12, 318–322.
- Hilgart, M. C., Sanishvili, R., Ogata, C. M., Becker, M., Venugopalan, N., Stepanov, S., Makarov, O., Smith, J. L. & Fischetti, R. F. (2011). J. Synchrotron Rad. 18, 717–722. [DOI] [PMC free article] [PubMed]
- Höfer, N., Aragão, D., Lyons, J. A. & Caffrey, M. (2011). Cryst. Growth Des. 11, 1182–1192. [DOI] [PMC free article] [PubMed]
- Holton, J. M. & Frankel, K. A. (2010). Acta Cryst. D66, 393–408. [DOI] [PMC free article] [PubMed]
- Kissick, D. J., Gualtieri, E. J., Simpson, G. J. & Cherezov, V. (2010). Anal. Chem. 82, 491–497. [DOI] [PMC free article] [PubMed]
- Lacomb, R., Nadiarnykh, O., Townsend, S. S. & Campagnola, P. J. (2008). Opt. Commun. 281, 1823–1832. [DOI] [PMC free article] [PubMed]
- Lavault, B., Ravelli, R. B. G. & Cipriani, F. (2006). Acta Cryst. D62, 1348–1357. [DOI] [PubMed]
- Luft, J. R., Wolfley, J. R. & Snell, E. H. (2011). Cryst. Growth Des. 11, 651–663. [DOI] [PMC free article] [PubMed]
- Madden, J. T., DeWalt, E. L. & Simpson, G. J. (2011). Acta Cryst. D67, 839–846. [DOI] [PMC free article] [PubMed]
- Maurus, R., Bogumil, R., Nguyen, N. T., Mauk, A. G. & Brayer, G. (1998). Biochem. J. 332, 67–74. [DOI] [PMC free article] [PubMed]
- Meents, A., Gutmann, S., Wagner, A. & Schulze-Briese, C. (2010). Proc. Natl Acad. Sci. USA, 107, 1094–1099. [DOI] [PMC free article] [PubMed]
- Nanao, M. H. & Ravelli, R. B. (2006). Structure, 14, 791–800. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Padayatti, P., Palczewska, G., Sun, W., Palczewski, K. & Salom, D. (2012). Biochemistry, 51, 1625–1637. [DOI] [PMC free article] [PubMed]
- Pothineni, S. B., Strutz, T. & Lamzin, V. S. (2006). Acta Cryst. D62, 1358–1368. [DOI] [PubMed]
- Sanishvili, R., Nagarajan, V., Yoder, D., Becker, M., Xu, S., Corcoran, S., Akey, D. L., Smith, J. L. & Fischetti, R. F. (2008). Acta Cryst. D64, 425–435. [DOI] [PMC free article] [PubMed]
- Sgarbossa, A., Lenci, F., Bergamini, E., Bizzarri, R., Cerbai, B., Signori, F., Gori, Z. & Maccheroni, M. (2003). Biogerontology, 4, 379–385. [DOI] [PubMed]
- Shen, Y., Markowicz, P., Winiarz, J., Swiatkiewicz, J. & Prasad, P. N. (2001). Opt. Lett. 26, 725–727. [DOI] [PubMed]
- Stepanov, S., Hilgart, M., Yoder, D. W., Makarov, O., Becker, M., Sanishvili, R., Ogata, C. M., Venugopalan, N., Aragão, D., Caffrey, M., Smith, J. L. & Fischetti, R. F. (2011). J. Appl. Cryst. 44, 772–778. [DOI] [PMC free article] [PubMed]
- Stoller, P., Celliers, P. M., Reiser, K. M. & Rubenchik, A. M. (2003). Appl. Opt. 42, 5209–5219. [DOI] [PubMed]
- Vernede, X., Lavault, B., Ohana, J., Nurizzo, D., Joly, J., Jacquamet, L., Felisaz, F., Cipriani, F. & Bourgeois, D. (2006). Acta Cryst. D62, 253–261. [DOI] [PubMed]
- Wampler, R. D., Kissick, D. J., Dehen, C. J., Gualtieri, E. J., Grey, J. L., Wang, H.-F., Thompson, D. H., Cheng, J.-X. & Simpson, G. J. (2008). J. Am. Chem. Soc. 130, 14076–14077. [DOI] [PMC free article] [PubMed]
- Wanapun, D., Kestur, U. S., Kissick, D. J., Simpson, G. J. & Taylor, L. S. (2010). Anal. Chem. 82, 5425–5432. [DOI] [PubMed]
- Weissleder, R. (2001). Nature Biotechnol. 19, 316–317. [DOI] [PubMed]
- Yi, J., Orville, A. M., Skinner, J. M., Skinner, M. J. & Richter-Addo, G. B. (2010). Biochemistry, 49, 5969–5971. [DOI] [PMC free article] [PubMed]
- Ying, J., Liu, F. & Alfano, R. R. (2000). Appl. Opt. 39, 509–514. [DOI] [PubMed]
- Yoo, K. M., Xing, Q. & Alfano, R. R. (1991). Opt. Lett. 16, 1019–1021. [DOI] [PubMed]
- Zheng, M.-L., Fujita, K., Chen, W.-Q., Duan, X.-M. & Kawata, S. (2011). J. Phys. Chem. C, 115, 8988–8993.
- Zoumi, A., Yeh, A. & Tromberg, B. J. (2002). Proc. Natl Acad. Sci. USA, 99, 11014–11019. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: thaumatin, 4dc5
PDB reference: 4dc6
PDB reference: myoglobin, 4dc7
PDB reference: 4dc8