

Abstract
The G coupled-protein receptor kinase 4 (GRK4) negatively regulates the dopaminergic system by desensitizing the dopamine-1-receptor (D1R). The expressional control of GRK4 has not been reported, but here, we show that the transcription factor c-Myc binds to the promoter of GRK4 and positively regulates GRK4 protein expression in human renal proximal tubule cells (RPTCs). Addition of phorbol esters (PMA) to RPTCs not only increased c-Myc binding to the GRK4 promoter, but also increased both phospho-c-Myc and GRK4 expression. The PMA-mediated increase in GRK4 expression was completely blocked by the c-Myc inhibitor, 10074-G5, indicating that GRK4 is downstream of phospho-c-Myc. The autocrine production of angiotensin II (Ang II) in RPTCs increased the phosphorylation and activation of c-Myc and subsequently GRK4 expression. 3-Amino-4-thio-butyl sulfonate (EC-33), an inhibitor of aminopeptidase A (APA), increased RPTC secretion of Ang II. EC-33 or Ang II increased the expression of both phospho-c-Myc and GRK4, which was blocked by 10074-G5. Blockade of the angiotensin II type 1 receptor (AT1R) with losartan decreased phospho-c-Myc and GRK4 expression. Both inhibition of c-Myc activity and blockade of AT1R restored the coupling of D1R to adenylyl cyclase (AC) stimulation in uncoupled RPTCs (uRPTCs) while PMA or Ang II caused the uncoupling of normally coupled RPTCs (nRPTCs). We suggest that the AT1R impairs D1R function via c-Myc activation of GRK4. This novel pathway may be involved in the increase in blood pressure in hypertension that is mediated by increased activity of the renin-angiotensin system and decreased activity of the renal dopaminergic system.
Keywords: angiotensin II, losartan, c-Myc, dopamine, promoters, cyclic AMP (cAMP), renin angiotensin system
INTRODUCTION
Blood pressure is maintained in the normal range, in part, by renal regulation of sodium balance. Under conditions of moderate sodium intake, over 50% of renal excretion is caused by the renal dopaminergic system.1 Dopamine, secreted from the renal proximal tubule cell (RPTC), acts as an intracrine, autocrine, and paracrine hormone in the renal proximal tubule (RPT) and other nephron segments to stimulate dopamine receptors (D1R, D2R, D3R, D4R, and D5R).2 Simultaneous stimulation of D1R and D5R with D1-like agonists leads to an increase in cAMP accumulation through adenylyl cyclase while stimulation of D2R, D3R, and D4R decreases adenylyl cylase activity.3 Stimulation of the D1R and D5R receptors leads to natriuresis through inhibition of sodium/hydrogen exchanger 3 (NHE3) and the sodium-potassium pump NaKATPase.1, 3–5
The G protein-coupled receptor kinase 4 (GRK4) is one of the 7 member GRK family, which are serine-threonine kinases that are involved in the desensitization of G protein-coupled receptors (GPCRs). GRK4 phosphorylates the D1R to cause D1R internalization, thus increasing the reabsorption of sodium.6 The regulation of GRK4 expression is particularly important since increased renal protein expression of GRK4, in rodents, may be implicated in hypertension. High renal GRK4 expression inhibits the ability of the D1R to increase sodium excretion. In the spontaneously hypertensive rats (SHR), basal levels of GRK4 are 90% higher in the kidney than in their normotensive control, Wistar-Kyoto Rats (WKY).7 Selective renal cortical inhibition of GRK4 expression with GRK4 antisense oligonucleotides increased sodium excretion and ameliorated the hypertension in SHR. The hypertensive phenotype was further attenuated when renal AT1R expression was also inhibited. 8
The molecular mechanisms involved in the regulation of GRK4 are not well understood. Promoter analyses of GRK4 revealed that the 2125 base pairs immediately upstream of the coding sequence are involved in regulating GRK4 expression and are sufficient for high transcriptional activity in transfected human embryonic kidney (HEK) cells.9 When successive regions of the 2125 promoter region were deleted, transcriptional activity was decreased or increased in a cell-specific manner. However, little is known of the actual transcription factors that activate or repress the expression of GRK4.
c-Myc, a human homolog of the avian myelocytomatosis viral oncogene v-Myc, is involved in cancer progression in greater than 50% of all cancers.10 With respect to hypertension, c-Myc has been implicated in hypertrophy and fibrosis of the heart11, 12 and atherosclerosis13 but not yet in kidney-mediated hypertension. In Burkitt’s lymphoma cells overexpressing c-Myc due to chromosomal translocation, c-Myc was shown to bind to the promoter region of the GRK4.14 This finding suggests that c-Myc may be a transcription factor for GRK4. In addition, phorbol esters can transiently increase c-Myc mRNA expression in human myeloid leukemia cells15 and have been shown to increase promoter activity of GRK4 in transfected HEK-293 cells.9 In cells other than renal proximal tubule, phorbol esters have also been shown to increase phospho-c-Myc, suggesting that phospho-c-Myc may activate GRK4 expression. Therefore, we hypothesized that c-Myc could negatively regulate the dopaminergic system by activating GRK4 expression via its promoter and may be involved in the pathogenesis of hypertension.
MATERIALS AND METHODS
Cell Culture
We selected several cell lines from our collection of human RPTC isolated from normal tissue of nephrectomies under an institutional review board-approved protocol according to the Declaration of Helsinki, Title 45, Part 46, U.S. Code of Federal Regulations. Primary cell lines were used for most of our experiments except in experiments involving the real-time intracellular cAMP biosensor. Immortalized RPTC lines were used in the cAMP assay due to low transfection efficiency of primary cells (>5 transfected cells per imaging field of view are needed for reproducible results). Previously characterized primary RPTC lines used were 19, a D1R/AC-uncoupled RPTC (uRPTC), and 22, a normally D1R/AC-coupled RPTC (nRPTC).4, 16 RPTCs for the cAMP assay were immortalized with hTERT, using the methods documented by Kowolik et al and Wieser et al.17, 18 Culture conditions are detailed in the Online Data Supplement (please see http://hyper.ahajournals.org).
Drug Treatment
The cells were incubated with different combinations of the following drugs: phorbol 12-myristate 13-acetate (PMA, Sigma, 100 nmol/L), 10074-G5 (c-Myc inhibitor, Sigma, 30 μmol/L), losartan (LOS, Ang II type 1 receptor antagonist, Sigma, 10 μmol/L), Ang II peptide (Sigma, 10 nmol/L), 3-Amino-4-thio-butyl sulfonate (EC-33, 500 nmol/L), and a fenoldopam mesylate (FEN, Hospira Inc., 1μmol/L). EC-33 was used to inhibit aminopeptidase A (APA, which converts Ang II to Ang III). The duration of incubation and combinations of drugs are indicated on the figure legends.
Angiotensin II ELISA
The angiotensin II ELISA was performed using a commercial Angiotensin II EIA Kit (Cayman Chemical). Details provided in the Online Data Supplement.
Angiotensin and CD13 Confocal Microscopy
Cells were fixed and permeabilized and stained with B93 anti-angiotensinogen antibody (made in house) and anti-CD13 antibody. See details in Online Data Supplement.
Determination of cAMP Accumulation
We quantified the accumulation of cAMP using a FRET biosensor as previously reported.5 Before stimulating cells with FEN, a specific agonist for D1R and D5R, we incubated the RPTCs for 3 hours with various drugs: Ang II (10 nmol/L), LOS (10 μmol/L), 10074-G5 (30 μmol/L), PMA (100 nmol/L).
Chromatin Immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed using Human c-Myc ExactaChIP Chromatin IP Kit (R&D Systems) to isolate c-Myc-bound DNA promoters. DNA isolated from the assay was amplified with qPCR using primers specific to the GRK4 promoter: forward primer (5′ – TCC CAA GGA ACA AGG TTA CG – 3′) and reverse primer (5′ – CCT TCC GCG TTT ACT TTG AG – 3′). See Online Data Supplement for details.
Immunoblotting
Proteins in cellular lysates were detected using the following primary antibodies: H70 rabbit polyclonal antibody to GRK4 (1:200 dilution, Santa Cruz sc-13079); rabbit monoclonal antibody to phospho-c-Myc (1:1000 dilution, Epitomics1203-1); and monoclonal mouse antibody to β-actin (1:30,000 dilution, Sigma). See Online Data Supplement for details.
GRK4 Confocal Microscopy and Plasma Membrane Localization
GRK4 localized on the plasma membrane was measured using RPTCs that were labeled with biotin, fixed and permeabilized, then stained with anti-GRK4 H70 antibody (1:200 dilution). See details in the Online Data Supplement
RESULTS
We evaluated the possibility of c-Myc binding to the GRK4 promoter by performing a chromatin immunoprecipitation. Primary cultures of RPTCs were stimulated with either DMSO vehicle (VEH) control or PMA (100 nmol/L) for 3 hours. Since PMA is known to activate phospho-c-Myc through a PKC-mediated pathway, we hypothesized that PMA stimulation should increase the amount of c-Myc bound to the GRK4 promoter. Following formaldehyde cross-linking, we purified the DNA bound to c-Myc using a biotinylated anti-c-Myc antibody provided in the kit and detected the presence of GRK4 gene promoter sequences using qPCR. As shown in Figure 1, c-Myc was bound to the GRK4 promoter under basal, pre-stimulatory conditions (VEH, 897.52 copies ± 170.29; n=3). Following the 3 hour PMA treatment, c-Myc binding increased > 3-fold (3036.09 copies ± 100.25; n=3; p<0.05).
Figure 1.
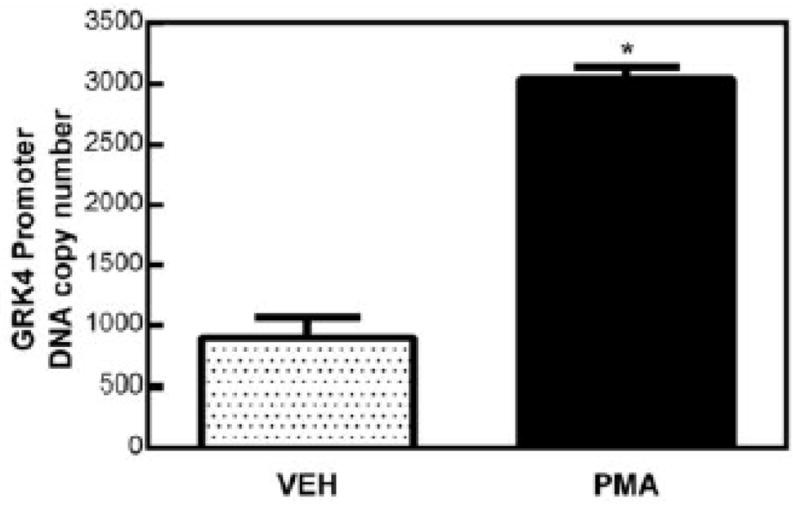
Chromatin immunoprecipitation of c-Myc binding to the GRK4 promoter. Cells were treated with PMA (100 nmol/L, 3 hr) or DMSO vehicle control (VEH). Degree of c-Myc binding was measured using qPCR of the GRK4 promoter. Copy number was greater for PMA-treated cells (3036.09 copies ± 100.29) than for VEH-treated cells (897.52 copies ± 170.29) indicating a higher degree of c-Myc binding in response to PMA (>3 fold increase over VEH, n=3, p<0.05).
We next determined if stimulation with PMA could increase total cellular phospho-c-Myc and GRK4 protein expression. Primary RPTCs treated with PMA (100 nmol/L, 24 hr) had a significant increase in phospho-c-Myc protein levels (127.78%±17.74; n=6; p<0.001) compared to the vehicle control (Figure 2a).
Figure 2.

(a) The expression of phospho-c-Myc was measured by western blot and graphed as the ratio of cellular actin after treatment with PMA (100 nmol/L, 24 hr) or DMSO vehicle (VEH). Phospho c-Myc expression was increased in response to PMA (127.78% ± 17.74, n=6, p<0.001). (b) Western blot analysis showing GRK4 expression was increased (40.51% ± 5.71, n=12, p<0.001) with PMA (100 nmol/L, 24 hr) and was blocked by the c-Myc inhibitor 10074-G5 (30 μmol/L, 24 hr). (+) control lane indicates lysates of HEK cells that have been transfected with a constitutively expressed GRK4. We used this lysate to confirm that the western blot bands we analyzed were GRK4.
Figure 2b shows that GRK4 protein expression also increased after 24 hour incubation with 100 nmol/L PMA (40.51%±5.71; n=12; p<0.001). Co-incubation of the c-Myc inhibitor, 10074-G5 (30 μmol/L, 24 hr), along with PMA blocked this increase, restoring GRK4 expression to basal levels. 10074-G5 is a compound that prevents the binding of c-Myc to its partner, Max, so that it cannot initiate transcription of target genes; 10074-G5, by itself had no effect on GRK4 expression. These results suggest that phospho-c-Myc is in the regulatory pathway for increased GRK4 expression.
We then investigated whether or not the AT1R may act through phospho-c-Myc to regulate GRK4. We first wanted to verify whether an autocrine Ang II signal existed in our primary RPTCs by quantifying Ang II levels in RPTC supernatants. Primary cultures of RPTCs were incubated with DMSO, the vehicle which served as a control for EC-33 (APA inhibitor). Supernatants were collected 3 hours after stimulation (Figure 3). We used EC-33 to block the activity of APA and therefore, the conversion of Ang II to Ang III. Under non-stimulatory conditions, cells treated with the DMSO vehicle control (VEH) showed an increase in the concentration of Ang II in the supernatants after 3 hours compared to serum free media (SFM) that was not introduced to cells (1.01pg/mL ± 0.036; n=6; p<0.0001), suggesting Ang II production by human RPTCs in primary culture.
Figure 3.

ELISA for Ang II. Cells were treated with EC-33 (500 nmol/L) or DMSO vehicle (VEH) for 3 hours. The level of Angiotensin II increased after three hours in VEH-treated cells compared to serum free media only (SFM) by 1.01pg/mL ± 0.036, n=6, #p<0.0001), and in EC-33 treated cells versus SFM (1.46pg/mL ± 0.050, n=6, #p<0.0001). Angiotensin II also increased in EC-33 treated cells compared to VEH treated cells (0.45pg/mL± 0.050; n=6; *p<0.0005)
Cells treated with EC-33 for 3 hours also showed a marked increase in the levels of Ang II in the supernatant relative to SFM (1.45pg/mL ± 0.050; n=6; p<0.0001). In addition, cells treated with EC-33 for 3 hours also showed a further increase in the concentration of Ang II compared to VEH (0.45pg/mL ± 0.050; n=6; p<0.0005), indicating that EC-33 is inhibiting the conversion of Ang II to Ang III. To further substantiate our findings, we stained our RPTCs for angiotensinogen protein (Figure S2), and found that all of our cells contain angiotensinogen protein in varying amounts, which can then be utilized for production of autocrine Ang II. Our results are consistent with the findings of Kamiyama et al. who found that in mouse19 and rat20 renal proximal tubule cells, the S1, S2 and S3 segments contain angiotensinogen protein.
After confirming the presence of Ang II production in primary RPTC culture, the cells were treated with Ang II (10 nmol/L), EC-33 (500 nmol/L), or a combination of both to determine if the AT1R could be stimulated with exogenously added or endogenously produced Ang II to affect phospho-c-Myc and GRK4 levels. Cells treated with Ang II showed an increase in the protein levels of phospho-c-Myc (86.86%± 15.21; n=6; p<0.01) (Figure 4a). These results are consistent with the findings of Naftilan et al., who observed an increase in the mRNA transcript of c-Myc which persisted for 6 hours after stimulation with Ang II (10 nmol/L).21 Figure 4b shows GRK4 protein levels increasing as well (43.63%± 10.11; n=6; p<0.05) but returning to VEH values when co-treated with the c-Myc inhibitor 10074-G5. Cells treated with EC-33 (500 nmol/L, 3 hr), had a similar increase in protein levels of both phospho-c-Myc (75.00%± 6.43; n=6; p<0.0001) (Figure 4a) and GRK4 (33.84%± 6.16; n=6; p<0.005) (Figure 4b). The combination of Ang II and EC-33 also caused phospho-c-Myc to increase by 79.54% ± 12.01 (n=6; p<0.001, Figure 4a) and GRK4 to increase by 34.12% ± 4.44 (n=6; p<0.05, Figure 4b). GRK4 levels were not altered by co-incubation of both EC-33 and 10074-G5. Treatment of primary RPTCs with EC-33 yielded the same effects as treatment with Ang II, suggesting that the effects were due to autocrine Ang II produced by RPTCs. We tested this hypothesis by inhibiting AT1R with losartan (an AT1R antagonist) (10 μmol/L, 24 hr). The expression of phospho-c-Myc (21.70% ± 4.10; n=6; p<0.05), as well as GRK4 (18.49%± 3.83; n=15; p<0.005) was reduced by losartan (Figure 5a and 5b respectively). These results further substantiated our findings suggesting human RPTCs are capable of producing autocrine Ang II that acts on AT1R to increase phospho-c-Myc and GRK4 expression.
Figure 4.

(a) Phospho-c-Myc expression was increased in response to EC-33 (500 nmol/L) by 75.00% ± 6.43 (n=6; p<0.0001) and ANG II (10 nmol/L, angiotensin receptor agonist) by 86.86% ± 15.21 (n=6; p<0.005). The combination of Ang II and EC-33 caused phospho-c-Myc to increase by 79.54% ± 12.01 (n=6; p<0.001). There was no synergistic activity between the combination of EC-33 and Ang II indicating that either agonist maximally stimulated phospho c-Myc levels. (b) GRK4 expression in response to EC-33, Ang II, and 10074-G5. The expression of GRK4 was increased by EC-33 (33.84% ± 6.16; n=6; p<0.005), Ang II (43.63% ± 10.11; n=6; p<0.05) and a combination of Ang II and EC-33 (34.12% ± 4.43; n=6; p<0.05). GRK4 expression was not increased with 10074-G5 alone or in combination with Ang II or all three agonists indicating that 10074-G5 blocked the GRK4 agonist effects of EC-33 and Ang II.
Figure 5.

(a) Phospho-c-Myc expression was decreased (21.70% ± 4.10; n=6; p<0.05) in response to losartan (10μM) (AT1R receptor antagonist) for 24 hours. (b) GRK4 expression decreased (18.49%± 3.82; n=15; p<0.005) in response to Losartan (10μM, 24 hours).
Using confocal microscopy, we determined that changes in GRK4 expression were not associated with changes in GRK4 plasma membrane localization (Figure S1). Treatment of cells with Ang II, losartan, 10074-G5, Ang II plus losartan, and Ang II plus 10074-G5 did not alter the ratio of GRK4 at the plasma membrane to GRK4 in the cytoplasm.
Previously, we have shown that in uRPTC, the D1R is uncoupled to AC and cannot cause a cAMP increase in response to FEN. This uncoupling is, in part, due to higher GRK4 activity, which can phosphorylate and densensitize the D1R, preventing it from reaction with FEN. Inhibition of GRK4 by siRNA in uRPTC, restored D1R coupling to AC and caused an increase in cAMP in response to FEN.5 We, therefore, wanted to determine if we could re-couple the D1R to AC in uRPTCs by inhibiting c-Myc to inhibit GRK4. Since we hypothesized that c-Myc positively regulates GRK4, we expected that an inhibition of c-Myc would also inhibit GRK4 expression, leading to a reversal of the uncoupled phenotype in uRPTCs. The cAMP response was measured using a cAMP FRET biosensor, ICUE3. Because we had to transiently transfect our cells with ICUE3 and primary cells do not have a high transfection efficiency or reproducibility, we used immortalized cells for this experiment.
Figure 6 shows lack of a cAMP accumulation upon FEN (1 μmol/L, 15 min) stimulation in uRPTCs (i19). However, an increase in cAMP accumulation in response to FEN was observed for nRPTCs (i22) (22.45% ± 17.89; n=14; p = 0.0004), compared to the FEN-induced response in uRPTCs. uRPTCs were then pre-treated with losartan or 10074-G5 before FEN stimulation. Either losartan (10 μmol/L, 3 hr) or 10074-G5 (30 μmol/L, 3 hr) pre-treatment completely rescued the uncoupling of AC in uRPTCs, causing a significant increase in intracellular cAMP levels when compared to uRPTCs that were not pre-treated. cAMP accumulation increased 17.33% ± 3.53(n=28; p <0.0001) for losartan-treated uRPTCs and 16.21% ± 0.97 (n=47; p<0.005) for 10074-G5-treated cells. FEN-stimulated cAMP accumulation was rescued to 98.7% ± 1.48 (losartan) and 97.9% ± 0.79 (10074-G5) of nRPTC levels and not significantly different from FEN-stimulated cAMP accumulation in nRPTCs. These data suggest that inhibiting c-Myc may provide an alternative mechanism to inhibit GRK4 and recouple cells to AC. In contrast, pre-treatment of nRPTCs with Ang II or PMA induced the expression of the uncoupled phenotype (FEN-stimulated AC). FEN-induced cAMP accumulation decreased by 24.40% ± 3.75 in Ang II-treated cells (n=16), and 18.18% ± 2.00 in PMA-treated cells (n=14, p<0.001). These data suggest that over-active c-Myc may lead to an increased GRK4 protein expression and uncoupling of D1R from AC.
Figure 6.

The lack of response to fenoldopam (FEN)-stimulated cAMP accumulation in uRPTC can be reverted to normal by incubation with the AT1R antagonist, losartan (LOS), or the c-Myc inhibitor 10074-G5. Intracellular cAMP accumulation was measured in real-time using a cAMP FRET biosensor, ICUE3. FEN stimulation (FEN, 1 μmol/L, 15 min) increased cAMP in nRPTC (*P<0.001 vs VEH; n=14) but not in uRPTC. Pretreatment of uRPTC with LOS (10 μmol/L, 3 hr) completely reverted the FEN stimulated cAMP accumulation of uRPTC to normal (#P<0.001 vs uRPTC FEN; n=28). Similarly, pre-treatment of uRPTC with the c-Myc inhibitor, 10074-G5 (30 μmol/L, 3 hr) also completely reverted the FEN stimulated cAMP accumulation of uRPTC to normal (#P<0.001 vs uRPTC FEN; n=47). Neither LOS nor 10074-G5 pre-stimulation significantly changed the response of nRPTC to FEN. Pre-stimulation of nRPTC with Ang II (Ang II, 10 nmol/L, 3 hr) caused the uncoupling of nRPTC (&P<0.001 vs VEH FEN, n=16). Similarly, pre-stimulation of nRPTC with PMA (100 nmol/L, 3 hr) also caused the uncoupling of nRPTC (&P<0.001 vs VEH FEN, n=14).
DISCUSSION
Stimulation of RPTCs with Ang II has shown c-Myc to be associated with increases in pro-inflammatory22 and pro-fibrotic23 signaling cascades mediating renal injury in many kidney disease states including hypertension. Loss of dopamine receptor function in the proximal tubule also leads to hypertension, including a mechanism involving an increased sensitivity of the RAS to angiotensin II.24, 25 Gene variants in members of the RAS pathway, leading to increased activity, have been demonstrated to be associated with hypertension: AGT rs2004776 (angiotensinogen) and ACE rs4305 (angiotensin converting enzyme).26 Ang II has also been shown to activate c-Myc, which is a transcription factor that activates many proliferative cellular pathways including renal proximal tubule cells.27 Ang II stimulation has also been associated with inactivation of the dopaminergic system in the kidney.24 PMA has been shown to activate c-Myc28 and has even been shown to increase the transcript of GRK4.9 In humans, single nucleotide polymorphisms in GRK4 are associated with hyperphosphorylation and inactivation of the D1R.29 Since the D1R has been shown to heterodimerize and negatively regulate the AT1R,30, 31 a negative feedback cycle may be initiated where lack of D1R signaling leads to loss of inhibition of AT1R, which in turn leads to an increase in c-Myc expression, GRK4 expression and further inhibition of D1R. An interruption of this negative feedback mechanism could lead to re-establishing D1R function.
Our previous studies have shown that the coupling of D1R to AC is tied to sodium transport in RPTCs. We showed that addition of FEN generated a cAMP response and led to decreased sodium transport in nRPTCs.5 Because RPTCs mainly reabsorb molecules from the lumen of the nephron, a decrease in sodium transport correlates to decreased sodium reabsorption and/or increased sodium excretion. We have also previously shown that uRPTCs fail to accumulate cAMP and fail to decrease sodium transport in response to FEN, suggesting greater sodium reabsorption and less sodium excretion.5
Our present study links c-Myc transcriptional regulation of GRK4 to coupling and possibly sodium transport. Importantly, we show that D1R can be uncoupled from AC stimulation with agents that activate c-Myc and increase expression of GRK4. Furthermore, both the c-Myc- induced D1R-AC uncoupling and innate D1R-AC uncoupling in RPTCs can be reversed to normal D1R-AC coupling by c-Myc inhibition. These experiments using nRPTCs and uRPTCs have identified a novel and critical signaling molecule that connects aberrant Ang II activation to D1R-AC uncoupling via c-Myc transcriptional control of GRK4. Although it would seem that using c-Myc inhibitors therapeutically may be ill advised for treatment of hypertension, the use of a dominant negative c-Myc construct in mice showed that they could nearly completely inhibit c-Myc activity in all tissues for extended periods of time, with minimal and reversible side effects.32 The use of a c-Myc inhibitor could likely be used at very low dosages, as many drugs tend to accumulate in the proximal tubule of the kidney. Thus, the inhibition of the c-Myc pathway may lead to a novel therapeutic modulation of GRK4 transcription and potentially hypertension.
PERSPECTIVES
In summary, we describe a mechanism whereby Ang II stimulation leads to the uncoupling of the D1R from AC via the c-Myc transcriptional activation of GRK4 (a kinase that inactivates the D1R) in human RPTCs. We also show that primary human RPTCs in culture synthesize and secrete Ang II and that blocking the conversion of Ang II to Ang III by addition of the APA inhibitor EC-33 leads to an increase in Ang II concentration, c-Myc activation, GRK4 transcriptional activation and D1R-AC uncoupling. Blockade of AT1R or inhibition of c-Myc prevents the Ang II-mediated uncoupling of D1R from AC. Addition of the diacylglycerol mimic PKC activator, PMA, also leads to stimulation of c-Myc and to D1R-AC uncoupling. This potentially deleterious phenotype is reversed by the addition of a c-Myc inhibitor. Inhibition of the AT1R or c-Myc inhibitor in RPTCs that are D1R-uncoupled leads to reversion of their uncoupled phenotype and enables the uRPTCs to regain the ability to stimulate AC upon D1R stimulation
Supplementary Material
Novelty and Significance.
What is new?
A novel role for c-Myc regulating GRK4 expression and thus modulating the renin angiotensin and dopaminergic systems
Autocrine Ang II feedback signal exists in cultured renal proximal tubule cells
Inhibition of c-Myc can restore the coupling defect in D1R AC uncoupled renal proximal tubule cells
What is relevant?
The blood pressure lowering effect of AT1R blockers may be mediated by the downregulation of c-Myc and GRK4
Novel compounds that inhibit c-Myc or GRK4 could be used to treat hypertension without the negative effects of general AT1R blockade
Summary.
This article is the first to show transcriptional regulation of GRK4 by c-Myc thus placing it as a key regulator of the renal renin angiotensin and dopaminergic systems
Acknowledgments
Helen E. McGrath for editorial assistance.
SOURCES OF FUNDING
NIH grants HL074940 and DK039308
Footnotes
DISCLOSURES
R.A.F was awarded US Patent (no 6,660,474) on “GRK mutants in essential hypertension,” assigned to Hypogen, Inc.
References
- 1.Pelayo JC, Fildes RD, Eisner GM, Jose PA. Effects of dopamine blockade on renal sodium excretion. Am J Physiol. 1983;245:F247–253. doi: 10.1152/ajprenal.1983.245.2.F247. [DOI] [PubMed] [Google Scholar]
- 2.Siragy HM, Felder RA, Howell NL, Chevalier RL, Peach MJ, Carey RM. Evidence that intrarenal dopamine acts as a paracrine substance at the renal tubule. Am J Physiol. 1989;257:F469–477. doi: 10.1152/ajprenal.1989.257.3.F469. [DOI] [PubMed] [Google Scholar]
- 3.Wang X, Villar VA, Armando I, Eisner GM, Felder RA, Jose PA. Dopamine, kidney, and hypertension: Studies in dopamine receptor knockout mice. Pediatr Nephrol. 2008;23:2131–2146. doi: 10.1007/s00467-008-0901-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gildea JJ, Israel JA, Johnson AK, Zhang J, Jose PA, Felder RA. Caveolin-1 and dopamine-mediated internalization of nakatpase in human renal proximal tubule cells. Hypertension. 2009;54:1070–1076. doi: 10.1161/HYPERTENSIONAHA.109.134338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gildea JJ, Shah I, Weiss R, Casscells ND, McGrath HE, Zhang J, Jones JE, Felder RA. Hk-2 human renal proximal tubule cells as a model for g protein-coupled receptor kinase type 4-mediated dopamine 1 receptor uncoupling. Hypertension. 2010;56:505–511. doi: 10.1161/HYPERTENSIONAHA.110.152256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rankin ML, Marinec PS, Cabrera DM, Wang Z, Jose PA, Sibley DR. The d1 dopamine receptor is constitutively phosphorylated by g protein-coupled receptor kinase 4. Mol Pharmacol. 2006;69:759–769. doi: 10.1124/mol.105.019901. [DOI] [PubMed] [Google Scholar]
- 7.Sanada H, Yatabe J, Midorikawa S, Katoh T, Hashimoto S, Watanabe T, Xu J, Luo Y, Wang X, Zeng C, Armando I, Felder RA, Jose PA. Amelioration of genetic hypertension by suppression of renal g protein-coupled receptor kinase type 4 expression. Hypertension. 2006;47:1131–1139. doi: 10.1161/01.HYP.0000222004.74872.17. [DOI] [PubMed] [Google Scholar]
- 8.Yatabe J, Sanada H, Midorikawa S, Hashimoto S, Watanabe T, Andrews PM, Armando I, Wang X, Felder RA, Jose PA. Effects of decreased renal cortical expression of g protein-coupled receptor kinase 4 and angiotensin type 1 receptors in rats. Hypertens Res. 2008;31:1455–1464. doi: 10.1291/hypres.31.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hasenkamp S, Telgmann R, Staessen JA, Hagedorn C, Dordelmann C, Bek M, Brand-Herrmann SM, Brand E. Characterization and functional analyses of the human g protein-coupled receptor kinase 4 gene promoter. Hypertension. 2008;52:737–746. doi: 10.1161/HYPERTENSIONAHA.108.114512. [DOI] [PubMed] [Google Scholar]
- 10.Buendia MA, Bourre L, Cairo S. Myc target mirs and liver cancer: Small molecules to get myc sick. Gastroenterology. 2012;142:214–218. doi: 10.1053/j.gastro.2011.12.023. [DOI] [PubMed] [Google Scholar]
- 11.Lee HG, Chen Q, Wolfram JA, Richardson SL, Liner A, Siedlak SL, Zhu X, Ziats NP, Fujioka H, Felsher DW, Castellani RJ, Valencik ML, McDonald JA, Hoit BD, Lesnefsky EJ, Smith MA. Cell cycle re-entry and mitochondrial defects in myc-mediated hypertrophic cardiomyopathy and heart failure. PLoS One. 2009;4:e7172. doi: 10.1371/journal.pone.0007172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bello Roufai M, Li H, Sun Z. Heart-specific inhibition of protooncogene c-myc attenuates cold-induced cardiac hypertrophy. Gene Ther. 2007;14:1406–1416. doi: 10.1038/sj.gt.3302995. [DOI] [PubMed] [Google Scholar]
- 13.de Nigris F, Balestrieri ML, Napoli C. Targeting c-myc, ras and igf cascade to treat cancer and vascular disorders. Cell Cycle. 2006;5:1621–1628. doi: 10.4161/cc.5.15.3138. [DOI] [PubMed] [Google Scholar]
- 14.Li Z, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, Ren B. A global transcriptional regulatory role for c-myc in burkitt’s lymphoma cells. Proc Natl Acad Sci U S A. 2003;100:8164–8169. doi: 10.1073/pnas.1332764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernstein SH, Kharbanda SM, Sherman ML, Stone RM, Kufe DW. Inhibition of protein kinase c is associated with a decrease in c-myc expression in human myeloid leukemia cells. FEBS Lett. 1991;294:73–76. doi: 10.1016/0014-5793(91)81346-a. [DOI] [PubMed] [Google Scholar]
- 16.Gildea JJ, Wang X, Shah N, Tran H, Spinosa M, Van Sciver R, Sasaki M, Yatabe J, Carey RM, Jose PA, Felder RA. Dopamine and angiotensin type 2 receptors cooperatively inhibit sodium transport in human renal proximal tubule cells. Hypertension. 2012;60:396–403. doi: 10.1161/HYPERTENSIONAHA.112.194175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kowolik CM, Liang S, Yu Y, Yee JK. Cre-mediated reversible immortalization of human renal proximal tubular epithelial cells. Oncogene. 2004;23:5950–5957. doi: 10.1038/sj.onc.1207801. [DOI] [PubMed] [Google Scholar]
- 18.Wieser M, Stadler G, Jennings P, Streubel B, Pfaller W, Ambros P, Riedl C, Katinger H, Grillari J, Grillari-Voglauer R. Htert alone immortalizes epithelial cells of renal proximal tubules without changing their functional characteristics. Am J Physiol Renal Physiol. 2008;295:F1365–1375. doi: 10.1152/ajprenal.90405.2008. [DOI] [PubMed] [Google Scholar]
- 19.Kamiyama M, Garner MK, Farragut KM, Kobori H. The establishment of a primary culture system of proximal tubule segments using specific markers from normal mouse kidneys. Int J Mol Sci. 2012;13:5098–5111. doi: 10.3390/ijms13045098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamiyama M, Farragut KM, Garner MK, Navar LG, Kobori H. Divergent localization of angiotensinogen mrna and protein in proximal tubule segments of normal rat kidney. J Hypertens. 2012;30:2365–2372. doi: 10.1097/HJH.0b013e3283598eed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naftilan AJ, Pratt RE, Dzau VJ. Induction of platelet-derived growth factor a-chain and c-myc gene expressions by angiotensin ii in cultured rat vascular smooth muscle cells. J Clin Invest. 1989;83:1419–1424. doi: 10.1172/JCI114032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruiz-Ortega M, Ruperez M, Lorenzo O, Esteban V, Blanco J, Mezzano S, Egido J. Angiotensin ii regulates the synthesis of proinflammatory cytokines and chemokines in the kidney. Kidney Int Suppl. 2002:S12–22. doi: 10.1046/j.1523-1755.62.s82.4.x. [DOI] [PubMed] [Google Scholar]
- 23.Chen J, Chen JK, Nagai K, Plieth D, Tan M, Lee TC, Threadgill DW, Neilson EG, Harris RC. Egfr signaling promotes tgfβ-dependent renal fibrosis. J Am Soc Nephrol. 2012;23:215–224. doi: 10.1681/ASN.2011070645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gildea JJ. Dopamine and angiotensin as renal counterregulatory systems controlling sodium balance. Curr Opin Nephrol Hypertens. 2009;18:28–32. doi: 10.1097/MNH.0b013e32831a9e0b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeng C, Jose PA. Dopamine receptors: Important antihypertensive counterbalance against hypertensive factors. Hypertension. 2011;57:11–17. doi: 10.1161/HYPERTENSIONAHA.110.157727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson AD, Newton-Cheh C, Chasman DI, Ehret GB, Johnson T, Rose L, Rice K, Verwoert GC, Launer LJ, Gudnason V, Larson MG, Chakravarti A, Psaty BM, Caulfield M, van Duijn CM, Ridker PM, Munroe PB, Levy D Consortium CfHaARiGE, Consortium GB Study WsGH. Association of hypertension drug target genes with blood pressure and hypertension in 86,588 individuals. Hypertension. 2011;57:903–910. doi: 10.1161/HYPERTENSIONAHA.110.158667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolf G, Neilson EG. Angiotensin ii induces cellular hypertrophy in cultured murine proximal tubular cells. Am J Physiol. 1990;259:F768–777. doi: 10.1152/ajprenal.1990.259.5.F768. [DOI] [PubMed] [Google Scholar]
- 28.Lindsten T, June CH, Thompson CB. Multiple mechanisms regulate c-myc gene expression during normal t cell activation. EMBO J. 1988;7:2787–2794. doi: 10.1002/j.1460-2075.1988.tb03133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Felder RA, Sanada H, Xu J, Yu PY, Wang Z, Watanabe H, Asico LD, Wang W, Zheng S, Yamaguchi I, Williams SM, Gainer J, Brown NJ, Hazen-Martin D, Wong LJ, Robillard JE, Carey RM, Eisner GM, Jose PA. G protein-coupled receptor kinase 4 gene variants in human essential hypertension. Proc Natl Acad Sci USA. 2002;99:3872–3877. doi: 10.1073/pnas.062694599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zeng C, Luo Y, Asico LD, Hopfer U, Eisner GM, Felder RA, Jose PA. Perturbation of d1 dopamine and at1 receptor interaction in spontaneously hypertensive rats. Hypertension. 2003;42:787–792. doi: 10.1161/01.HYP.0000085334.34963.4E. [DOI] [PubMed] [Google Scholar]
- 31.Khan F, Spicarová Z, Zelenin S, Holtbäck U, Scott L, Aperia A. Negative reciprocity between angiotensin ii type 1 and dopamine d1 receptors in rat renal proximal tubule cells. Am J Physiol Renal Physiol. 2008;295:F1110–1116. doi: 10.1152/ajprenal.90336.2008. [DOI] [PubMed] [Google Scholar]
- 32.Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, Evan GI. Modelling myc inhibition as a cancer therapy. Nature. 2008;455:679–683. doi: 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.