

Abstract
The major mammalian exonuclease TREX1 has been proposed to play a role in DNA repair and drug resistance. However, no cellular evidence substantiates this claim. Recent reports indicate TREX1’s involvement in autoimmunity. To further understand its role, we studied TREX1 expression and functionality in anticancer drug-treated tumor cells. We report that the expression and localization of TREX1 are cell-type dependent. Camptothecin and other DNA damaging agents induced both TREX1 protein and its mRNA in a dose- and time-dependent manner. Using a TREX1-inducible cell line, we performed clonogenic assays and found no change in sensitivity of the cells to the agents upon TREX1 induction, suggesting that TREX1 may not play a role in DNA repair or drug sensitivity. Nevertheless, TREX1 serves as a key enzyme in the degradation of DNA from dying cells leading to less cellular DNA. Ubiquitously expressed in normal tissues, TREX1 may act in degrading DNA in all cell types undergoing a dying process before phagocytosis occurs.
Keywords: Autoimmunity, camptothecin, DNA degradation, dying cells, TREX1
1. Introduction
TREX1 is the major mammalian 3′–5′ DNA exonuclease [1]. It excises bases from 3′ end of single- and double-stranded DNA with a preference for mismatched nucleotides [2–5]. TREX1 and its homolog TREX2 (with 44% homology, but lacks a unique C-terminal region present in TREX1 [3]) are members of the DnaQ family with three conserved exonuclease motifs. The crystal structures of homodimeric TREX1 and TREX2 reveal their structure-function correlations [6–8].
TREX1 and TREX2 may have DNA-editing function during DNA replication, or repair due to that TREX1 is able to edit 3′ mismatched bases or nucleotide analogs such as araCMP in reconstituted systems containing DNA pol α or β [2,5], whereas TREX2 could interact with DNA pol δ and increase its accuracy [9]. TREX1-null mice, however, did not show an increase in spontaneous mutation frequency or cancer incidence but displayed reduced survival and developed inflammatory myocarditis [10]. This observation suggests that TREX1 deficiency may lead to an inflammatory response through the activation of innate immunity by the accumulated nucleic acids.
Dominant mutations in human TREX1 gene mapped to chromosome 3p21 are linked to the development of autoimmune diseases including: (i) Aicardi-Goutières syndrome (AGS) [11–13], a severe neurological brain disease mimicking congenital viral infection with the features of demyelination and calcification of the basal ganglia through the elevated levels of interferon (IFN)-α (ii) Retinal vasculopathy and associated diseases such as migraine with cerebral leukodystrophy accompanied by visual loss, stroke and dementia [14,15], (iii) Systemic lupus erythematosus (SLE), a chronic inflammatory disorder of the internal organs characterized by the presence of antinuclear autoantibodies [16], and (iv) Familial chilblain lupus with ulcerating skin lesions in acral locations [17–19]. TREX1 in dominant mutations associated with autoimmune diseases is reported to show defective exonuclease acitivities on double-stranded DNA degradation [20]. Moreover, single nucleotide polymorphisms of TREX1 are associated with the development of autoantibodies in SLE patients [6].
Embryo fibroblast from TREX1-null mice was reported to accumulate single-stranded DNA species of 60–65 bases in the cytoplasm during DNA replication, accompanied by chronic ATM-dependent checkpoint activation [21], leading to the hypothesis that the accumulated single-stranded DNA could activate NF-κB pathway and the downstream inflammatory genes such as IFN-α via toll-like receptors (TLRs) including TLR9 within the endosomal compartment (also comprehensively reviewed in [22–24]). More recently, it was reported that TREX1’s deficiency leads to the accumulation of endogenous retroelement-derived DNA and the dysregulation of the IFN-stimulatory DNA pathway, which could be the major cause of autoimmunity in TREX1-null mice [25].
Furthermore, TREX1, but not TREX2, is found in an endoplasmic reticulum-associated complex (SET) and acts with NM23-H1 endonuclease to degrade DNA during granzyme A-mediated cell death. TREX1 is therefore implicated in the action of cytotoxic T and NK cells [26,27].
In this study, we addressed the role of TREX1 in anti-cancer drug-treated tumor cells. We first examined the expression and localization of TREX1 in different tumor cell lines followed by treatment with DNA damaging agents. Several drugs with different mechanisms of action were selected: camptothecin, a DNA topoisomerase (Topo) I inhibitor; etoposide, a DNA Topo II inhibitor; cisplatin and oxaliplatin, the platinum-based DNA-crosslinking compounds with anti-cancer activities; hydrogen peroxide (H2O2), a DNA oxidizer; doxorubicin, an anthracycline antibiotic with DNA-interacting capability used for cancer treatment; arsenite, an amphoteric oxide used to treat leukemia; as well as Ara C, gemcitabine and troxacitabine (L-OddC), the cytidine analogs with D- or L-configuration used in various carcinomas. We also developed a TREX1-inducible cell line to study the fate of cellular DNA following DNA damaging agents-induced cell death. We demonstrated that TREX1 acts as a key enzyme in the degradation of DNA in drug-treated dying cells.
2. Materials and methods
2.1. Reagents
CPT, cisplatin, oxaliplatin, arsenite, AraC, doxorubicin, etoposide and propidium iodide were purchased from Sigma (St. Louis, MO, USA). L-OddC was provided by Shire Biochem, Inc. (Laval, Quebec, Canada). Gemcitabine (Gemzar) was purchased from Eli Lilly (Indianapolis, IN, USA). H2O2 was purchased from J.T. Baker (Phillipsburg, NJ, USA). Hygromycin B and doxycycline were purchased from Clontech (Palo Alto, CA, USA).
2.2. Cell lines
The human cell lines used were either of lymphoid or epithelial tumor cells origin. KB, H9, HepG2 and RKO cell lines from nasopharyngeal carcinoma, T-cell lymphoid tumor cells, hepatocellular carcinoma and colorectal cancer cells, respectively, were employed for further studies. A detailed description is provided in the Supplementary information.
2.3. Cloning, expression of the human TREX1 protein, and generation of TREX1 monoclonal antibody
For the generation of TREX1 monoclonal antibody, human TREX1 encoding sequence was first cloned for the expression of TREX1 protein. As detailed in the Supplementary information, TREX1 protein was then used to immunize 4-week old BALB/c mice, following the procedures described previously [28]. The antibody was confirmed to recognize the N-terminal region of TREX1 and applied to the immunoblotting and immunoprecipitation assays.
2.4. Confocal microscopy and subcellular fractionation
The confocal microscopy analysis was performed as previously described [28]. For subcellular fractionation, different centrifugal forces were chosen as detailed in the Supplementary information.
2.5. Immunoblotting
The resolved proteins by SDS-PAGE were transferred to nitrocellulose membrane and detected by primary antibodies, followed by horseradish peroxidase-conjugated secondary antibodies. A detailed description of the antibodies used in this study is provided in the Supplementary information.
2.6. Immunoprecipitation and exonuclease activity assay
KB and RKO cells treated with different concentrations of CPT for 20 h were lysed by RIPA buffer (1× PBS, 2 mM DTT, 1 mM PMSF, 0.5 mM EDTA, 0.5% NP40, 0.005% SDS, and protease inhibitor cocktail). The cell lysates were subjected to immunoprecipitation by TREX1 mAb (1:100), followed by addition of protein A agarose (Invitrogen). The immunoprecipitated TREX1 protein was assayed for its exonuclease activity following the procedures as described [29]. Briefly, the reaction mixture (10 μl) contained 2.5 nM of 5′ γ-[32P]ATP labeled oligonucleotide 21mer (5′-GTGGCGCGGAGACTTAGAGAC-3′), 50 mM Tris-HCl, pH 8.0, 1 mM MgCl2, 1 mM DTT, 0.1 mg/ml BSA, and 2 μl of the immunoprecipitated TREX1 protein in appropriate enzyme dilution. Radiolabeled bands were visualized by autoradiography.
2.7. Quantitative real-time RT-PCR assay
Cellular RNA from different cell lines was harvested using the RNeasy Mini Kit (Qiagen). Primers and Taqman probe for TREX1 mRNA detection were designed using Beacon Designer (V.3.0) (Premier BioSoft) and the sequences are as follows:
5′-GGTGCCTTCTGTGTGGATAGC-3′ (sense), (nt 445–465)
5′-CTTCCTTGGGCCGTGTTCTG-3′ (anti-sense), (nt 525-506)
and 5′-TGCTTGCTCGCTCCAGGGCCTTCA-3′ (Taqman probe). (nt 476–499) The Taqman probe was modified at its 5′-end with 6-FAM and 3′-end with BHQ-1 (Biosearch Technologies, Inc, Novato, CA).
β-actin was used as an internal control. The sequences are as follows:
5′-ATTGCCGACAGGATGCAGAA-3′ (sense), (nt 925–944)
5′-GCTGATCCACATCTGCTGGAA-3′ (anti-sense), (nt 1074-1054)
and 5′-CAAGATCATTGCTCCTCCTGAGCGCA-3′ (Taqman probe). (nt 981–1006). The Taqman probe was modified at the 5′-end with TexasRed and 3′-end with BHQ-1. The iCycler iQ system (from Bio-Rad) and the Platinum qRT-PCR ThermoScript OneStep System (Invitrogen) were used for RNA detection.
2.8. Northern blot detection of TREX1 mRNA
Total RNA from KB and RKO cells treated with different concentrations of CPT for 14 h was isolated and subjected to Northern blot analysis. Using α-[32P]dCTP, a TREX1 probe was prepared by random labeling the full length TREX1-encoding fragment cleaved from the plasmid pET28a(+)-TREX1 (see Supplementary materials). RNA samples (30 μg each) were resolved by using 1.2% agarose gel and transferred to a nitrocellulose membrane followed by autoradiography according to the manufacturer’s instructions (Qiagen).
2.9. Establishment of a doxycycline-regulated RKO cell line with inducible expression of TREX1 protein
RKO cells grown in RPMI 1640 medium with 10% (tetracycline-free) FBS (Clontech) were transfected with pcDNA 6/TR vector (Invitrogen) and selected by 5 μg/ml blasticidin. The stable cell line was further transfected with the plasmid pcDNA 5/TO (Invitrogen) containing TREX1-encoding sequence and selected by 150 μg/ml hygromycin B. The level and homogeneity of protein expression were determined by immunoblot and confocal microscopy, respectively.
2.10. In situ DNA agarose gel electrophoresis assay
RKO/TREX1 cells (2×106 cells/well/6-well plate) were added with doxycycline overnight and then treated with different doses of CPT for 18 h. After treatment, the cells were washed with PBS, pelleted down by centrifugation at 300 ×g, and resuspended in 50 μl of sample buffer (1×TBE, 4 μl RNase A (10 mg/ml), 0.25% bromophenol blue, 0.25% xylene cyanol, 40% (w/v) sucrose) at room temperature for 30 min before being loaded onto a 1% horizontal agarose slab gel [30]. The area surrounding the wells for loading cells contained 2% SDS and 1 mg/ml proteinase K. After loading, the gel was run at 0.4 V/cm for 3 h followed by 3 V/cm for 48 h at 4°C, and stained with ethidium bromide.
2.11. Clonogenic assay
Clonogenic assay was performed as previously described [31]. RKO/TREX1 cells (400 cells/well/6-well plate) with or without added doxycycline overnight were exposed to serial dilutions of chemotherapeutic drugs for 24 h, and grown in fresh RPMI 1640 for 10 to 14 days. The cells were fixed and stained by 0.5% methylene blue in 50% ethanol for 2 h. The cloning efficiency for each drug was determined by colony counts.
2.12. Cellular DNA labeling by incorporation of [14C]thymidine
Cellular DNA was labeled as previously described [32]. Briefly, RKO cells (5×106 cells/10-cm2 plate) were labeled by 0.25 μCi/ml [14C]thymidine for 40 h, in which doxycycline was added together to induce TREX1 expression. At the end of labeling, the medium was replaced with phenol red-free RPMI 1640 containing 10% dialyzed FBS and the cells were chased with 1 μM cold thymidine for 2 h. The cells were then washed twice with PBS and treated with different doses of CPT for another 18, 24 or 30 h as indicated. Doxycycline was maintained at the same concentration. The harvested cell pellet and culture medium were subjected to 70% methanol treatment. The methanol-soluble fractions were analyzed using HPLC SAX column coupled with β-counter. Thymidine incorporation was normalized in a duplicate experiment by calculating the ratio of each sample, subjected to β-counter analysis, over its total volume. The s.d. was obtained by three independent experiments. The methanol-insoluble fractions were treated with 1 mg/ml proteinase K at 50°C overnight and subjected to 12% SDS-PAGE followed by autoradiogram.
2.13. Propidium iodide flow cytometric assay
The DNA content of RKO/TREX1 cells was analyzed by flow cytometry. Upon CPT treatment at 1,000 nM for 24 h RKO/TREX1 cells with or without added doxycycline were harvested, centrifuged at 300 g for 5 min, washed twice with PBS, and fixed in 100% methanol on ice for at least 1 h. The cells were then washed once with PBS and resuspended in 1 ml PBS containing 250 μg/ml RNase A (type I-A; Sigma-Aldrich) and propidium iodide (50 μg/ml). Flow cytometry was carried out on BD FACSCalibur and data was analyzed using Flow Jo software.
3. Results
3.1. TREX1 expression and localization
TREX1 mRNA is expressed in all human tissues using RT-PCR [33]. Whether TREX1 protein is concomitantly expressed with its mRNA is unknown. To study the expression of TREX1 protein, different tumor cell lines were examined by immunoblot. All the lymphoid tumor cells including B-cell lineage (BJAB, BL5, BL8, L5 and H1) and T-cell lineage (H9 and CEM) expressed TREX1 protein (Fig. 1A). In BJAB cells, an additional smaller protein of 30 kD was recognized. Among the epithelial tumor cells, TREX1 was expressed in nasopharyngeal (KB and C666) and cervical (HeLa), but not detected even by loading more (over 40 μg) of the cellular extracts in breast (MCF7), liver (Huh7 and HepG2) or colon (HCT8 and RKO) tumor cells (Fig. 1B). The lack of TREX1 expression in these cell lines suggests that TREX1 is not essential in at least some tumor cells of epithelial lineage.
Fig. 1.
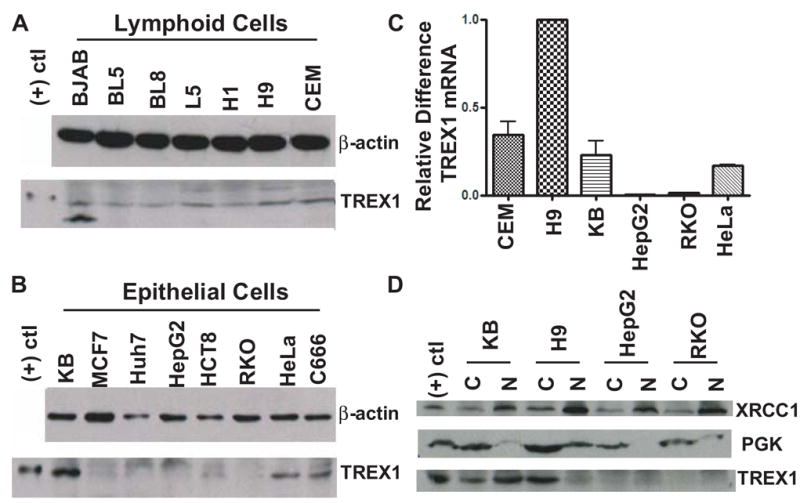
TREX1 expression and localization. (A) Immunoblot analysis of TREX1 expression in lymphoid cell lineage. β-Actin served as internal control. (B) TREX1 expression in epithelial cell lineage. (C) Detection of TREX1 mRNA using quantitative real-time RT-PCR. The s.d. shown on the bars were obtained from two independent experiments. (D) Localization of TREX1 using subcellular fractionation assay. XRCC1 and PGK served as markers of the nuclear and cytosolic fractions, respectively.
TREX1 mRNA was examined by quantitative real-time RT-PCR. KB and HeLa cells expressed similar levels of TREX1 mRNA, whereas CEM cells expressed approximately one third more TREX1 mRNA than KB and HeLa cells. H9 cells expressed the highest level, whereas HepG2 and RKO cells showed almost undetectable levels of TREX1 mRNA (Fig. 1C). This was further confirmed by semi-quantitative RT-PCR (Fig. S1). The relatively low expression of TREX1 mRNA in HepG2 and RKO cells explained why TREX1 protein was undetected. The difference in TREX1 mRNA expression among different tumor cell lines could be due to a genomic or epigenomic variation of the regulatory elements of TREX1 mRNA.
To study the localization of TREX1, subcellular fractionation and confocal microscopy were applied. In the fractionation study, XRCC1 and PGK served as nuclear and cytoplasmic markers, respectively, to monitor a separation of those two compartments. TREX1 was detected in both the cytosolic and nuclear fractions of KB cells. In contrast, TREX1 was primarily detected in the cytosolic fraction of H9 cells, but undetected in HepG2 and RKO cells (Fig. 1D). More TREX1 was found in the nuclear fraction of KB cells, but more was in the cytoplasm using confocal microscopy (compare Fig. 1D and Fig. S2). The localization of TREX1 in HeLa cells showed a similar pattern to KB cells using cellular fractionation and confocal microscopy (Fig. S3, A and C). Contrary to TREX1 being primarily found in the cytosolic fraction of H9 cells, in CEM cells, it was primarily localized in the nuclear fraction (Fig. S3, B). Using four cell lines (H9, KB, CEM and HeLa), we have demonstrated that the intracellular distribution of TREX1 is cell line specific. However, the reason why TREX1 shows different subcellular localizations between different cell types is not clear. Further studies in examining the distribution of TREX1 in other cells that express different amounts of TREX1 may provide more insight.
Whether less confocal detection of TREX1 in the nucleus came from the masking of TREX1 epitope through its association with nuclear proteins requires further investigation. The presence of TREX1 in the cytoplasm of KB and H9 cells confirmed that TREX1 is at least a cytoplasmic protein [26] and that the cellular localization of TREX1 is as a minimum dependent on tumor-cell types.
3.2. CPT induced both TREX1 protein and its mRNA
We investigated the effect of camptothecin (CPT) on the expression of TREX1 protein using different cell lines. TREX1 was induced by CPT in a dose-dependent manner in KB cells at concentrations higher than the IC50 value (less than 10 nM) (Fig. 2A). The increases of TREX1 were calculated as 2.4-, 2.6-, and 4.2-fold for 100, 300 and 1,000 nM CPT, respectively. KB100 and KB100R cells with 32- and 2.5-fold more CPT resistance than KB parental cells also expressed induced TREX1 protein upon CPT treatment (Fig. S4, A). TREX1 mRNA increased as well in KB and KB-derived cells with CPT treatment in a dose-dependent manner (Fig. S4, B). Other cell lines such as H9 and CEM also showed TREX1 induction (data not shown).
Fig. 2.

CPT induced TREX1 protein and its mRNA in KB cells. (A) Immunoblot analysis of TREX1. (B) Time-course study of the induction of TREX1 protein. (B, top-inlay, shows the induction of TREX1 protein and its mRNA in a time-course manner). (C) TREX1 distribution in KB cells after CPT treatment. (D) Detection of TREX1 protein in KB and RKO cells. (E) Exonuclease activity assay of the immunoprecipitated TREX1 from CPT-treated KB and RKO cell lysates. (F) Northern blot detection of TREX1 mRNA from CPT-treated KB and RKO cells. Upper panel showed the TREX1 mRNA detected by the TREX1 probe, whereas lower panel showed the ribosomal RNA (28S and 18S with 5 kb and 1.9 kb in size, respectively) which served as an RNA quality and loading control.
In addition, we investigated the effect of the duration of CPT treatment on the expression of TREX1 protein and its mRNA. TREX1 protein was induced at 6 h, and reached a plateau at 12 h (Fig. 2B). 1,000 nM CPT treatment increased TREX1 expression by 1.3-, 2.9- and 2.3-fold at 6 h, 12 h, and 24 h, respectively. TREX1 mRNA was induced at 3 h, and reached a plateau at 6 h that was maintained till 12 h (Fig. 2B, top-inlay). The induction of TREX1 mRNA was 3 h prior to that of TREX1 protein, suggesting that the increase of TREX1 mRNA is partly responsible for that of TREX1 protein. Since actinomycin D suppressed the effect of CPT on TREX1 mRNA (Fig. S5), transcriptional activation could be involved for TREX1 mRNA’s upregulation.
To study the cellular distribution of TREX1, KB cells were treated with CPT for 24 h and then fractionated. TREX1 was induced by CPT treatment in a dose-dependent manner (Fig. 2C). The protein levels were compared. More TREX1 appeared in the nucleus than in the cytoplasm after CPT treatment, signifying an accelerated TREX1 nuclear translocation and the suggestive role of TREX1 in DNA metabolism or inducible apoptosis after DNA damage. Topo I, which is down-regulated by CPT probably due to the induced destabilization of Topo I mRNA [34], served as a control. p53, a DNA damage sensor, was induced, consistent with a previous report [35]. In contrast, TREX1 was not induced in RKO cells, despite the induction of p53 and the truncation of another DNA damage sensor, Poly (ADP-ribose) polymerase-1 (PARP-1) [36], from 113 kD to 85 kD (Fig. 2D). Concentrations of CPT over 300 nM caused severe truncation of PARP-1, indicating that the cells were in dying (apoptotic) [37] state. Moreover, TREX1 was induced in KB but not in RKO cells, suggesting that its induction is independent of the functional p53 levels. Since the effect of CPT on TREX1 protein was examined using antibodies, the specificity of the antibody used in this study was given in the full size immunoblot of the cell lysates from KB and RKO cells treated with different doses of CPT (Fig. S6).
The exonuclease activity of TREX1 was not abolished by using antibodies. To assess if TREX1 activity was increased with TREX1 induction, the activity of the immunoprecipitated TREX1 from total cell lysate was examined. The activity was shown in KB but not in RKO cell lysate, confirming the presence of TREX1 in KB but not in RKO cells (Fig. 2E). The increased activity in KB cells indicated that the induced TREX1 by CPT treatment is enzymatically active. In addition, the immunoprecipitated TREX1 from the cytosolic fraction of KB cells also showed exonuclease activity (data not shown), indicating that TREX1 can exert its function in the cytoplasm, a finding consistent with single-stranded DNA accumulation in the cytoplasm when TREX1 is absent [21].
We used Northern blot analysis to further confirm that TREX1 mRNA was induced by CPT treatment in a dose-dependent manner in KB but not in RKO cells (Fig. 2F), which is comparable to the increase of TREX1 exonuclease activity in KB but not in RKO cells using immunoprecipitation methodology (Fig. 2E). No detection of TREX1 protein in RKO cells was due to undetectable expression of TREX1 mRNA (Fig. 2F, right), consistent with the results shown previously (Fig. 1C) using quantitative RT-PCR.
Furthermore, other anti-cancer drugs including cisplatin, etoposide and L-OddC at higher dosages induced TREX1 protein and its mRNA in KB cells (Fig. S7). H2O2 also induced TREX1 protein in RKO cells in which a TREX1-expressing plasmid was stably or transiently transfected (Fig. S8).
3.3. TREX1 degrades DNA from CPT-treated RKO/TREX1 cells
We investigated the action of TREX1 using an established RKO/TREX1 cell line. The expression of TREX1 was increased with increasing doxycycline (Fig. 3A). In the fractionation study, PARP-1, Topo I and XRCC 1 served as markers for fractionation. Despite TREX1 being detectable in all the fractions, the nuclear matrix showed the largest amount (Fig. 3B). PARP-1 and XRCC 1 served as controls for nuclear matrix. PARP-1 is reported to be with PARP-2, XRCC 1 and related proteins in rat testis nuclear matrix [38], whereas Topo I is a nucleolar protein involved during transcription (for a review, see [39]). Confocal microscopy revealed that TREX1 was localized in the cytoplasm and the nuclear matrix-containing perinuclear region overlapping with endoplasmic reticulum (Fig. 3C). Since the C-terminus of TREX1 is highly hydrophobic and predicted to be membrane-embedded [8,16], whether TREX1 could localize in all the membrane-containing organelles requires to be examined. The nuclear matrix framework is a distinct subnuclear compartment, dynamically and spatially supporting the processes of DNA transcription, replication, and repair [40–42]. The abundance of TREX1 in the nuclear matrix suggests the existence of functional TREX1 complexes, which are probably formed through the interaction of its N-terminal polyproline helix with other proteins [6,8]. As in KB cells, a discrepancy was also observed in TREX1-induced RKO/TREX1 cells with respect to the relative amount of TREX1 in using two different techniques, namely the confocal and subcellular fractionation. This observation further supports the possible association of TREX1 with nuclear proteins leading to the masking of its epitope and less confocal detection in the nucleus.
Fig. 3.

TREX1 degrades DNA from CPT-treated RKO/TREX1 cells. (A) Immunoblot analysis of TREX1 expression by Doxycline (Dox). (B) Localization of TREX1 using subcellular fractionation assay. To normalize the samples for loading, lysis buffer added to each fraction was proportional to 3 volumes of the size of the cell pellet (Cyto: cytoplasm; NP: nucleoplasm; NM: Nuclear matrix; Chrom: Chromosome). Nuclear proteins PARP-1, Topo I and XRCC 1 served as fractionation controls. (C) Confocal microscopy study. F-actin, specifically labeled by phalloidin, was a counterstaining marker for cytoplasm. 100 ng/ml Dox was added. Bars, 10 μm. (D) In situ DNA agarose gel electrophoresis of RKO/TREX1 cells treated with or without CPT and stained by ethidium bromide.
To study the action of TREX1 on cellular DNA following CPT treatment, in situ DNA agarose electrophoresis was performed by loading the same numbers of RKO/TREX1 cells. Without CPT treatment, even higher TREX1 expression showed no effect on cellular DNA, but with CPT treatment, less DNA remained in the wells accompanied by DNA smearing (Fig. 3D). This indicates that DNA damage is required for TREX1 to exert its function and that TREX1 functions to degrade damaged DNA. The presence of DNA smearing could be due to severe DNA breaks and cross-linking, and DNA degradation may occur in those drug-treated dying cells. Under constant TREX1 expression by doxycycline, DNA degradation is correlated with the concentration of CPT (Fig. 3D, middle). Little to no difference in DNA smearing was observed when 1,000 nM CPT was added, regardless of the presence or absence of doxycycline (Fig. 3D, right). A discrete band (over 10 kb in size) was observed using this method in the cells treated with CPT and might be a result of the accumulated damaged DNA. Contrary to CPT-treated KB cells (data not shown), RKO cells did not show any significant DNA smearing, further confirming TREX1’s role in DNA degradation in KB cells after CPT treatment. As in situ DNA agarose electrophoresis was applied in this study for measuring damaged DNA, but there are other useful methods like the Comet Assay or pulse-field gel electrophoresis. To get more insight of the role of TREX1 on damaged DNA, these different methods could be applied in future studies.
3.4. TREX1 may not play a role in damaged DNA repair
TREX1 has been suggested to play a role in DNA repair. If this is true, the chemosensitivity of the cells to the treatment of DNA damaging agents should decrease upon TREX1 expression. To address this issue, RKO/TREX1 cells pretreated with or without doxycycline were assessed using a clonogenic assay with respect to their 24 h drug sensitivity. A variety of DNA damaging agents with different mechanisms of action were used in the experiments. As shown in Table 1, TREX1 expression did not alter sensitivities of the cells to the DNA damaging agents examined, suggesting that TREX1 may not play a role in DNA repair. However, this needs to be further clarified due to the concern that TREX1 may not act alone but together with other proteins as a repair complex. The rest of the complex might be the rate-determining step for DNA repair if only TREX1 was overexpressed, leading to no decrease in chemosensitivity.
Table 1.
IC50 (nM) for anti-clonogenicity
| Reagent | RKO/TREX1 Cells | |
|---|---|---|
| Dox (−) | Dox (+)(100ng/ml) | |
| Camptothecin (CPT) | 3.7 ± 1.3 | 3.1 ± 0.9 |
| Gemcitabine | 1.9 ± 0.2 | 1.8 ± 0.4 |
| Doxorubicin | 7.7 ± 1.7 | 7.5 ± 1.5 |
| AraC | 37.5 ± 22.0 | 37.0 ± 23.0 |
| L-OddC | 165.0 ± 35.0 | 194.5 ± 25.5 |
| Oxaliplatin | 179.5 ± 29.5 | 229.0 ± 79.0 |
| Etoposide | 540.0 ± 40.0 | 570.0 ± 30.0 |
| Arsenite | 3,800.0 ± 300.0 | 3,900.0 ± 400.0 |
The IC50 values standing for the inhibition of clonogenicity are shown as ranges of the results from three experiments. ± stands for the difference between the highest and the average values.
3.5. TREX1 determines the fate of damaged DNA
Based on the observation (Fig. 3D) that TREX1 functions in the degradation of damaged DNA, we used thymidine incorporation and methanol fractionation to further study the fate of damaged DNA. RKO/TREX1 cells labeled by [14C]thymidine followed by CPT treatment were assessed. The cells and culture medium were fractionated by 70% methanol. The methanol-soluble fractions contained labeled thymine, thymidine, thymidine metabolites, and thymidine-incorporated oligonucleotides. The methanol-insoluble fractions contained DNA and other macromolecules. Without CPT treatment, TREX1 expression showed no effect on the distribution of the labeled thymidine (Fig. 4, A and B, left). With 1,000 nM CPT treatment, the increase of TREX1 expression by doxycycline had an apparent effect on the relative ratio of the labeled thymidine in the methanol-soluble and -insoluble medium fraction, as more was in the methanol-soluble than in the -insoluble fraction (Fig. 4A, middle). A slight increase in the exonuclease activity was observed in the medium (data not shown), suggesting that although TREX1 exerts its function primarily inside the cells, an extracellular function of TREX1 cannot be excluded. Whether the labeled thymidine appearing in the medium was through passive diffusion or a transporter-mediated mechanism is unclear. CPT at 1,000 nM triggered release of the labeled thymidine into medium at a higher efficiency than CPT at 100 nM, consistent with the observation that CPT at 1,000 nM caused more DNA degradation than CPT at 100 nM (Fig. 4, A and B, right; also compare Fig. 3D). The increase of the labeled thymidine in the medium was time-dependent on TREX1 expression, resulting in a 3- to 4-fold increase of its release (Fig. 4C, compare 0/1,000 and 10/1,000). CPT treatment for only 12 h showed no significant release (data not shown). There was a time difference concerning the induction of TREX1 protein and the release of degraded DNA into medium (compare Fig. 2B and Fig. 4C), suggesting a network of intracellular signaling, leading to DNA degradation and its release.
Fig. 4.

TREX1 determines the fate of damaged DNA. (A) Distribution of the labeled [14C]thymidine in RKO/TREX1 cells fractionated by 70% methanol into methanol-soluble and -insoluble fractions. Detected by β-counter, the results are presented as percentage of the total counts. The s.d. shown on the bars were obtained from three independent experiments. (B) Methanol-insoluble cell extract fraction. (C) Time-course study of the labeled thymidine in the medium using HPLC. (D and E) Distribution of the labeled thymidine in RKO cells.
With parental RKO cells, only a mild change of the labeled thymidine was observed in the methanol-insoluble fractions of medium and cell extract (Fig. 4, D and E). Notably, p53 also possesses 3′-5′ exonuclease activity [43]. Its induction by CPT treatment in RKO cells did not show any effect on cellular DNA, suggesting that DNA degradation is primarily attributed to the work of TREX1. Using a CPT-treated RKO/TREX1-mutant cell line with abolished TREX1 exonuclease activity due to double mutations (D18A/E20A) in the conserved Exo I motif, as expected, there was no release of the labeled thymidine (data not shown), confirming the role of TREX1 in DNA degradation.
The damaged DNA was studied using SDS-PAGE followed by autoradiogram and HPLC analysis. There was a reverse correlation of the labeled thymidine in the methanol-soluble and -insoluble fractions (Fig. 5A, compare Fig. 4, A and B). The more labeled thymidine there was in the methanol-soluble fraction, the less there was in the methanol-insoluble fraction. The labeled thymidine in the medium was found to be thymidine, and thymine possibly, with no detection of mono- (TMP), di- (TDP) or tri- (TTP) phosphate metabolites of thymidine (Fig. 5B). The lack of detection of TMP, the TREX1 product studied in vitro [3], could be due to subsequent action of cellular nucleotidase and thymidine phosphorylase.
Fig. 5.

Study the damaged DNA. (A) SDS-PAGE analysis of the methanol-insoluble cell extract and medium fractions of RKO/TREX1 cells, followed by auto-radiogram. λ/HindIII markers stained by ethidium bromide and γ-[32P]ATP labeled 21mer oligonucleotide used in the exonuclease activity assay were used as the size markers. (B) HPLC analysis of the methanol-soluble fraction of medium. (Thy, thymine; Thd, thymidine; TMP, thymidine monophosphate; TDP, thymidine diphosphate; TTP, thymidine triphosphate). (C) Immunoblot analysis.
The effect of CPT on TREX1-induced RKO/TREX1 cells was assessed. TREX1 was present in both the cytosolic and nuclear fractions. Together with the partially truncated forms, TREX1 was induced as seen in KB cells after CPT treatment (Fig. 5C). No induction of TREX1 mRNA was detected in this cell line (bearing CMV promoter), the induction of TREX1 protein by CPT treatment could therefore be mediated through posttranscriptional processes as well.
3.6. TREX1 degrades apoptotic DNA in drug-treated dying cells
To further characterize TREX1’s role in cellular DNA degradation, the DNA content of RKO/TREX1 cells under a combination of doxycycline and CPT treatment for 24 h was analyzed by flow cytometry. The gated cells in 1.5×104 events were divided into two populations: the live and apoptotic cells with shrinkage through a combination of forward scatter properties and propidium iodide uptake (Fig. 6A, a–d). The cells were primarily alive in normal condition without doxycycline and CPT treatment (Fig. 6A, a), in which a diminutive number of cells underwent apoptosis, consistent with the observation of an extent of DNA smearing in the absence of doxycycline (Fig. 3A, left).
Fig. 6.


TREX1 clears apoptotic DNA in drug-treated dying cells. Analyzed by flow cytometry, the DNA content of CPT-treated RKO/TREX1 cells for 24 h with or without doxycycline was studied. (A, a–d) The cells in 1.5×104 events were gated and divided into two populations: the live (FSC-H: 550–1,000) and the apoptotic cells (FSC-H: 200–530). (B, a–d). The DNA content of the live cell population was distributed into subG1 and G1-S-G2-M areas. (C, a–d). The DNA content in the apoptotic cell population under log format was distinctly distributed into two sub-areas. Data represent typical experiments that were repeated three times with similar results.
The addition of doxycycline increased around 10% apoptotic cells (Fig. 6A, b) whereas CPT treatment at 1,000 nM caused 40% apoptotic cells (Fig. 6A, c). The expression of TREX1 did not significantly change the percentages of the live and apoptotic cell populations with around 55% versus 40%, respectively (Fig. 6A, compare c and d). In the live cell population, CPT treatment induced cell cycle arrest at the G1 phase (Fig. 6B, compare c and d with a and b), and only a small amount of DNA was found in the sub-G1 phase. With TREX1 expression by doxycycline, the DNA content was slightly decreased in the sub-G1 (0.7%) but slightly increased (0.7%) in the G1 phase (Fig. 6B, compare c and d). In the apoptotic cell population, the increase of DNA content depended on CPT treatment (Fig. 6C, compare c and d with a and b), and the DNA in CPT-treated cells was distributed into two sub-areas (Fig. 6C, c and d), caused probably by the severity of DNA damage and ploidy. The DNA content in TREX1-induced apoptotic cells by doxycycline showed obvious difference as more than 20% DNA was degraded, preserving presumably the more intact DNA (Fig. 6C, compare c and d). This experiment confirmed that TREX1 functions in apoptotic DNA degradation in drug-treated dying cells.
4. Discussion
Anti-cancer drugs with varying mechanisms of action on DNA induced TREX1 protein and its mRNA, suggesting a role of TREX1 in DNA metabolism or inducible apoptosis. Notably, its homolog TREX2 is depleted in cells by cisplatin treatment [44], implying distinct roles played by both proteins. Exonuclease induction from cytotoxic insult is not exclusively limited to TREX1. The ribonuclease interferon-stimulated gene 20 (ISG20) could be induced by interferons or double-stranded RNA and also belongs to the DnaQ family [45]. It is very unlikely that TREX1’s induction is dependent on a unique type of DNA damage caused by anti-cancer drugs. For instance, CPT provokes double-stranded DNA breaks through the stabilization of transient DNA-Topo I cleavage complexes that collide with the DNA replication fork and induce apoptosis [46,47]. This is different from the lesions caused by the chain-terminator L-OddC. Regardless how they are formed, all the DNA breaks generated by treatment with anti-cancer drugs are accessible to TREX1-mediated 3′-5′ DNA cleavage. These DNA breaks could serve as initiation sites where TREX1 starts its DNA degradation. To exert its role, TREX1 may require the coordination of the endonuclease NM23-H1 [26]. As the effect of TREX1 on DNA degradation could be clearly seen when cells were under CPT treatment for over 18 h, it is likely that other apoptotic nucleases such as caspase-activated DNase (CAD) and Endo G, localized in the cytoplasm and mitochondria, respectively, could also be involved during apoptosis to first lead to nucleosomal DNA cleavage [48]. In addition to chromosomal DNA, TREX1 may also degrade episomal DNA arising from DNA replication, viral infection, or endogenous retroviruses, consistent with the finding that TREX1 is able to metabolize single-stranded DNA derived from endogenous retroelements [25]. Actinomycin D could suppress the inducible effect of CPT treatment on TREX1 mRNA, implying that the increase of TREX1 mRNA is not due to the increase of its stability, but due to transcriptional activation of its synthesis. CPT is reported to activate the transcriptional factors including NF-κB [49], c-Myb and Rfx1 [50], whether the synthesis of TREX1 mRNA is directly regulated by these transcriptional factors requires further investigation. Interestingly, bone marrow-derived macrophages from mice transfected with IFN-stimulatory DNA also resulted in the upregulation of TREX1 mRNA [25], raising a question whether a convergent pathway exists through which TREX1 is induced by the treatment of both CPT and IFN-stimulatory DNA. Based on recent advances on intracellular pathways for nucleic acid recognition, DNA damage caused by CPT treatment may lead to the activation of NF-κB and the downstream proinflammatory genes through the endosomal pathway including TLR7 and TLR9. The cytosolic pathway, including the activation of the transcriptional factor IRF3 pathway leading to type I-IFNs activation (such as IFN-α and IFN-β), however, is not ruled out, as reviewed by others [51,52].
DNA damaging agents can induce apoptosis. At higher dosages, CPT induced TREX1 expression; the accompanied DNA damage under such condition could be severe and irreparable. Our results suggested that TREX1 may not play a role in DNA repair, the induced TREX1, however, dramatically changed the distribution and proportion of the apoptotic DNA in the flow cytometric study, indicating that signaling from apoptotic cells could be responsible for TREX1’s induction and at least a fraction of the apoptotic DNA was susceptible to TREX1’s degradation. This induction could have physiological importance, such as it speeds up the clearance of potentially immunogenic DNA in dying cells. From this perspective, TREX1 plays a protective role in human tissues to prevent the occurrence of autoimmune or neurological disease. Signaling from apoptotic cells that trigger TREX1 may also prove important (i.e., cross-talk) in the activation of other cellular components to facilitate DNA clearance. The mechanism of CPT-induced apoptosis is recently suggested to be regulated by ATM-mediated NF-κB activation, involving degradation of Akt1, a family of serine/threonine kinase. Akt1 degradation is presumably mediated by caspase-3, breaking the balance between cell survival and apoptosis [53]. As caspases are usually activated during apoptosis, whether the induction of TREX1 is caspase-dependent and how cells differentiate the reparability of DNA damage remain to be clarified. Furthermore, whether the induction of TREX1 by cisplatin, etoposide and L-OddC is through the same mechanism and whether other types of cell death including autophagy and necrosis could also lead to TREX1 induction and DNA degradation warrant further study. In addition to anti-cancer drugs, whether other DNA mutagenic agents such as UV, γ-irradiation and mitomycin C that are able to induce DNA damage could all lead to DNA degradation mediated by the upregulation of TREX1 remain to be elucidated.
Impaired clearance of dying cells could contribute to the development of SLE and other autoimmune disorders [54,55]. Under healthy conditions, phagocytes, including macrophages, quickly remove dying cells from tissues. In SLE patients, the number and phagocytic capabilities of macrophages are significantly reduced [56]. It is interesting to know the activity of TREX1 in those impaired macrophages. Our experiment employing tumor cells indicates that TREX1 degrades apoptotic DNA in dying cells before the phagocytosis occurs. As TREX1 is ubiquitously expressed in normal tissues, it is possible that TREX1 plays the role of DNA degradation in all cell types undergoing a dying process.
Tumor cells lacking TREX1 may mimic TREX1-null fibroblasts, displaying inflammatory status in vivo, and become more dangerous under chemotherapy from the accumulation of DNA damage to induce autoimmune disease. For tumor cells expressing TREX1, chemotherapy could lead to more TREX1 expression and less serious inflammation. In this sense, TREX1 expression determines the inflammatory potential of tumor cells undergoing chemotherapeutic treatment. For those patients with autoimmune or neurological diseases due to immunogenic cellular DNA, genotoxic stress may aggravate their severity. The treatment with a combination of DNA exonuclease and endonuclease should help control or alleviate severity of these types of autoimmune disorders.
It is unknown why TREX1 was truncated in RKO/TREX1, but not in KB cells, while no loss of TREX1’s exonuclease activity was observed. Presumably, the truncation occurred at the C-terminal region without damaging the conserved motifs. The induced TREX1 expression in RKO/TREX1 cells probably altered the complex arrangement of TREX1/cellular protein(s), leading to a breakdown of its protection.
In conclusion, TREX1 plays the role of DNA degradation, especially in dying cells before their phagocytosis by macrophages. Possibly, TREX1 also participates in the pathway of cell death. Failure of TREX1 in cellular DNA degradation could result in autoimmune disorders, which partly explain why mutations in human TREX1 gene are linked to Goutieres syndrome and SLE. TREX1 may have other function(s), particularly in immune cells. Notably, all the lymphoid tumor cells examined have higher TREX1 expression. Studies on the interaction between TREX1 and cellular proteins may delineate novel pathways or mechanisms involved in the host response to DNA damage.
Supplementary Material
Appendix A. Supplementary data
Acknowledgments
We thank E. Gullen and C.-H. Hsu for assistance in cell culture; G. Yang for discussion; E. Paintsil as well as Y. Cheng for critical reading of the manuscript. We also thank Dr. F.W. Perrino for his advice in the beginning of this project. Y.-C. C is a Fellow of the National Foundation for Cancer Research. The work was supported by National Cancer Institute, National Institutes of Health and Yale Liver Center [grant numbers CA-63477, DK P30 -34989].
Footnotes
Conflict of interest
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lindahl T, Gally JA, Edelman GM. Properties of deoxyribonuclease 3 from mammalian tissues. J Biol Chem. 1969;244:5014–5019. [PubMed] [Google Scholar]
- 2.Hoss M, Robins P, Naven TJ, Pappin DJ, Sgouros J, Lindahl T. A human DNA editing enzyme homologous to the Escherichia coli DnaQ/MutD protein. EMBO J. 1999;18:3868–3875. doi: 10.1093/emboj/18.13.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mazur DJ, Perrino FW. Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3′-->5′ exonucleases. J Biol Chem. 1999;274:19655–19660. doi: 10.1074/jbc.274.28.19655. [DOI] [PubMed] [Google Scholar]
- 4.Mazur DJ, Perrino FW. Excision of 3′ termini by the Trex1 and TREX2 3′-->5′ exonucleases, characterization of the recombinant proteins. J Biol Chem. 2001;276:17022–17029. doi: 10.1074/jbc.M100623200. [DOI] [PubMed] [Google Scholar]
- 5.Perrino FW, Miller H, Ealey KA. Identification of a 3′-->5′-exonuclease that removes cytosine arabinoside monophosphate from 3′ termini of DNA. J Biol Chem. 1994;269:16357–16363. [PubMed] [Google Scholar]
- 6.de Silva U, Choudhury S, Bailey SL, Harvey S, Perrino FW, Hollis T. The Crystal Structure of TREX1 Explains the 3′ Nucleotide Specificity and Reveals a Polyproline II Helix for Protein Partnering. J Biol Chem. 2007;282:10537–10543. doi: 10.1074/jbc.M700039200. [DOI] [PubMed] [Google Scholar]
- 7.Perrino FW, Harvey S, McMillin S, Hollis T. The human TREX2 3′ -> 5′-exonuclease structure suggests a mechanism for efficient nonprocessive DNA catalysis. J Biol Chem. 2005;280:15212–15218. doi: 10.1074/jbc.M500108200. [DOI] [PubMed] [Google Scholar]
- 8.Brucet M, Querol-Audi J, Serra M, Ramirez-Espain X, Bertlik K, Ruiz L, Lloberas J, Macias MJ, Fita I, Celada A. Structure of the dimeric exonuclease TREX1 in complex with DNA displays a proline-rich binding site for WW Domains. J Biol Chem. 2007;282:14547–14557. doi: 10.1074/jbc.M700236200. [DOI] [PubMed] [Google Scholar]
- 9.Shevelev IV, Ramadan K, Hubscher U. The TREX2 3′-->5′ exonuclease physically interacts with DNA polymerase delta and increases its accuracy. ScientificWorldJournal. 2002;2:275–281. doi: 10.1100/tsw.2002.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morita M, Stamp G, Robins P, Dulic A, Rosewell I, Hrivnak G, Daly G, Lindahl T, Barnes DE. Gene-targeted mice lacking the Trex1 (DNase III) 3′-->5′ DNA exonuclease develop inflammatory myocarditis. Mol Cell Biol. 2004;24:6719–6727. doi: 10.1128/MCB.24.15.6719-6727.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, Voit T, Lebon P, Bonthron DT, Jackson AP, Barnes DE, Lindahl T. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 2006;38:917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 12.Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, Artuch R, Montalto SA, Bacino CA, Barroso B, Baxter P, Benko WS, Bergmann C, Bertini E, Biancheri R, Blair EM, Blau N, Bonthron DT, Briggs T, Brueton LA, Brunner HG, Burke CJ, Carr IM, Carvalho DR, Chandler KE, Christen HJ, Corry PC, Cowan FM, Cox H, D’Arrigo S, Dean J, De Laet C, De Praeter C, Dery C, Ferrie CD, Flintoff K, Frints SG, Garcia-Cazorla A, Gener B, Goizet C, Goutieres F, Green AJ, Guet A, Hamel BC, Hayward BE, Heiberg A, Hennekam RC, Husson M, Jackson AP, Jayatunga R, Jiang YH, Kant SG, Kao A, King MD, Kingston HM, Klepper J, van der Knaap MS, Kornberg AJ, Kotzot D, Kratzer W, Lacombe D, Lagae L, Landrieu PG, Lanzi G, Leitch A, Lim MJ, Livingston JH, Lourenco CM, Lyall EG, Lynch SA, Lyons MJ, Marom D, McClure JP, McWilliam R, Melancon SB, Mewasingh LD, Moutard ML, Nischal KK, Ostergaard JR, Prendiville J, Rasmussen M, Rogers RC, Roland D, Rosser EM, Rostasy K, Roubertie A, Sanchis A, Schiffmann R, Scholl-Burgi S, Seal S, Shalev SA, Corcoles CS, Sinha GP, Soler D, Spiegel R, Stephenson JB, Tacke U, Tan TY, Till M, Tolmie JL, Tomlin P, Vagnarelli F, Valente EM, Van Coster RN, Van der Aa N, Vanderver A, Vles JS, Voit T, Wassmer E, Weschke B, Whiteford ML, Willemsen MA, Zankl A, Zuberi SM, Orcesi S, Fazzi E, Lebon P, Crow YJ. Clinical and molecular phenotype of aicardi-goutieres syndrome. Am J Hum Genet. 2007;81:713–725. doi: 10.1086/521373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stephenson JB. Aicardi-Goutieres syndrome (AGS) Eur J Paediatr Neurol. 2008;12:355–358. doi: 10.1016/j.ejpn.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 14.Richards A, van den Maagdenberg AM, Jen JC, Kavanagh D, Bertram P, Spitzer D, Liszewski MK, Barilla-Labarca ML, Terwindt GM, Kasai Y, McLellan M, Grand MG, Vanmolkot KR, de Vries B, Wan J, Kane MJ, Mamsa H, Schafer R, Stam AH, Haan J, de Jong PT, Storimans CW, van Schooneveld MJ, Oosterhuis JA, Gschwendter A, Dichgans M, Kotschet KE, Hodgkinson S, Hardy TA, Delatycki MB, Hajj-Ali RA, Kothari PH, Nelson SF, Frants RR, Baloh RW, Ferrari MD, Atkinson JP. C-terminal truncations in human 3′-5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet. 2007;39:1068–1070. doi: 10.1038/ng2082. [DOI] [PubMed] [Google Scholar]
- 15.Stam AH, van den Maagdenberg AM, Haan J, Terwindt GM, Ferrari MD. Genetics of migraine: an update with special attention to genetic comorbidity. Curr Opin Neurol. 2008;21:288–293. doi: 10.1097/WCO.0b013e3282fd171a. [DOI] [PubMed] [Google Scholar]
- 16.Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, de Silva U, Bailey SL, Witte T, Vyse TJ, Kere J, Pfeiffer C, Harvey S, Wong A, Koskenmies S, Hummel O, Rohde K, Schmidt RE, Dominiczak AF, Gahr M, Hollis T, Perrino FW, Lieberman J, Hubner N. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007;39:1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 17.Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, Robins P, Harvey S, Hollis T, O’Hara A, Herrick AL, Bowden AP, Perrino FW, Lindahl T, Barnes DE, Crow YJ. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;80:811–815. doi: 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee-Kirsch MA, Gong M, Schulz H, Ruschendorf F, Stein A, Pfeiffer C, Ballarini A, Gahr M, Hubner N, Linne M. Familial chilblain lupus, a monogenic form of cutaneous lupus erythematosus, maps to chromosome 3p. Am J Hum Genet. 2006;79:731–737. doi: 10.1086/507848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee-Kirsch MA, Chowdhury D, Harvey S, Gong M, Senenko L, Engel K, Pfeiffer C, Hollis T, Gahr M, Perrino FW, Lieberman J, Hubner N. A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J Mol Med. 2007;85:531–537. doi: 10.1007/s00109-007-0199-9. [DOI] [PubMed] [Google Scholar]
- 20.Lehtinen DA, Harvey S, Mulcahy MJ, Hollis T, Perrino FW. The TREX1 double-stranded DNA degradation activity is defective in dominant mutations associated with autoimmune disease. J Biol Chem. 2008;283:31649–31656. doi: 10.1074/jbc.M806155200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang YG, Lindahl T, Barnes DE. Trex1 Exonuclease Degrades ssDNA to Prevent Chronic Checkpoint Activation and Autoimmune Disease. Cell. 2007;131:873–886. doi: 10.1016/j.cell.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 22.Coscoy L, Raulet DH. DNA mismanagement leads to immune system oversight. Cell. 2007;131:836–838. doi: 10.1016/j.cell.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brooks PJ, Cheng TF, Cooper L. Do all of the neurologic diseases in patients with DNA repair gene mutations result from the accumulation of DNA damage? DNA Repair (Amst) 2008;7:834–848. doi: 10.1016/j.dnarep.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Driscoll M. TREX1 DNA exonuclease deficiency, accumulation of single stranded DNA and complex human genetic disorders. DNA Repair (Amst) 2008;7:997–1003. doi: 10.1016/j.dnarep.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 25.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chowdhury D, Beresford PJ, Zhu P, Zhang D, Sung JS, Demple B, Perrino FW, Lieberman J. The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol Cell. 2006;23:133–142. doi: 10.1016/j.molcel.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 27.Chowdhury D, Lieberman J. Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol. 2008;26:389–420. doi: 10.1146/annurev.immunol.26.021607.090404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lam W, Chen C, Ruan S, Leung CH, Cheng YC. Expression of deoxynucleotide carrier is not associated with the mitochondrial DNA depletion caused by anti-HIV dideoxynucleoside analogs and mitochondrial dNTP uptake. Mol Pharmacol. 2005;67:408–416. doi: 10.1124/mol.104.007120. [DOI] [PubMed] [Google Scholar]
- 29.Chou KM, Cheng YC. The exonuclease activity of human apurinic/apyrimidinic endonuclease (APE1). Biochemical properties and inhibition by the natural dinucleotide Gp4G. J Biol Chem. 2003;278:18289–18296. doi: 10.1074/jbc.M212143200. [DOI] [PubMed] [Google Scholar]
- 30.Nutter LM, Grill SP, Li JS, Tan RS, Cheng YC. Induction of virus enzymes by phorbol esters and n-butyrate in Epstein-Barr virus genome-carrying Raji cells. Cancer Res. 1987;47:4407–4412. [PubMed] [Google Scholar]
- 31.Gao W, Lam W, Zhong S, Kaczmarek C, Baker DC, Cheng YC. Novel mode of action of tylophorine analogs as antitumor compounds. Cancer Res. 2004;64:678–688. doi: 10.1158/0008-5472.can-03-1904. [DOI] [PubMed] [Google Scholar]
- 32.Nutter LM, Grill SP, Cheng YC. The sources of thymidine nucleotides for virus DNA synthesis in herpes simplex virus type 2-infected cells. J Biol Chem. 1985;260:13272–13275. [PubMed] [Google Scholar]
- 33.Mazur DJ, Perrino FW. Structure and expression of the TREX1 and TREX2 3′ --> 5′ exonuclease genes. J Biol Chem. 2001;276:14718–14727. doi: 10.1074/jbc.M010051200. [DOI] [PubMed] [Google Scholar]
- 34.Beidler DR, Cheng YC. Camptothecin induction of a time- and concentration-dependent decrease of topoisomerase I and its implication in camptothecin activity. Mol Pharmacol. 1995;47:907–914. [PubMed] [Google Scholar]
- 35.Deptala A, Li X, Bedner E, Cheng W, Traganos F, Darzynkiewicz Z. Differences in induction of p53, p21WAF1 and apoptosis in relation to cell cycle phase of MCF-7 cells treated with camptothecin. Int J Oncol. 1999;15:861–871. doi: 10.3892/ijo.15.5.861. [DOI] [PubMed] [Google Scholar]
- 36.Althaus FR, Kleczkowska HE, Malanga M, Muntener CR, Pleschke JM, Ebner M, Auer B. Poly ADP-ribosylation: a DNA break signal mechanism. Mol Cell Biochem. 1999;193:5–11. [PubMed] [Google Scholar]
- 37.Simizu S, Takada M, Umezawa K, Imoto M. Requirement of caspase-3(-like) protease-mediated hydrogen peroxide production for apoptosis induced by various anticancer drugs. J Biol Chem. 1998;273:26900–26907. doi: 10.1074/jbc.273.41.26900. [DOI] [PubMed] [Google Scholar]
- 38.Tramontano F, Di Meglio S, Quesada P. Co-localization of poly(ADPR)polymerase 1 (PARP-1) poly(ADPR)polymerase 2 (PARP-2) and related proteins in rat testis nuclear matrix defined by chemical cross-linking. J Cell Biochem. 2005;94:58–66. doi: 10.1002/jcb.20295. [DOI] [PubMed] [Google Scholar]
- 39.Laurincik J, Maddox-Hyttel P. Nucleolar remodeling in nuclear transfer embryos. Adv Exp Med Biol. 2007;591:84–92. doi: 10.1007/978-0-387-37754-4_6. [DOI] [PubMed] [Google Scholar]
- 40.Anachkova B, Djeliova V, Russev G. Nuclear matrix support of DNA replication. J Cell Biochem. 2005;96:951–961. doi: 10.1002/jcb.20610. [DOI] [PubMed] [Google Scholar]
- 41.Stein GS, Zaidi SK, Braastad CD, Montecino M, van Wijnen AJ, Choi JY, Stein JL, Lian JB, Javed A. Functional architecture of the nucleus: organizing the regulatory machinery for gene expression, replication and repair. Trends Cell Biol. 2003;13:584–592. doi: 10.1016/j.tcb.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 42.Zaidi SK, Young DW, Choi JY, Pratap J, Javed A, Montecino M, Stein JL, van Wijnen AJ, Lian JB, Stein GS. The dynamic organization of gene-regulatory machinery in nuclear microenvironments. EMBO Rep. 2005;6:128–133. doi: 10.1038/sj.embor.7400337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mummenbrauer T, Janus F, Muller B, Wiesmuller L, Deppert W, Grosse F. p53 Protein exhibits 3′-to-5′ exonuclease activity. Cell. 1996;85:1089–1099. doi: 10.1016/s0092-8674(00)81309-4. [DOI] [PubMed] [Google Scholar]
- 44.Chen MJ, Dumitrache LC, Wangsa D, Ma SM, Padilla-Nash H, Ried T, Hasty P. Cisplatin depletes TREX2 and causes Robertsonian translocations as seen in TREX2 knockout cells. Cancer Res. 2007;67:9077–9083. doi: 10.1158/0008-5472.CAN-07-1146. [DOI] [PubMed] [Google Scholar]
- 45.Degols G, Eldin P, Mechti N. ISG20, an actor of the innate immune response. Biochimie. 2007;89:831–835. doi: 10.1016/j.biochi.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 46.Hsiang YH, Hertzberg R, Hecht S, Liu LF. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem. 1985;260:14873–14878. [PubMed] [Google Scholar]
- 47.Li TK, Liu LF. Tumor cell death induced by topoisomerase-targeting drugs. Annu Rev Pharmacol Toxicol. 2001;41:53–77. doi: 10.1146/annurev.pharmtox.41.1.53. [DOI] [PubMed] [Google Scholar]
- 48.Nagata S, Nagase H, Kawane K, Mukae N, Fukuyama H. Degradation of chromosomal DNA during apoptosis. Cell Death Differ. 2003;10:108–116. doi: 10.1038/sj.cdd.4401161. [DOI] [PubMed] [Google Scholar]
- 49.Hellweg CE, Arenz A, Bogner S, Schmitz C, Baumstark-Khan C. Activation of nuclear factor kappa B by different agents: influence of culture conditions in a cell-based assay. Ann N Y Acad Sci. 2006;1091:191–204. doi: 10.1196/annals.1378.066. [DOI] [PubMed] [Google Scholar]
- 50.Fan J, Zhan M, Shen J, Martindale JL, Yang X, Kawai T, Gorospe M. En masse nascent transcription analysis to elucidate regulatory transcription factors. Nucleic Acids Res. 2006;34:1492–1500. doi: 10.1093/nar/gkj510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krug A. Nucleic acid recognition receptors in autoimmunity. Handb Exp Pharmacol. 2008:129–151. doi: 10.1007/978-3-540-72167-3_7. [DOI] [PubMed] [Google Scholar]
- 52.Bhoj VG, Chen ZJ. Linking retroelements to autoimmunity. Cell. 2008;134:569–571. doi: 10.1016/j.cell.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 53.He L, Kim BY, Kim KA, Kwon O, Kim SO, Bae EY, Lee MS, Kim MS, Jung M, Moon A, Bae K, Ahn JS. NF-kappaB inhibition enhances caspase-3 degradation of Akt1 and apoptosis in response to camptothecin. Cell Signal. 2007;19:1713–1721. doi: 10.1016/j.cellsig.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 54.Gaipl US, Munoz LE, Grossmayer G, Lauber K, Franz S, Sarter K, Voll RE, Winkler T, Kuhn A, Kalden J, Kern P, Herrmann M. Clearance deficiency and systemic lupus erythematosus (SLE) J Autoimmun. 2007;28:114–121. doi: 10.1016/j.jaut.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 55.Tanaka M, Miyake Y. Apoptotic cell clearance and autoimmune disorder. Curr Med Chem. 2007;14:2892–2897. doi: 10.2174/092986707782360006. [DOI] [PubMed] [Google Scholar]
- 56.Gaipl US, Voll RE, Sheriff A, Franz S, Kalden JR, Herrmann M. Impaired clearance of dying cells in systemic lupus erythematosus. Autoimmun Rev. 2005;4:189–194. doi: 10.1016/j.autrev.2004.10.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix A. Supplementary data