

Abstract
A common strategy for preparing tryptophan-derived epidithiodioxopiperazine (ETP) natural product containing a hydroxyl substituent adjacent to a quaternary carbon stereocenter is reported. This strategy is exemplified by enantioselective total syntheses of four heptacyclic ETP natural products — gliocladine C (6), leptosin D (7), T988C (8), and bionectin A (9)—starting with the di-(tertbutoxycarbonyl) derivative 17 of the trioxopiperazine natural product gliocladin C, which is readily available by enantioselective chemical synthesis. In addition, total syntheses of the enantiomer of gliocladine C (ent-6) and gliocladin A (11), the di(methylthio) congener of bionectin A, are reported. These syntheses illustrate a synthetic strategy wherein diversity in the dioxopiperazine unit of ETP natural products is introduced at a late stage in a synthetic sequence. In vitro cytotoxicity of compounds in this series against invasive human prostrate (DU145) and melanoma (A2058) cancer cell lines is described and compared to that of chaetocin A (4).
Introduction
Epipolythiodioxopiperazine (ETP) toxins are a class of fungal metabolites (Figure 1) that possess unique molecular architectures and potent activities against parasites, viruses, bacteria and cancer cells.1,2 In recent studies, epidithiodioxopiperzines have shown selectivity against myeloma and solid tumors,1,3 with a number of molecular targets now identified.4 The biological activity of these natural products is associated with the di- or polysulfide-bridged dioxopiperazine subunit,1 which can form covalent adducts with cysteine residues of proteins, generate reactive oxygen species by a redoxcycling mechanism,5 or sequester zinc from protein targets.4d In spite of their promising biological properties, the therapeutic potential of molecules containing ETP fragments is largely unexplored, possibly a result of the scarcity of ETP natural products and the difficulty in preparing this unique fragment.1
Figure 1.

Representative epidithiodioxopiperazine (ETP) and related di(methylthio) dioxopiperazine natural products.
Both the structure and chemical lability of ETPs pose a number of challenges for chemical synthesis. The ground breaking syntheses of sporidesmin A (2)6 and gliotoxin (1)7 reported by Kishi and co-workers in the 1970s were achieved by introducing the ETP fragment in masked form as a nucleophilic unit.8 This imaginative method has been employed subsequently to assemble simplified ETP9a structures and in the total synthesis of additional ETP natural products.9b,c Beginning with Movassaghi's pioneering total synthesis of dideoxyverticillin A (3) in 2009, enantioselective total syntheses of several dimeric, C2-symmetric, ETP natural products have been accomplished recently.10–14 Several of these recent total syntheses improved methods introduced by earlier workers for incorporating the disulfide fragment of epidithiodioxopiperazine by both electrophilic and nucleophilic sulfenylation.15 In addition, strategies for controlled introduction of 2–4 sulfur atoms into the bridge of these natural products have been developed.16
The largest group of ETP toxins is tryptophan-derived and contains an ETP fragment fused to a cyclotryptamine moiety. Many of these naturally occurring compounds possess oxygenation in the pyrrolidine ring adjacent to the quaternary carbon stereocenter (e.g., 5–11), and a majority of the dimeric structures, illustrated by leptosin K (5), are C1-symmetric. The presence of a hydroxyl substituent adjacent to the quaternary carbon stereocenter markedly increases the lability of molecules of this type to both acids and bases.17,18 We report herein the first approach for preparing cyclotryptophan ETP natural products containing both hydroxyl substitution at C11 of the pyrrolidine ring and a C10b quaternary stereocenter. Our strategy is exemplified by enantioselective total syntheses of gliocladine C (6),2a,19 leptosin D (7),20 T988C (8),21 and bionectin A (9).2b In addition, the total synthesis of a di(methylthio) congener gliocladin A (11,22 also called bionectin C2b) is reported.23 In vitro cytotoxicity of compounds in this series against invasive human prostrate and melanoma cancer cell lines is described also and compared to that of chaetocin A.
Results and Discussion
Synthesis Plan
Central to our synthetic strategy was the recognition that functionality embedded in the di-(tert-butoxycarbonyl) derivative 1719 of the trioxopiperazine natural product gliocladin C22,24 could be manipulated to access various members of the family of ETP natural products exemplified by 6–9, in addition to di(methylthio) analogues such as 11. We envisaged fashioning the epidisulfide by displacement of the oxygen substituents at C3 and C11a of precursor 15, which in turn should be available from stereoselective dihydroxylation of the C11,C11a double bond of alkylidene dioxoxopiperazine 16.25 Alternatively, stereoselective epoxidation of the double bond of intermediate 16 could also provide a suitably oxidized precursor for fashioning the ETP fragment. In both cases, we expected the oxidant to approach the double bond from the C11-Re face, as the X-ray structure of (+)- gliocladin C19 suggests that the fused-indoline ring would shield the C11-Si face (Figure 2). Tertiary alcohol 16 would result from chemoselective addition of an appropriate organometallic reagent to the most electrophilic carbonyl group of trioxopiperazine 17.26 Recently, we reported an efficient second-generation total synthesis of (+)-gliocladin C that proceeds via the intermediacy of 17.19 The central feature of this convergent synthesis was the late stage coupling of enantioenriched pro-di-electrophile 18 (or its enantiomer) with pro-dinucleophile 19, which allows access to gram quantities of either di-(tert-butoxycarbonyl)gliocladin C (17) or its enantiomer.
Figure 2.

X-Ray model of gliocladin C19 showing steric shielding of the C11, C11a double bond by the fused indoline fragment.
Enantioselective Total Synthesis of (+)-Gliocladine C and ent-Gliocladine C
We began our synthesis endeavors by targeting gliocladine C (6), which contains a methyl substituent at C3.2a Selective addition of methylmagnesium chloride to (+)-di-(tert-butoxycarbonyl)gliocladin C (17) at –78 °C gave alcohol product 20 as a 9:1 mixture of alcohol epimers (Scheme 2). It was critical in this reaction to maintain the temperature at −78 °C, as over methylation occurred at higher reaction temperatures. When the 9:1 mixture of epimers 20 was exposed to excess Et3N in DMF at room temperature, epimerization of the alcohol stereocenter by reversible opening of the dioxopiperazine ring resulted in a ~2.5:1.0 mixture of alcohol epimers. Acetylation of the 9:1 mixture of alcohol epimers 20 upon reaction with acetic anhydride and 4-dimethylaminopyridine (DMAP) at room temperature provided 21 as a separable mixture of acetate epimers in ratios that ranged from 3:1—9:1. As a distinction is necessary throughout this discussion, the protected alcohol epimers are classified as either top (T) or bottom (B) based upon their corresponding retention factors on silica gel thin layer chromatography, with (T) being the least polar. Dihydroxylation of the alkylidene double bond of the separated acetate epimers 21T (minor diastereomer) and 21B was achieved using the Sharpless ADmix-α conditions.27 Because of the hindered nature of the double bond, these reactions required the presence of additional K2OsO4•2H2O (0.25 equiv) and (DHQ)2PHAL (0.13 equiv) to proceed at a convenient rate. To our surprise, the major product isolated from the oxidation of 21T, diol 22, was determined to have the undesired β-configuration (vide infra). In contrast, oxidation of epimer 21B took place with modest facial selectivity from C11-Re face to give the ring-contracted oxazolidinone 23 (2:1 dr) in 61% yield. This product being the result of acetate cleavage under the basic dihydroxylation conditions to generate ketone 24.28,29
Scheme 2.

CD spectrum of 9 in EtOH (c = 2 × 10−4 M).
We turned to examine a sequence in which the alcohol substituent of intermediate 20 was masked with a base-stable tert-butyldimethylsiloxy group (Scheme 3). Silylation of the 9:1 mixture of alcohol epimers 20 provided a 3:2 mixture of siloxy epimers, the erosion in configuration at C3 undoubtedly arising from triethylamine-promoted epimerization of the tertiary alcohol prior to silylation. In our initial experiments, these siloxy epimers were separated on silica gel to provide pure samples of 25T (major diastereomer) and 25B. Epimers 25T and 25B were individually dihydroxylated with OsO4/NMO, AD-mix-α or AD-mix-β, with additional K2OsO4•2H2O and (DHQ)2PHAL [or (DHQD)2PHAL] being added to the latter reactions to increase the rate of dihydroxylation. The α-diol product was formed with high selectivity from oxidation of 25B, independent of the oxidant. In contrast, dihydroxylation of 25T using AD-mix-α yielded the α-diol selectively (dr = 14:1), whereas both AD-mix-β (dr = 5:1) and OsO4 afforded significant amounts of the β-diol product. The relative configuration of diol products 26 and 27 was assigned from diagnostic nuclear Overhauser enhancement (NOE) of the C11 methine hydrogen, as has been documented for diastereomers such as 28 and 29 (see Scheme 3).2a,21,30 For example, irradiation of the C11-H of diol 26 leads to an observable NOE signal with the aromatic C10-H resonance. In these rigid structures, a β-hydrogen at C11 is in close proximity to both C10-H and C4’-H, whereas, an α-hydrogen at C11 is held far from the C10-H such that the dominant NOE signals observed upon selective irradiation of this hydrogen occur with the C2’-H, C4’-H, and C5a-H.31 The diminished facial selectivity seen in dihydroxylation of siloxy epimer 25T from the C11-Re face appears not to be the result of steric interactions with the siloxy substituent, as the lowest selectivity was observed with the smallest oxidant OsO4.32
Scheme 3.

CD spectrum of 9 in EtOH (c = 2 × 10−4 M).
We were now set to address the critical incorporation of the epidithio bridge. Initially, we attempted to access gliocladine C (6) directly from diol 26B. However, the acid lability of a pyrrolidinoindoline containing a C11-hydroxyl substituent proved problematic, with 3,3’-biindole being the only product isolated upon exposure of 26B to excess H2S and BF3•OEt2 at room temperature. As this fragmentation undoubtedly arises by cleavage via a retro enamine-aldol process of the C10b–C11 bond,17,18 it was apparent that the C11-hydroxyl substituent would require protection prior to introducing the disulfide (Scheme 4). Therefore, the initially produced 3:2 mixture of siloxy epimers 25 was dihydroxylated with AD-mix-α and the crude diol products were acetylated to provide α-diacetates 30 in 76% yield over the two steps; this intermediate contained ~5% of the corresponding β-diacetates. Diacetate 30B was transformed into a crystalline intermediate, which allowed assignment of the relative configuration of the siloxy stereocenters of the epimers of intermediates 25, 26 and 30.33
Scheme 4.

CD spectrum of 9 in EtOH (c = 2 × 10−4 M).
Diacetates 30 were successfully transformed to (+)-gliocladine C in two additional steps. Exposure of intermediate 30 to 10 equiv of BF3•OEt2 in a 1:1 mixture of condensed H2S and CH2Cl2 initially at −78 °C with slow warming to room temperature, followed by oxidation with either iodine or air provided a single ETP product 32.34 Precisely how the oxidation step was carried out proved to be important, because the first step gave rise to a BF3 complex of a dithiol intermediate—presumably 31—even after aqueous work up.35 This intermediate was characterized by a combination of low-resolution mass spectrometry, 11B NMR, and 19F NMR analysis. After some experimentation, it was found that allowing this unpurified intermediate to react at room temperature with either iodine and Et3N in EtOAc, or an oxygen atmosphere in MeOH/EtOAc, provided ETP 32 in 53–70% yield from diacetates 30.36 All that remained was deacetylation, which was best accomplished by heating 32 at 45 °C in methanol in the presence of an excess of La(OTf)3.37 In this way, enantiopure (+)-gliocladine C (6), [α]23D + 505 (c 0.47 pyridine), was formed in 75% yield; this product exhibited spectroscopic and optical rotation data in agreement with those reported for the natural sample.2a,38 Starting with (−)-di-(tert-butoxycarbonyl)gliocladin C, (−)-17,39 ent-(+)-gliocladine C (ent-6), [α]23D −489 (c 0.16 pyridine), was prepared in identical fashion in 24% overall yield over six steps.40
The facial selectivity in the introduction of the epidisulfide bridge to form ETP 32 requires further comment. Diastereoselection in the addition of sulfur to C11a of an intermediate N-acyliminium ion intermediate is believed to result from steric shielding by the angular 3-indolyl substituent. An alternative explanation in which the C11 acetate plays a decisive role—for example, by formation of a dioxolanium ion intermediate—appears less likely in light of our earlier report of the transformation summarized in eq 1.37
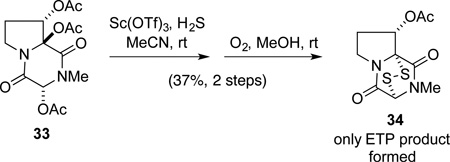 |
(1) |
Enantioselective Total Synthesis of (+)-Leptosin D
In order to establish the generality of the synthetic sequence we had developed, as well as to provide samples for biological evaluation, we turned to the synthesis of the ETP natural products 7–9 that differ in substitution at C3. Following the chemistry defined for the synthesis of gliocladine C, potential leptosin D precursor 37 was obtained in 46% yield over four steps from (+)-17 (Scheme 5). In this series, dihydroxylation of both siloxy epimers of intermediate 36 with AD-mix-α gave rise to the α-configured diacetates 37 in high distereoselectivity (20:1 dr). However, upon attempted formation of the corresponding ETP from 37, only monothiol 38 was obtained.
Scheme 5.

CD spectrum of 9 in EtOH (c = 2 × 10−4 M).
We postulated that a better leaving group at C3 would be required to overcome the increased A1,3 strain associated with generation of the C3 N-acyliminium ion intermediate in the isopropyl series. Thus, tertiary alcohol intermediate 35 was acetylated, providing acetate 39 as a 1.2:1.0 mixture of epimers. Upon dihydroxylation with AD-mix-α, 39T afforded the β-diol 40 with high stereoselectivity (20:1 dr), whereas 39B provided the α-diol 41 also with high stereoselectivity (20:1 dr).41 Fortunately, the formation of ring-contracted products analogous to 23 was not observed during Sharpless dihydroxylation of 39, presumably as a result of the larger isopropyl group mitigating cleavage of the C3 acetate. The relative configurations of diols 40 and 41 were assigned using NOE data in the manner previously discussed.31 Furthermore, NOE enhancements between the C11a hydroxyl proton of 41 and the isopropyl group indicated that these substituents were cis.
The elaboration of diol 41 to leptosin D is summarized in Scheme 7. Diacetylation of 41 with a large excess of acetic anhydride and DMAP provided diacetate 42 in 91% yield. As observed in the gliocladine C series, exposure of 42 to excess H2S in the presence of BF3•OEt2 at −78 °C to room temperature, followed by oxidation, provided a single epidisulfide product, 43, in 70% yield. Lewis acid-promoted deacetylation of 43 gave (+)-leptosin D (7) in 78% yield. The optical rotation of synthetic 7, [α]22D +423 (c 0. 225, CHCl3), compared well with the value reported for the natural sample, [α]21D +436 (c 0.51, CHCl3), as did all spectroscopic data.20
Scheme 7.

CD spectrum of 9 in EtOH (c = 2 × 10−4 M).
In a similar fashion, β-diol 40 was elaborated to ETP intermediate 45 in 62% overall yield (Scheme 8). Deacetylation of 45 gave 11-epi-leptosin D (46) in 80% yield. The absence of 1H NMR NOE enhancement between H11 and H10 and the presence of an NOE between H11 with both H2’ and H4’, confirmed the relative configuration of the secondary alcohol of ETP 46. 1H NMR spectral data for synthetic 46 were essentially identical to that reported for 48, a degradation product of leptosin K1 (eq 2).30,42 The absolute configuration of 48 had been established by CD spectroscopy, thus 48 and synthetic 11-epi-leptosin D (46) should be enantiomers. However, the rotation of 46, ([α]D 23 +212 (c 0.13, CHCl3) is of the same sign and similar magnitude to that reported for leptosin K1 degradation product 48, [α]D23 +179 (c 0.17, CHCl3).
Scheme 8.
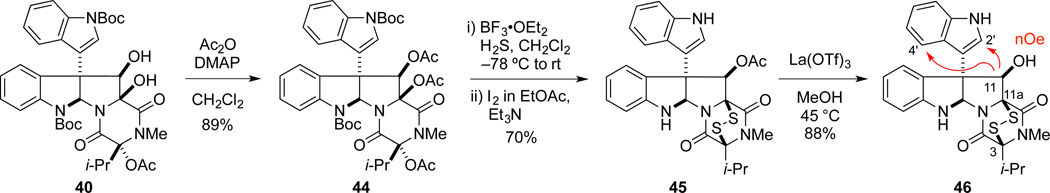
CD spectrum of 9 in EtOH (c = 2 × 10−4 M).
 |
(2) |
We turned to CD spectroscopy to clarify the structural ambiguity associated with synthetic 11-epi-leptosin D (46) and leptosin K1 degradation product 48. Epidisulfides have been the subject of a number of theoretical and experimental CD studies.30,43 The sign of the bands at approximately 230 and 270 nm—with more emphasis on the 270 nm band—can be used to assign the absolute configuration of the epidisulfide bridge in ETPs of the type considered here. The contribution of transitions arising from both the disulfide bonding orbitals to the antibonding σ-orbitals of the dioxopiperazine (n to σ* transition) and the peptide n to π* transition in the dioxopiperazine ring dominate at about 230 nm for disulfides with small C–S–S–C dihedral angles, with a positive band typically correlating with the 3S,11aS configuration.43d,44 Whereas a charge-transfer band arising from an interaction of the bonding disulfide orbitals with the antibonding π-orbitals of the dioxopiperazine (n to π* charge-transfer) dominates at about 270 nm, with a negative band corresponding to the 3S,11aS epidisulfide configuration.43d
In our studies, the negative CD band at 265 nm for (+)-11-epi-leptosin D (46) and the similar band observed for (+)-leptosin D (7) provide strong support for our assignment of the 3S,11aS absolute configuration for these products (Figure 3). Similarly, the CD data reported by Numata and coworkers for epidisulfide 48 are in good agreement with the assigned 3R,11aR configuration for this product.30 As NOE data for synthetic 11-epi-leptosin D (46) support the relative configuration at C11, we surmise that the optical rotation for 48 was reported incorrectly by the earlier authors.
Figure 3.

CD spectrum of 7 and 46 in EtOH (c = 2 × 10−4 M).
Enantioselective Total Synthesis of (+)-T988C
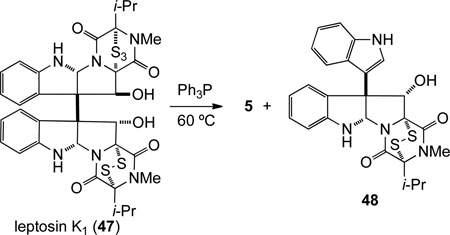
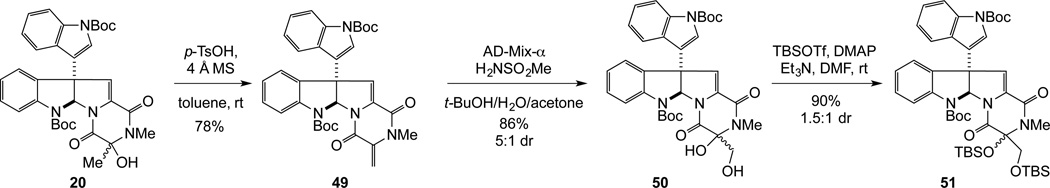
The total synthesis of T988C (8) posed an additional synthetic challenge: an efficient method for installing the hydroxymethyl side chain, preferably protected as its tert-butyldimethylsilyl ether to allow concomitant deprotection during formation of the epidisulfide, would be required. Initial efforts focused on SmI2-mediated45 addition of (tert-butyldimethylsiloxy)methyl chloride46 to the C3 carbonyl group of trioxopiperazine 17. However, addition of this organometallic reagent to 17 was not observed. Therefore, our attention turned to the formation of an exomethylene, which could be oxidized to provide the hydroxymethyl functionality. In this vein, the Wittig reaction of (+)-17 with methyltriphenylphosphorane was examined and found to be capricious. However, acid mediated dehydration of hemiaminal 20 provided dialkylidene dioxopiperazine 49 in 78% yield (Scheme 9). The exomethylene unit of 49 could be selectively epoxidized using dimethyldioxirane;47 however, the product was not stable and only diol 50 was isolated in low yield. After some experimentation, selective dihydroxylation of the exocyclic methylidene of 49 was achieved using AD-mix-α without additional potassium osmate and (DHQ)2PHAL, giving diol 50 in 86% yield.48 Protection of the 5:1 mixture of epimeric diols 50 as their corresponding tert-butyldimethylsilyl ethers provided 51 a separable 1:1.5 mixture of disilylepimers.
Scheme 9.
CD spectrum of 9 in EtOH (c = 2 × 10−4 M).
Fortunately, both epimeric disilyl ethers 51 could be elaborated to (+)-T988C (8). Oxidation of 51T or 51B using our augmented AD-mix-α conditions took place with high diastereoselectivity to provide 52 in good yield as predominantly the α-diol stereoisomers (20:1 dr) (Scheme 10). Acetylation of the combined diols 52 (1:1.5 with respect to the C3 center) afforded α-diacetate 53 as a mixture of siloxy epimers.49 Treatment of this mixture of silylether epimers 53 with excess H2S in the presence of BF3•OEt2 at −78 °C to room temperature, followed by oxidation with iodine and Et2N, provided a single epidisulfide product, 54, in 70% yield. To realize this yield, it was essential in the first step to increase the amount of BF3•OEt2 to 30 equiv, otherwise a separable mixture of 54 and the corresponding product in which the primary silyl ether had not been removed was isolated. Deacetylation of 54 as before, provided (+)-T988C (8). The optical rotation of synthetic 8, [α]23D +282 (c 0. 024, MeOH), compared well with the value reported for the natural sample, [α]21D +277 (c 0.0006, MeOH), as did all spectroscopic data.21
Scheme 10.

CD spectrum of 9 in EtOH (c = 2 × 10−4 M).
Enantioselective Total Synthesis of (+)-Bionectin A and (+)-Gliocladin A
Focus then turned toward the preparation of the bionectin A (9)2b and gliocladin A (11), which lack an alkyl substituent at C3.22,2b Reduction of the trioxopiperazine (+)-17 with NaBH4 at −42 °C gave a 1:1 mixture of epimeric alcohol products (Scheme 11). In contrast, reduction with lithium tri-sec-butylborohydride (L-selectride) at −78 °C was highly stereoselective, providing alcohol 55 as a 14:1 mixture of alcohol epimers. Silylation of this 14:1 mixture of epimers 55 took place without epimerization at C3 to give 56 as a separable 14:1 mixture of C3-siloxy epimers. The relative configurations of these epimers were tentatively assigned by their relative polarities. In general, we have found that epimers possessing a β-oriented C3 oxygen substituent (cf. 25B and 39B) were more polar on silica gel. Dihydroxylation of 56, and subsequent diacetylation of the major α-diol product 57 afforded intermediate 58 in 75% yield over two steps.49
Scheme 11.

CD spectrum of 9 in EtOH (c = 2 × 10−4 M).
Intermediate 58 could be employed to introduce either the epidisulfide or the di(methylthio) functionality of 9 and 11, respectively (Scheme 12). To this end, 58 was exposed to BF3•OEt2 and MeSH to afford a separable 3:1 mixture of di(methylthio) derivative 61 and a methylthio stereoisomer. It was readily established by 1D NOE analysis that these products differ in configuration at C3, with the C11a methylthio substituent being β-oriented in each stereoisomer.50 Therefore, a two-step sequence was used to stereoselectively introduce the di(methylthio) functionality. In this sequence, the unpurified dithiol intermediate 59 formed from BF3•OEt2-promoted reaction of 58 with H2S was allowed to react with excess MeI and K2CO3 in acetone to yield 61 as a single stereoisomer.51 The acetyl group was then removed to provide (+)-gliocladin A (11). In this step, the addition of DMAP was necessary to increase the rate of acetyl cleavage, otherwise decomposition pathways appeared to dominate because of instability of the product to the reaction conditions. Under the best conditions found, the reaction returned a 1.5:1.5:1.0 mixture of dithioether 61, gliocladin A (11), and 3,3’-biindole. The optical rotation of synthetic 11, [α]23D +266 (c 0. 34, MeOH), compared well with the value reported for the natural sample, [α]21D +244 (c 0.2, MeOH), as did spectroscopic data.22,2b
Scheme 12.

CD spectrum of 9 in EtOH (c = 2 × 10−4 M).
Dithiol intermediate 59 was converted also to the corresponding epidisulfide by exposure iodine and Et3N. To our surprise, the resulting product 60 was somewhat unstable and was best transformed immediately to the corresponding alcohol product. This deacylation had to be carried out under carefully prescribed conditions; judicious choice of both substrate concentration and temperature were paramount, as increasing either afforded significant quantities of 3,3’-biindole. The product 9 isolated after acetyl cleavage exhibited spectroscopic and mass spectrometric data expected for this ETP product. However, NMR data for synthetic bionectin A (9) did not agree as closely with the data reported by Zheng and coworkers2b as similar comparisons of the other natural products prepared in this study.52,53 The structural assignment for synthetic 9 was based upon 1D and 2D NMR data, including NOE data confirming that the C11-OH is α-oriented. In addition, a positive CD band at 230 nm and negative band at 270 nm) indicated that the ETP moiety possessed the 3S,11aS configuration (Figure 4). We also determined that the small differences in 1H NMR shifts between synthetic 9 and those reported for bionectin A were not an artifact of different protonation states by showing that the 1H NMR signals of 9 were unchanged upon sequentially adding 0.1–2 equiv of trifluoroacetic acid.54 As the largest difference in 1H NMR resonances was seen for C5a-H and N(6)-H (Δδ = 0.21 for each), we speculate that the natural product contained a small amount of a metal impurity bound between N(6) and the proximal carbonyl group.55
Figure 4.

CD spectrum of 9 in EtOH (c = 2 × 10−4 M).
Biological Evaluation
Having synthesized a number of epidithiodioxopiperazines and related structures, their activity against two invasive cancer cell lines, DU145 (human prostate cancer) and A2058 (melanoma), were determined (Table 1). Analysis of the data reveals a number of trends. Most notably a disulfide is required for activity, with di(methylthio) congener 11 (entry 12) being inactive within the detection limits of the assay.1,56–58 In regard to the active compounds, potency against the two cancer cell lines varies little whether the C11-hydroxyl group is acetylated (entries 5–7) or not (entries 1–3). However, the relative configuration of the C11-oxygen does affect activity (entries 2 and 6 vs 8 and 9), such that a 4–10 fold decrease in activity is seen when the C11-oxygen is cis to the epi-disulfide (entries 8 and 9 compared with entries 1–7). Furthermore, the data suggest that the relative configuration of the epi-disulfide bridge relative to the C10b quaternary carbon center is important for activity, with glioclatine (10) being about 2–3 fold more potent than diastereomer 62 (entries 10 and 11).59 Absolute configuration does not appear to be important for activity, as gliocladine C and its enantiomer possess similar activities against DU145 and A2058 (entries 1 and 13). The presence of a hydroxyl substituent at C11 has only a minor influence, with glioclatine being about 2-fold less active than gliocladine C (entries 1 and 10). As ETP natural products 6–9 had been isolated by different coworkers and screened in different biological assays, the data in Table 1 allow the first insight into the role—minor as it turns out—of the C3 substituent (entries 1–7). We also had the opportunity to test chaetocin A (4), which possess two epidithiodioxopiperazine fragments, in the same assays. When compared to T988C, which has the same C3 hydroxymethyl substituent, chaetocin A is 7 times more active against DU145 than T988C and 16 times more active against A2058 (entries 3 and 14).60 However it merits note that chaetocin A is only 3.5 times more active against A2058 melanoma than the acetoxy derivative 32 of gliocladine C or leptosin D (entries entries 2, 5, and 14).
Table 1.
Activity against invasive prostate cancer (DU145) and melanoma (A2508) cell lines.a
| Entry | Structure | DU145 IC50 |
A2058 IC50 |
|
|---|---|---|---|---|
| 1 | 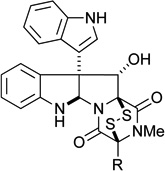 |
R = Me (6) gliocladine C |
0.43 μM | 0.68 μM |
| 2 | R = i-Pr (7) leptosin D |
0.29 μM | 0.85 μM | |
| 3 | R = CH2OH (8) T988C |
0.53 μM | 0.96 μM | |
| 4 | R = H (9) | 0.70 μM | 2.0 μM | |
| 5 | 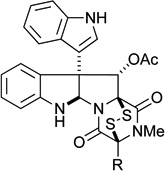 |
R = Me (32) | 0.26 μM | 0.96 μM |
| 6 | R = i-Pr (43) | 0.39 μM | 1.0 μM | |
| 7 | R = CH2OH (54) | 0.50 μM | 1.2 μM | |
| 8 | 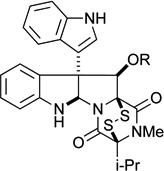 |
R = H (46) | 2.8 μM | 4.0 μM |
| 9 | R = Ac (45) | 2.5 μM | 4.2 μM | |
| 10 | 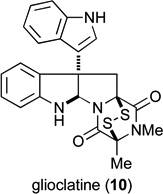 |
0.85 μM | 1.8 μM | |
| 11 | 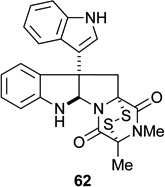 |
2.3 μM | 3.8 μM | |
| 12 | 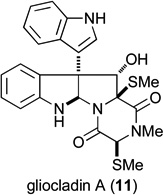 |
>5 μM | >5 μM | |
| 13 | ent-gliocladine C | 0.68 μM | 0.35 μM | |
| 14 | 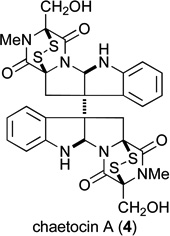 |
0.073 μM | 0.061 μM | |
Conclusions
The first total syntheses of the epidithiodioxopiperazine natural products gliocladine C (6), leptosin D (7), T988C (8), and bionectin A (9) are reported, as well as total syntheses of ent-gliocladin C and the di(methylthio)-containing natural product gliocladin A (11). In these syntheses, the α-ketoimide carbonyl and alkylidene double bond of the trioxopiperazine unit of di-(tertbutoxycarbonyl) gliocladin C (17) are exploited to incorporate the C3 substituent and stereoselectively introduce the C11 hydroxyl group and the disulfide bridge of the ETP natural products 6–9. Pivotal steps in the syntheses are; (a) introduction of the C3 substituent by nucleophilic addition to the C3 carbonyl group, (b) stereoselective dihydroxylation of the C11-C11a double bond, and (c) stereoselective introduction of sulfur substituents at C3 and C11a by BF3•OEt2-promoted reaction of H2S with precursors containing an acetoxy leaving group at C11a and a siloxy (or acetoxy) leaving group at C3, followed by mild oxidation. Stereoselection in forming the disulfide bridge is suggested to result from initial iminium ion formation at C11a, followed by kinetically controlled trapping with H2S from the face opposite the angular 3-indolyl substituent.
The concise chemical synthesis of (+)- or (−)-17 (10 steps and 15% overall yield from isatin)19 and the divergent sequences developed in this study for elaborating these intermediates (6–8 steps, 14–25% yield) provided ample quantities of each targeted compound for in vitro cytotoxicity evaluations using DU145 prostrate and A2058 melanoma cells. As would be expected, the disulfide was required for activity.1,56 As reported for chaetocin A and its enantiomer in inhibiting histone methyltranseferase G9a,11 gliocladine C and its enantiomer exhibited similar activities against both cancer cell lines. The role of the C3 substituent, which was systematically varied in this study, was found to be minor, as was the effect of a C11 oxygen substituent. Synthetic T988C, which has the same C3 hydroxymethyl substituent as the ETP fragments of chaetocin A yet a single ETP fragment, was ~10 times less active than chaetocin A. However, the presence of two ETP fragments in not essential to realize good activity, because chaetocin A is only 3.5 times more active against A2058 melanoma than the acetoxy derivative 32 of gliocladine C or leptosin D.
Supplementary Material
Scheme 1.

CD spectrum of 9 in EtOH (c = 2 × 10−4 M).
Scheme 6.

CD spectrum of 9 in EtOH (c = 2 × 10−4 M).
Acknowledgement
Support from the NIH National Institutes of General Medical Sciences (5R01GM030859 to LEO), the A. Gary Anderson Family Foundation (to DH), and postdoctoral fellowship support to JED (GM090473), SMM (GM082113), and FLZ (Swiss NSF) is gratefully acknowledged. NMR and mass spectra were determined at UC Irvine using instruments purchased with the assistance of NSF and NIH shared instrumentation grants. We thank Dr. John Greaves, Department of Chemistry, UC Irvine, for assistance with mass spectrometric analyses, and Professor Atsushi Numata for providing spectral data files for leptosin D.
Footnotes
Supporting Information. Experimental details for synthesis of new compounds and their cytotoxicity assay, copies of 1H and 13C NMR spectra of new compounds, and CD spectra for ETP products. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.(a) Vigushin DM, Mirsaidi N, Brooke G, Sun C, Pace P, Inman L, Moody CJ, Coombes RC. Med. Oncol. 2004;21:21–30. doi: 10.1385/MO:21:1:21. [DOI] [PubMed] [Google Scholar]; (b) Waring P, Eichner RD, Müllbacher A. Med. Res. Rev. 1988;8:499–524. doi: 10.1002/med.2610080404. [DOI] [PubMed] [Google Scholar]; (c) Jiang CS, Guo YW. Mini-Rev. Med. Chem. 2011;11:728–745. doi: 10.2174/138955711796355276. [DOI] [PubMed] [Google Scholar]; (d) Cornacchia C, Cacciatore I, Baldassarre L, Mollica A, Feliciani F, Pinnen F. Mini-Rev. Med. Chem. 2012;12:2–12. doi: 10.2174/138955712798868959. [DOI] [PubMed] [Google Scholar]; (e) Borthwick AD. Chem. Rev. 2012;112:3641–3716. doi: 10.1021/cr200398y. [DOI] [PubMed] [Google Scholar]
- 2.Dong J-Y, He HP, Shen Y-M, Zhang KQ. J. Nat. Prod. 2005;68:1510–1513. doi: 10.1021/np0502241. [DOI] [PubMed] [Google Scholar]; (b) Zheng C-J, Kim C-J, Bae KS, Kim Y-H, Kim W-G. J. Nat. Prod. 2006;69:1816–1819. doi: 10.1021/np060348t. [DOI] [PubMed] [Google Scholar]; (c) Chen Y, Du HG, Liu X-Z, Che Y, Ye X. Cell. Prolif. 2009;42:838–847. doi: 10.1111/j.1365-2184.2009.00636.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Erkel G, Gehrt A, Anke T, Sterner O. Z. Naturforsch. 2002;57C:759–767. doi: 10.1515/znc-2002-7-834. [DOI] [PubMed] [Google Scholar]; (b) Isham CR, Tibodeau JD, Jin W, Xu R, Timm MM, Bible KC. Blood. 2007;109:2579–2588. doi: 10.1182/blood-2006-07-027326. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, Cornell–Kennon S, Lee J, Wang B, Wang J, Memmert K, Naegeli HU, Petersen F, Eck MJ, Bair KW, Wood AW, Livingston DM. Cancer Cell. 2004;6:33–43. doi: 10.1016/j.ccr.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 4. Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Nat. Chem. Biol. 2005;1:143–145. doi: 10.1038/nchembio721. Yanagihara M, Sasaki–Takahashi N, Sugahara T, Yamamoto S, Shinomi M, Yamashita I, Hayashida M, Yamanoha B, Numata A, Yamori T, Andoh T. Cancer Sci. 2005;96:816–824. doi: 10.1111/j.1349-7006.2005.00117.x. Block KM, Wang H, Szabó LZ, Polaske NW, Henchey LK, Dubey R, Kushal S, László CF, Makhoul J, Song Z, Meuillet EJ, Olenyuk BZ. J. Am. Chem. Soc. 2009;131:18078–18088. doi: 10.1021/ja807601b. Cook KM, Hilton ST, Mecinovi J, Motherwell WB, Figg WD, Schofield CJ. J. Biol. Chem. 2009;284:26831–26838. doi: 10.1074/jbc.M109.009498. Tibodeau JD, Benson LM, Isham CR, Owen WG, Bible KC. Antioxid. Redox Signaling. 2009;11:1097–1106. doi: 10.1089/ars.2008.2318. Teng Y, Iuchi K, Iwasa E, Fujishino S, Hamashima Y, Dodo K, Sodeoka M. Biorg. Med. Chem. Lett. 2010;20:5085–5088. doi: 10.1016/j.bmcl.2010.07.032. Park HB, Kim Y-J, Park JS, Yang HO, Lee KR, Kwon HC. J. Nat. Prod. 2011;74:2309–2312. doi: 10.1021/np200563x. Lee Y-M, Lim JH, Yoon H, Chun YS, Park JW. Hepatology. 2011;53:171–180. doi: 10.1002/hep.24010. Chaib H, Nebbioso A, Prebet T, Castellano R, Garbit S, Restouin A, Vey N, Altucci L, Colllette Y. Leukemia. 2012;26:662–674. doi: 10.1038/leu.2011.271.; and references 1 and 3
- 5.Gardiner DM, Waring P, Howlett BJ. Microbiology. 2005;151:1021–1032. doi: 10.1099/mic.0.27847-0. [DOI] [PubMed] [Google Scholar]
- 6.Kishi Y, Nakatsuka S, Fukuyama T, Havel M. J. Am. Chem. Soc. 1973;95:6493–6495. doi: 10.1021/ja00800a079. [DOI] [PubMed] [Google Scholar]
- 7. Fukuyama T, Kishi Y. J. Am. Chem. Soc. 1976;98:6723–6724. doi: 10.1021/ja00437a063.. For a full account, see: Fukuyama T, Nakatsuka SI, Kishi Y. Tetrahedron. 1981;37:2045–2078.
- 8.Kishi Y, Fukuyama T, Nakatsuka S. J. Am. Chem. Soc. 1973;95:6490–6492. doi: 10.1021/ja00800a078. [DOI] [PubMed] [Google Scholar]
- 9.(a) Kishi Y, Fukuyama T, Nakatsuka S. J. Am. Chem. Soc. 1973;95:6492–6493. doi: 10.1021/ja00800a078. [DOI] [PubMed] [Google Scholar]; (b) Wu Z, Williams LJ, Danishefsky SJ. Angew. Chem. Int. Ed. 2000;39:3866–3868. doi: 10.1002/1521-3773(20001103)39:21<3866::AID-ANIE3866>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 10.Kim J, Ashenhurst JA, Movassaghi M. Science. 2009;324:238–241. doi: 10.1126/science.1170777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iwasa E, Hamashima Y, Fujishiro S, Higuchi E, Ito A, Yoshida M, Sodeoka M. J. Am. Chem. Soc. 2010;132:4078–4079. doi: 10.1021/ja101280p. [DOI] [PubMed] [Google Scholar]
- 12.Nicolaou KC, Totokotsopoulos S, Giguère D, Sun YP, Sarlah D. J. Am. Chem. Soc. 2011;133:8150–8153. doi: 10.1021/ja2032635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Codelli JA, Puchlopek ALA, Reisman SE. J. Am. Chem. Soc. 2012;134:1930–1933. doi: 10.1021/ja209354e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fujiwara H, Kurogi T, Okaya S, Okana K, Tokoyama H. Angew. Chem. Int. Ed. 2012;51:13062–13065. doi: 10.1002/anie.201207307. [DOI] [PubMed] [Google Scholar]
- 14.Nicolaou KC, Lu M, Totokotsopoulos S, Heretsch P, Giguerè D, Sun YP, Sarlah D, Nguyen TH, Wolf IC, Smee DF, Day CW, Bopp S, Winzeler SA. J. Am. Chem. Soc. 2012;134:17320–17332. doi: 10.1021/ja308429f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.For a summary of methods for constructing the polysulfide fragments of ETPs, see reference 12.
- 16.Kim J, Movassaghi M. J. Am. Chem. Soc. 2010;132:14376–14378. doi: 10.1021/ja106869s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minato H, Matsumoto M, Katayama T. J. Chem. Soc. Perkin Trans. 1973;1:1819–1825. doi: 10.1039/p19730001819. [DOI] [PubMed] [Google Scholar]
- 18.Overman LE, Shin Y. Org. Lett. 2007;9:339–341. doi: 10.1021/ol062801y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.For the preliminary communication of the total synthesis of (+)-gliocladine C, see: DeLorbe JE, Jabri SY, Mennen SM, Overman LE, Zhang FL. J. Am. Chem. Soc. 2011;133:6549–6552. doi: 10.1021/ja201789v.
- 20.Takahashi C, Numata A, Ito Y, Matsumura E, Araki H, Iwaki H, Kushida K. J. Chem. Soc. Perkin Trans. 1994;1:1859–1864. [Google Scholar]
- 21.Feng Y, Blunt JW, Cole ALJ, Munro MHG. J. Nat. Prod. 2004;67:2090–2092. doi: 10.1021/np030326w. [DOI] [PubMed] [Google Scholar]
- 22. Usami Y, Yamaguchi J, Numata A. Heterocycles. 2004;63:1123–1129. (b) Gliocladin C was recently isolated from a terrestrial fungus, see: Bertinetti BV, Rodriguez MA, Godeas AM, Cabrera GM. J. Antibiot. 2010;63:681–683. doi: 10.1038/ja.2010.103.
- 23.Boyer and Movassaghi recently reported the synthesis of the di(methylthio)-containing natural product, gliocladin B (12), see: Boyer N, Movassaghi M. Chem. Sci. 2012;3:1798–1803. doi: 10.1039/C2SC20270K.
- 24.For syntheses of (+)-gliocladin C, see references: 18, 19, 23; Furst L, Narayanam JMR, Stephenson CRJ. Angew. Chem. Int. Ed. 2011;50:9655–9659. doi: 10.1002/anie.201103145.
- 25.The numbering system for leptosin D used by Numata and coworkers is employed.20 For a discussion of the various positional numbering systems used in this area, see p S3 of ref 10.
- 26.Person D, Le Corre M. Bull. Soc. Chim. Fr. 1989;5:673–676. [Google Scholar]
- 27.Hentges SG, Sharpless KB. J. Am. Chem. Soc. 1980;102:4263–4265. [Google Scholar]
- 28.The pH of the reaction medium was measured to be approximately 11
- 29.Exposure at room temperature of 21T to K2CO3 in acetone/H2O predominantly afforded 20 as a mixture of alcohol epimers (~3:1). In addition 21B was found to slowly undergo elimination of acetic acid to form a dienamide (49, vide infra) upon standing neat, whereas this decomposition pathway was not detected for 21T, suggesting that multiple pathways might lead to 23
- 30.Takahashi C, Minoura K, Yamada T, Numata A, Kushida K, Shingu T, Hagishita S, Nakai H, Sato T, Harada H. Tetrahedron. 1995;51:3483–3498. [Google Scholar]
- 31.One-dimensional NOE data obtained by irradiation of the C11-H of diol products 22, 23, 26T, 26B, 40, 41 is summarized in corresponding experimental procedures for the preparation of each compound (see Supporting Information).
- 32.The origin of diminished selectivity in dihydroxylation of the C11,C11a double bond in 25T from the C11-Re face is unclear.
- 33.Salman Jabri, unpublished studies at UC Irvine.
- 34.For an early construction of epidithiodioxopiperazines from the acid-promoted reaction of H2S with dioxopiperazines having leaving groups at C3 and C6, see: Ottenheijm HCJ, Kerkhoff GPC, Bijen JWHA, Spande TF. J. Chem. Soc. Chem. Commun. 1975:768–769.
- 35.This intermediate could be a mixture of C3 thiol epimers or BF3 complexes, as one major and at least one minor species are apparent in the 11B NMR and 19F NMR spectra (see Supporting Information).
- 36.The relative configuration of this product was confirmed by single crystal X-ray diffraction of the corresponding racemate, CCDC 814557
- 37.Overman LE, Sato T. Org. Lett. 2007;9:5267–5270. doi: 10.1021/ol702518t. [DOI] [PubMed] [Google Scholar]
- 38.The relative configuration of 6 was in accord with single-crystal X-ray analysis; however, the data set did not refine to a high level.
- 39.Prepared analogously to (+)-17 by use of (+)-4- pyrrolidinopyridinyl(pentamethylcyclopentadienyl)iron catalyst in the enantiodetermining step of the synthesis, see: reference 19
- 40.See Supporting Information.
- 41.It is of note that dihydroxylation of both 21T and 39T, which both have acetate substituents at C3, preferentially provided the β-diol products.
- 42.The relative configuration of leptosin K (5, see Fig 1), which was chemically correlated with leptosin K1, was established by single-crystal X-ray analysis, see: reference 30
- 43. Carmack M, Neubert LA. J. Am. Chem. Soc. 1967;89:7134–7136. Hauser D, Weber HP, Sigg HP. Helv. Chim. Acta. 1970;53:1061–1073. doi: 10.1002/hlca.19700530521. Minato H, Matsumoto M, Katayama T. J. Chem. Soc. D. 1971. 1971:44–45. Nagarajan R, Woody RW. J. Am. Chem. Soc. 1973;95:7212–7222. doi: 10.1021/ja00803a005. Woody RW. Tetrahedron. 1973;29:1273–1283.. (f) Reference 20
- 44.In some cases, the presence of other chromophores may make the use of the 230 nm band problematic, see: reference 43d.
- 45.Antonsen Ø, Benneche T, Undheim K. Acta Chem. Scand. 1992;46:757–760. [Google Scholar]
- 46.(a) Benneche T, Gundersen LL, Undheim K. Acta Chem. Scand. 1988;42B:384–389. [Google Scholar]; (b) Gundersen LL, Benneche T, Undheim K. Acta Chem. Scand. 1989;43:706–709. [Google Scholar]
- 47.Bartels A, Jones PG, Liebscher J. Synthesis. 2003:67–72. [Google Scholar]
- 48.Little chemoselectivity was realized using OsO4 and N-methylmorpholine N-oxide (NMO).
- 49.After purification, this product contained <5% β-diacetate.
- 50.See Supporting Information for 1D NOE data.
- 51.We have observed similar increases in stereoselectivity using this two-step sequence in the synthesis of other structurally related di(methylthio)dioxopiperazines. Whether the increase in stereoselectivity results form thermodynamic equilibration of dithiol intermediates, or involvement of the β-C11a-thiol intermediate in delivery of the second equivalent of H2S, or other factors is unclear at present.
- 52.See Supporting Information for spectral comparisons of NMR spectra of synthetic and isolated epidisulfide natural products.
- 53.Mean |Δδ|=0.073 ppm (1H NMR) and 0.57 ppm (13C NMR).
- 54.See Supporting Information for experimental details and spectral data.
- 55.Unfortunately we were unable to secure a comparison sample of natural bionectin A for direct comparison.
- 56.For a recent study of other gliotoxin and related natural products, see: Takahashi M, Takemoto Y, Shimazu T, Kawasaki H, Tachibana M, Shinkai Y, Takagi M, Shin-ya K, Igarashi Y, Ito A, Yoshida M. J. Antibiot. 2012;65:263–265. doi: 10.1038/ja.2012.6.
- 57.While this article was in review, Movassaghi, Hergenrother and coworkers reported in vitro cytotoxicity activity against several cancer cell lines (all different from the invasive cell lines employed in our studies) of 50 molecules synthesized in the Movassaghi laboratory, many containing ETP fragments or related dithio substituents.58 The "monomeric" members of this set were derived largely from sarcosine, thus containing a hydrogen substituent at C3, and none contained oxidation at C11. Although this impressive study and the one we report here largely targeted different sites for variation, two common new themes emerged: the importance of the configuration of the disulfide bridge and the minimal change in activity brought about by variations in the C3 substituent.
- 58.Boyer N, Morrison KC, Kim J, Hergenrother PJ, Movassaghi M. Chem. Sci. 2013 doi: 10.1039/C3SC50174D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Characterization data for these structures are provided in the Supporting Information.
- 60.Similar levels of cytotoxicity of T988C against five additional cancer cell lines has been reported recently, see: Li L, Luan Y, Gu Q, Zhu T. J. Nat. Prod. 2012;75:920–927. doi: 10.1021/np3000443.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.