

Abstract
Nanocrystals have drawn increasing interest in pharmaceutical industry because of the ability to improve dissolution of poorly water-soluble drugs. Nanocrystals can be produced by top-down and bottom-up technologies and have been explored for a variety of therapeutic applications. Here we review the methods of nanocrystal production and parenteral applications of nanocrystals. We also discuss remaining challenges in the development of nanocrystal products.
Keywords: Nanocrystals, nanosuspension, parenteral applications, poorly water-soluble drugs
1. Introduction
Approximately 40% of active pharmaceutical ingredients in the discovery stage have poor water-solubility [1]. In order to attain adequate bioavailability of poorly soluble drugs, special formulation strategies are employed to increase their dissolution in aqueous medium [2]. Traditionally, organic solvents are used as co-solvents [3] or a part of emulsion [4] to formulate the poorly soluble drugs as aqueous dosage forms. Alternatively, the drug is fitted in cyclodextrins, which have a hydrophobic interior and a hydrophilic exterior, and made soluble in water [5]. Another way of enhancing the solubility of poorly soluble drugs is to produce nanoparticulate formulations, such as liposomes [6], micelles [7], nanoemulsion [8], solid lipid nanoparticles [9], and polymeric nanoparticles [10]. However, relatively low drug loading efficiency, concerns for the safety of excipients, and complicated manufacturing process are noted as potential disadvantages of these strategies.
Nanocrystallization is a technique to produce crystalline particles of poorly soluble drugs in the nanometer range (i.e., nanocrystals). Due to the size and, thus, the high surface area to volume ratio, nanocrystals can increase the saturation solubility of a drug and the dissolution rate of drug particles [11]. Nanocrystals have gained increasing interest in the pharmaceutical industry because of the simple structures and compositions. They have been explored for a variety of therapeutic applications including oral [12], dermal [13], pulmonary [14], systemic administration [15], as well as targeted drug delivery [16] and intraperitoneal chemotherapy [17]. The objective of this article is to review the production of nanocrystals and their therapeutic applications focusing on parenteral use. We will also discuss the remaining challenges in the development of nanocrystal products.
2. Production of Nanocrystals
Nanocrystals of poorly soluble drugs can be created by “top-down” or “bottom-up” technologies (Fig. 1), or combinations of the two [18]. Nanocrystals are produced from the drug itself, with surfactants or polymeric stabilizers on the surface; thus, the drug content in nanocrystals approaches 100% [18]. The production of nanocrystals is relatively easy to scale up and transfer to industry as compared to other formulations on the market, such as liposomes [19, 20] and albumin-based nanoparticles [21]. Several nanocrystal products, produced by wet milling and high-pressure homogenization, have been approved by the US Food and Drug Administration as oral products (Table 1).
Fig. 1.
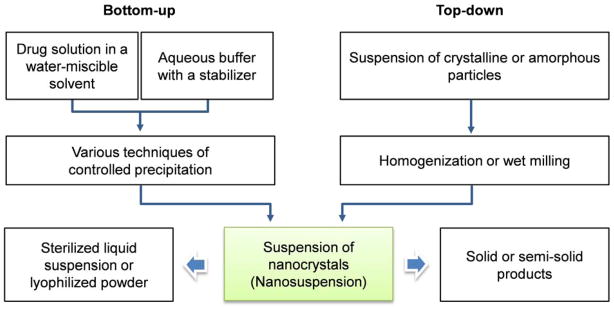
Schematics of bottom-up and top-down production of nanocrystals. Suspensions of nanocrystals can be further processed into sterile products or other dosage forms.
Table 1.
| Trade name (drug) | Manufacturing techniques | Indications | Company |
|---|---|---|---|
| Rapamune® (Sirolimus) | Top-down, wet milling | Immunosuppressive | Wyeth Pharmaceuticals |
| Emend® (Aprepitant) | Top-down, wet milling | Antiemetic | Merck & Co. |
| Tricor® (Fenofibrate) | Top-down, wet milling | Hypercholesterolemia | Abbott Laboratories |
| Triglide® (Fenofibrate) | Top-down, high-pressure homogenization | Hypercholesterolemia | Skye Pharma |
| Megace ES® (Megestrol acetate) | Top-down, wet milling | Antianorexic | Par Pharmaceutical |
| Avinza® (Morphine sulfate) | Top-down, wet milling | Psychostimulant drug
|
King Pharmaceuticals |
| Focalin® XR (Dexmethyl-phenidate HCl) | Top-down, wet milling | Attention deficit hyperactivity disorder | Novartis |
| Ritalin® LA (Methylphenidate HCl) | Top-down, wet milling | Attention deficit hyperactivity disorder | Novartis |
| Zanaflex Capsules™ (Tizanidine HCl) | Top-down, wet milling | Muscle relaxant | Acorda |
2.1 Bottom-up technologies
The bottom-up approach refers to methods that create small drug particles from drug molecules dissolved in an organic solution. Small drug particles are formed as drug molecules precipitate from solution in the presence of an agent and/or a condition that induces nucleation of the molecules. For example, a non-solvent, which is miscible with the solvent but does not dissolve the drug, is used to induce the nanocrystal formation, in conjunction with various methods to mix the drug solution with non-solvents such as rotation, liquid jets, or multi-inlet vortex mixing [22]. Alternatively, supercritical fluid, ultrasonic waves, or controlled solvent evaporation are employed to induce drug precipitation. These technologies are discussed in detail in a recent review article [22].
Particles produced by the bottom-up approach can be crystalline or amorphous. Amorphous nanoparticles produced by a technique called Nanomorph™ achieve higher saturation solubility and faster dissolution rate than nanocrystals [23, 24]. However, they are prone to partial or complete re-crystallization, which may lead to decreased bioavailability. Due to the stability and consistent performance, nanocrystals are usually favored over amorphous particles. On the other hand, the production of nanocrystals within a desired size range depends critically on precise control of the precipitation and prevention of the crystal growth during the production [18]. The complexity of the process control and potential risk of residual organic solvents have discouraged the development of commercial products [25]. Recently, spray-drying [26] and freeze-drying [27, 28] processes have been used to achieve continuous control of the crystallization at large scales.
2.2 Top-down technologies
Top-down approach is based on two basic size reduction methods: wet milling [29] and high-pressure homogenization [30]. The wet milling process applies shear stress on large drug particles by grinding an aqueous suspension that contains a drug and a surface stabilizer using beads or pearls in a milling chamber [31]. The outcome of the milling process is determined by the hardness of the drug, energy input, milling time, and stabilizer concentration [31]. Microfluidization and piston-gap homogenization are examples of high-pressure homogenization [18]. Microfluidization is based on the jet stream principle, where the size diminution is achieved by collision of two fluid streams of particle suspension in a Y-type chamber under high pressure [25, 30]. The piston-gap homogenizer forces a particle suspension to pass a small gap (~5 μm) under pressure. The high shear forces, turbulent flow, and cavitation generated during this passage can reduce the particle size to the nanometer range [25, 30]. The performance of this process depends on the number of cycles, power density, and temperature [30]. These techniques are widely used in industry.
Compared to bottom-up methods, top-down methods require higher energy consumption and a longer operation time. The risk of contamination due to the erosion of milling beads is also a disadvantage of wet milling [32]. Moreover, the high-energy process may induce phase transition of a drug, which may compromise the in-vivo performance of the products [33].
2.3 Combined technologies
A pre-treatment step (bottom-up) and particle size reduction step (top-down) may be combined. For example, precipitates are first obtained from anti-solvent precipitation, spray-drying, or lyophilization (pre-treatment step), followed by high-pressure homogenization (particle-size reduction) [18, 25]. One of the roles of the high-pressure homogenization step is to anneal the initial precipitates, which are often thermodynamically unstable, into an ordered crystal structure [25]. Two well-established techniques, NANOEDGE™ and smartcrystals®, have been discussed elsewhere in detail [18, 25].
2.4 Nanocrystal Stabilization
Due to the high surface energy generated by nanonization, surface stabilizers are needed to prevent aggregation and precipitation of nanocrystals. Examples of stabilizing systems are summarized in a recent review article [34]. For drug nanocrystals with no surface charge, anionic surfactants such as sodium cholate, sodium deoxycholate, and sodium lauryl sulfate are often used to keep the particles separated via electrostatic repulsion. Another way of stabilizing nanocrystals is to apply polymeric stabilizers on their surface and establish a steric barrier against aggregation. Polymers used for this purpose are derivatives of cellulose, polyvinyl alcohol, polyvinyl pyrrolidone, Polysorbates (polyoxyethylene sorbitan fatty acid esters), and Pluronics (or Poloxamers, triblock-copolymers of polyoxyethylene and polyoxypropylene) [35, 36]. Some of the stabilizers, such as arginine, amphiphilic amino acid copolymers, and vitamin E polyethylene glycol succinate (TPGS), are biologically active and provide additional functions to the nanocrystals [29]. For example, TPGS, an effective P-glycoprotein inhibitor [37], enables paclitaxel (PTX) nanocrystals to overcome multidrug resistance [38]. The effectiveness of PTX nanocrystals stabilized with TPGS was demonstrated in a nude mouse model bearing multidrug-resistant NCI/ADR-RES human ovarian cancer cells [38].
The effectiveness of a nanocrystal stabilizer depends on its affinity for a drug, the concentration, and the stabilizer to drug ratio in suspension. Relatively hydrophobic stabilizers have higher affinity for drug crystals and a greater stabilizing effect [39]. There is a positive correlation between particle size and the hydrophilic lipophilic balance (HLB) value of a non-ionic surfactant in bottom-up approaches; thus, the HLB value can be a useful guideline for the selection of a stabilizer [40]. Typically, a surfactant with a low HLB value (lipophilic surfactant) is a good stabilizer of hydrophobic nanocrystals. Adequate surface coverage by stabilizers, irrespective of their mechanisms, is critical to the stabilization of nanocrystals. However, it does not necessarily mean that the stability increases in proportion to the concentration of a stabilizer. When the concentration of a surfactant exceeds the critical micelle concentration (CMC), the excessive surfactant has a negative effect on the stability of the nanocrystal suspension (nanosuspension) because micelle formation begins to compete with adsorption to the nanocrystal surface [41–43].
3. Application of Nanocrystals
Nanocrystals are used as is or further processed into various dosage forms. Most nanocrystal products on the market are oral dosage forms and thoroughly reviewed elsewhere [25, 34]. Here we discuss emerging use of nanocrystals in parenteral applications, local and systemic, with a specific focus on the rationale and therapeutic outcomes.
3.1 Dermal application
Nanocrystals are used in dermal applications to enhance the dissolution of poorly soluble drugs in aqueous phase, thereby increasing the concentration gradient between the formulation and the skin and, thereby, transdermal penetration of the drug [25]. For example, nanocrystal formulations of poorly soluble antioxidative agents such as hesperetin [44] and lutein [13] were developed as anti-aging cosmetic products. Hesperetin nanocrystals were prepared by high-pressure homogenization using stabilizers suitable for dermal use, such as Poloxamer 188, Tween 80, Inutec SP1 (inulin lauryl carbamate) and Plantacare 2000 (alkyl polyglycoside) [44]. A short-term stability test showed that all nanosuspensions remained reasonably stable at different temperatures [44]. Similarly, a lutein nanocrystal suspension stabilized with Plantacare 2000 was produced by high-pressure homogenization [13]. With the reduction of particle size, lutein nanocrystals showed a saturation solubility 26.3 fold higher than that of coarse powder. The lutein nanocrystals were able to penetrate through cellulose nitrate membranes, used as an in-vitro model of a penetration barrier, 14 times better than coarse powder. However, no permeation through pig ear skin was observed, which indicates that the lutein entering the skin remained there due to the lipophilicity [13]. Another example is a solid-in-oil nanosuspension of diclofenac sodium [45]. The nanosuspension was developed for transdermal delivery of diclofenac, which induces severe gastric damages when administered orally [46]. The solid-in-oil nanosuspension was produced by suspending a freeze-dried emulsion mixture of diclofenac sodium and sucrose esters (surfactants) in isopropyl myristate [45]. The resulting nanosuspensions increased the flux of diclofenac sodium across the Yucatan micropig skin model by 3.8 fold as compared with a surfactant-free control [45].
3.2 Ocular application
The potential of nanosuspension for optical application has been relatively overlooked due to the popularity of mucoadhesive polymer nanoparticles [47–49]. However, clinical development of mucoadhesive formulations has been slow [50]; thus, nanocrystal formulations are gaining interest as a commercially viable alternative. Because nanosuspensions can be produced with a small amount of stabilizers generally regarded as safe (GRAS), they can quickly translate to commercial products once the proof of concept is demonstrated [25]. Nanosuspensions of several poorly soluble glucocorticoid drugs, such as hydrocortisone, prednisolone, and dexamethasone, have been produced to obtain better ocular bioavailability [51]. Compared to solutions and micro-crystalline suspensions, the nanosuspensions exhibited higher rates and extents of ophthalmic absorption and greater intensity and duration of the drug effect [51]. Another hydrocortisone nanosuspensions were produced by microfluidic precipitation (bottom-up) or wet milling (top-down) [52, 53]. Differential scanning calorimeter analysis and X-ray powder diffraction measurements indicated that hydrocortisone particles produced by microfluidic precipitation were amorphous, whereas the milled particles were crystalline and relatively more stable during storage. Both nanosuspensions showed higher ocular bioavailability than that of hydrocortisone solution [52].
3.3 Pulmonary application
Nanosuspensions can be nebulized for pulmonary drug delivery. In addition to the contribution to drug dissolution and diffusion, nanocrystals are known to have good tissue adhesiveness and prolonged residence time at the absorption site [11, 25], because of the dramatically increased contact area (per mass) with mucosa [18]. A nanocrystal form of baicalein, a bioactive flavonoid, was prepared by anti-solvent re-crystallization followed by high-pressure homogenization [14]. The baicalein nanosuspension (10 mg/kg) instilled into the lungs demonstrated a significantly faster onset time and higher concentration than oral baicalein nanocrystals (121 mg/kg) and almost identical pharmacokinetic parameters to those of intravenous (IV) injection (10 mg/kg) in a rat model [14]. Budesonide, an anti-inflammatory corticosteroid, was formulated as nanosuspensions using high-pressure homogenization for the local therapy of asthma [54, 55]. Stabilized by a combination of lecithin (electrostatic stabilizer) and tyloxapol (steric stabilizer), budesonide nanosuspension was stable during one year storage at room temperature [54] and proved to be safe and effective in healthy volunteers [55].
3.4 Intravenous (IV) application
Nanocrystals with a size in the range of 100–300 nm can take advantage of the enhanced permeability and retention (EPR) effect to reach solid tumors with high vascular densities. Several anti-cancer drugs, such as paclitaxel [38, 42, 56, 57], camptothecin [16, 58], deacety mycoepoxydiene [59], oridonin [41, 60, 61], cucurmin [62] and asulacrine [15], were formulated as injectable nanocrystal formulations. Nanocrystals have advantages over other nanoparticle formulations since the drug content is relatively high (~100%) and the formulation is less dependent on solubilizing agents that may have dose-limiting side effects. On the other hand, tumor accumulation of nanocrystals is predicated on prolonged circulation, which requires protection of the nanocrystal surface from opsonin adsorption and recognition by the reticuloendothelial system. For this purpose, nanocrystals may be coated with protective polymers like other nanoparticle systems. Nanocrystal form of nevirapine, an anti-retroviral drug, was surface-modified with serum albumin, dextran, and polyethylene glycol (PEG) by physical adsorption [63]. Of these, PEG protected nanocrystals from phagocytic uptake best; dextran and albumin coating rather increased the macrophage uptake as compared to bare nanocrystals [63]. The PEG-coated nanocrystals showed the longest mean residence time in blood as compared to bare- and the other coated nanocrystals [63].
When cellular uptake of nanocrystals at the target tissues is desired, the surface is further decorated with cell-specific ligands. For example, folate-receptor (FR) targeted nanocrystals were produced using a conjugate of folic acid and Pluronic F127 as a stabilizer of PTX nanocrystals [57]. The FR-targeted nanocrystal formulation showed a greater cytotoxicity than non-targeted one [57]. On the other hand, the addition of a targeting ligand did not always improve nanocrystal transport across the cell membrane. Nanocrystals of atovaquone, an antiparasitic agent, were produced using Tween 80, poloxamers, or sodium dodecyl sulfate [64, 65]. Apolipoprotein E (apoE), which binds to the low-density lipoprotein receptor (LDLr) abundant in the brain capillary endothelial cells, was added to improve their passage across the blood-brain barrier (BBB) [64]. However, there was no additional nanocrystal passage across the in vitro BBB model or increase in the brain uptake of nanocrystals attributable to apoE ligands [64]. Interestingly, nanocrystals stabilized with Tween 80 and Poloxamer 184 showed relatively good BBB passage irrespective of the presence of apoE and corresponding therapeutic effect in murine toxoplasmosis [64]. A later study showed that oral atovaquone nanocrystals stabilized with sodium dodecyl sulfate showed greater brain uptake and therapeutic effect than those with Poloxamer 188 [65]. The contribution of these surfactants to nanocrystal transport across BBB may be explained by several mechanisms including blocking of efflux pumps, reduction of drug uptake by phagocytes, opening of tight junctions, or (unknown) receptor-binding processes [64].
3.5 Intraperitoneal (IP) application
Recently, nanocrystals were used for IP chemotherapy of ovarian cancer. IP drug administration has gained increasing interest due to a number of preclinical and clinical results that demonstrated relatively good therapeutic effects compared to IV [66]. The potential benefits of IP drug delivery are multifaceted. A drug delivered IP can achieve a higher concentration and a longer half-life in the peritoneal cavity compared to those observed with IV [67–76] and, thus, has a greater opportunity for locoregional effects [77–79]. Moreover, an IP-administered drug is partly absorbed to systemic circulation, getting access to regions of organs and tissues that are not in direct contact with peritoneal fluid [80, 81]. Finally, if delivered as nanoparticles with a specific size, a drug can be trafficked through lymph nodes [76, 82, 83], which provides an opportunity to treat diseases spreading via the lymphatics. An example of IP application of paclitaxel nanocrystals was reported in conjunction with hyperthermic therapy [17]. Here, PTX nanocrystals were produced using Pluronic F127 as a stabilizer and used for hyperthermic IP chemotherapy (HIPEC). The PTX nanocrystals showed similar anti-tumor activity as Taxol with relatively low apparent toxicity [17]. Unlike Taxol, the blood level of PTX continued to increase even after the discontinuation of nanocrystal HIPEC, which suggests the long-term residence of nanocrystals in the peritoneal cavity [17].
4. Remaining Challenges in Nanocrystal Development for Parenteral Applications
4.1 Instability during storage
Ostwald ripening refers to a phenomenon that small particles gradually dissolve and redeposit on the surface of larger particles over time (Fig. 2) [84]. It occurs when the particle size in a dispersion system is heterogeneous and the dispersed phase (drug) has a limited solubility in the medium (water) [85], which, unfortunately, are the conditions frequently encountered in pharmaceutical suspensions [86, 87]. Ostwald ripening leads to the particle size growth and physical instability of a dispersion system during storage; therefore, there is a strong need for preventing this process.
Fig. 2.
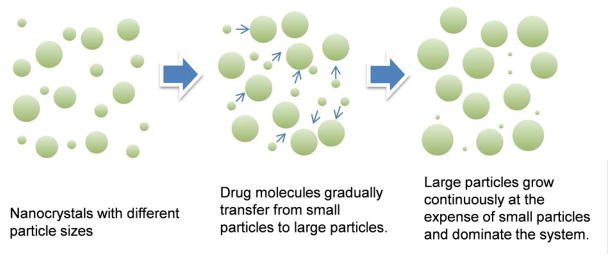
Schematic representation of Ostwald ripening in nanosuspension.
Surfactants and polymers are commonly used as stabilizers to delay detachment and attachment of drug molecules at the surface of dispersed particles [85]. Polymers are believed to be more effective than small molecular-weight surfactants because they tend to adhere to the nanocrystal surface less dynamically than surfactants [88]. Another way of preventing particle size growth is to produce uniform nanocrystals, thus eliminating one of the conditions for Ostwald ripening. Optimizing the process parameters, such as the number of high-pressure homogenization cycles [62], milling time [33, 52] and milling speed [89], can eliminate large particles and obtain narrow size distribution. Processing nanosuspensions into solid products is another way of avoiding dynamic changes in the medium and thus Ostwald ripening [34, 90].
4.2 Instability during applications
Due to the high surface area to volume ratio, complete dissolution of nanocrystals can occur quickly (in less than an hour) as long as a sink condition is maintained (Table 2). On the other hand, in locations with a limited volume of fluid, such as the peritoneal cavity, stomach, or the lungs, nanocrystals may be exposed to a non-sink condition for an extended period of time. This may limit the dissolution rate of the nanocrystals and provide an opportunity for sustained drug release [16]. However, with the gradual surface erosion and loss of stabilizing agents, the nanocrystals may become increasingly unstable and, thus, undergo agglomeration and Ostwald ripening over time. When this occurs, drug dissolution slows down significantly, to an extent that the drug is no longer bioavailable. We have experienced a consequence of particle agglomeration in an animal model of IP tumors [91]. Here, we produced PTX particles with precipitation followed by sonication and administered the particles IP into mice bearing ovarian tumors in the peritoneal cavity, using an in-situ crosslinkable hyaluronic acid-based hydrogel as a carrier [91]. The PTX precipitates delivered with the hydrogel were best retained in the peritoneal cavity as compared to other formulations, which included multiple injections of Taxol, a bolus injection of Taxol, PTX precipitates alone, and Taxol delivered with the hydrogel. Despite the prolonged IP retention, the anti-tumor effect of hydrogel-embedded PTX precipitates was not significantly different from the others, which were cleared from the peritoneal cavity much earlier. One of the possible explanations is the agglomeration of PTX precipitates, which led to incomplete dissolution of PTX [91].
Table 2.
Examples of nanocrystal dissolution in a sink condition
| Drug | Particle size (nm) | Dissolution rate | Reference |
|---|---|---|---|
| Oridonin | 322.7 | 98% dissolved in 24 min | [41] |
| Asulacrine | d(v; 0.5)* 133 ± 20 | 42% dissolved in 6 h | [15] |
| Celecoxib | d(v; 0.5) 360 | 91.8% dissolved in 50 min | [92] |
| Meloxicam | d(v; 0.5) 530 ± 110 | 100% dissolved in 10 min | [93] |
| Artemisinin | 100–360 | 75.9% dissolved in 4 h | [94] |
| Nitrendipine | 209 ± 9 | 90% dissolved in 2 min | [95] |
| Quercetin | 213.6 ± 29.3 | 73.2% dissolved in 20 min | [96] |
| Itraconazole | ~300 | 85% dissolved in 90 min | [12] |
| Camptothecin | 200–700 | 50% dissolved in 2 h | [16] |
Note: size of the particles for which 50% of the same volume contains particles smaller than d (v; 0.5).
4.3 Lack of target specificity
The surface of nanocrystals is decorated with specific ligands for target-specific delivery. A few examples with physically adsorbed ligands were introduced in section 3.4; however, continuous surface erosion poses a challenge to the longevity of the targeting effect. In this regard, it is worthwhile to note a recent approach to produce a co-crystal of a drug and functional molecules [56]. Here, Li et al. produced hybrid crystals by co-crystallization of PTX and fluorescent dyes, where guest dye molecules were integrated in PTX nanocrystals [56]. Since the dye was embedded throughout the matrix, the PTX-dye hybrid nanocrystals could be located via real-time imaging during their lifetime in the body. The same principle is applicable to producing drug-ligand hybrid nanocrystals with the prolonged target specificity.
5. Future Perspectives
While the design of nanoparticulate formulations has become increasingly sophisticated, structurally and conceptually simple nanocrystals have a unique advantage with respect to the development of commercial products. Nanocrystals can greatly enhance the saturation solubility and dissolution rate of poorly soluble drugs with simple production technologies and compositions. The large contact area of nanocrystals can allow for a greater interaction with tissue or cell surfaces and enhance drug absorption. Several oral nanocrystal products are available on the market, and dermal and IV products are actively explored. However, the potential of nanocrystals has not been thoroughly investigated for different applications such as targeted or local drug delivery. For example, nanocrystals can be combined with implantable delivery systems to attain a higher local concentration for a prolonged period of time. In addition, modification with a ligand presents an opportunity to deliver nanocrystals in a target-specific manner. However, the current methods of nanocrystal production and modification do not adequately address the challenges in development of such products, and new approaches to engineer nanocrystals are strongly awaited. In addition, much remains to be understood with respect to the mechanisms of intracellular transport and distribution of nanocrystals.
Highlights.
Introduce nanocrystals as commercially viable pharmaceutical products.
Review different methods of producing nanocrystals.
Review parenteral applications of nanocrystals.
Discuss remaining challenges in the development of nanocrystal products.
Acknowledgments
This work was supported by NSF DMR-1056997, NIH R21 CA135130, and a grant from the Lilly Endowment, Inc. to College of Pharmacy, Purdue University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Merisko-Liversidge EM, Liversidge GG. Drug Nanoparticles: Formulating Poorly Water-Soluble Compounds. Toxicol Pathol. 2008;36:43–8. doi: 10.1177/0192623307310946. [DOI] [PubMed] [Google Scholar]
- 2.Rabinow BE. Nanosuspensions in drug delivery. Nat Rev Drug Discov. 2004;3:785–96. doi: 10.1038/nrd1494. [DOI] [PubMed] [Google Scholar]
- 3.Squillante I, Emilio, Sethia S. Solid Dispersions: Revival with Greater Possibilities and Applications in Oral Drug Delivery. Crit Rev Ther Drug Carrier Syst. 2003;20:34. doi: 10.1615/critrevtherdrugcarriersyst.v20.i23.40. [DOI] [PubMed] [Google Scholar]
- 4.He S, Cui Z, Mei D, Zhang H, Wang X, Dai W, et al. A Cremophor-Free Self-Microemulsified Delivery System for Intravenous Injection of Teniposide: Evaluation In Vitro and In Vivo. AAPS PharmSciTech. 2012;13:846–852. doi: 10.1208/s12249-012-9809-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bouquet W, Ceelen W, Fritzinger B, Pattyn P, Peeters M, Remon JP, et al. Paclitaxel/β-cyclodextrin complexes for hyperthermic peritoneal perfusion – Formulation and stability. Eur J Pharm Biopharm. 2007;66:391–7. doi: 10.1016/j.ejpb.2006.11.025. [DOI] [PubMed] [Google Scholar]
- 6.Chen H, Tang L, Qin Y, Yin Y, Tang J, Tang W, et al. Lactoferrin-modified procationic liposomes as a novel drug carrier for brain delivery. Eur J Pharm Biopharm. 2010;40:94–102. doi: 10.1016/j.ejps.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 7.Shao K, Huang R, Li J, Han L, Ye L, Lou J, et al. Angiopep-2 modified PE-PEG based polymeric micelles for amphotericin B delivery targeted to the brain. J Control Release. 2010;147:118–26. doi: 10.1016/j.jconrel.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 8.Gong Y, Wu Y, Zheng C, Fan L, Xiong F, Zhu J. An Excellent Delivery System for Improving the Oral Bioavailability of Natural Vitamin E in Rats. AAPS PharmSciTech. 2012;13:961–966. doi: 10.1208/s12249-012-9819-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dodiya SS, Chavhan SS, Sawant KK, Korde AG. Solid lipid nanoparticles and nanosuspension formulation of Saquinavir: preparation, characterization, pharmacokinetics and biodistribution studies. J Microencapsul. 2011;28:515–27. doi: 10.3109/02652048.2011.590612. [DOI] [PubMed] [Google Scholar]
- 10.Gullotti E, Yeo Y. Beyond the imaging: Limitations of cellular uptake study in the evaluation of nanoparticles. J Control Release. 2012 doi: 10.1016/j.jconrel.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patravale VB, Date AA, Kulkarni RM. Nanosuspensions: a promising drug delivery strategy. J Pharm Pharmacol. 2004;56:827–40. doi: 10.1211/0022357023691. [DOI] [PubMed] [Google Scholar]
- 12.Sun W, Mao S, Shi Y, Li LC, Fang L. Nanonization of itraconazole by high pressure homogenization: Stabilizer optimization and effect of particle size on oral absorption. J Pharm Sci. 2011;100:3365–73. doi: 10.1002/jps.22587. [DOI] [PubMed] [Google Scholar]
- 13.Mitri K, Shegokar R, Gohla S, Anselmi C, Müller RH. Lutein nanocrystals as antioxidant formulation for oral and dermal delivery. Int J Pharm. 2011;420:141–6. doi: 10.1016/j.ijpharm.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J, Lv H, Jiang K, Gao Y. Enhanced bioavailability after oral and pulmonary administration of baicalein nanocrystal. Int J Pharm. 2011;420:180–8. doi: 10.1016/j.ijpharm.2011.08.023. [DOI] [PubMed] [Google Scholar]
- 15.Ganta S, Paxton JW, Baguley BC, Garg S. Formulation and pharmacokinetic evaluation of an asulacrine nanocrystalline suspension for intravenous delivery. Int J Pharm. 2009;367:179–86. doi: 10.1016/j.ijpharm.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 16.Zhang H, Hollis CP, Zhang Q, Li T. Preparation and antitumor study of camptothecin nanocrystals. Int J Pharm. 2011;415:293–300. doi: 10.1016/j.ijpharm.2011.05.075. [DOI] [PubMed] [Google Scholar]
- 17.De Smet L, Colin P, Ceelen W, Bracke M, Van Bocxlaer J, Remon J, et al. Development of a Nanocrystalline Paclitaxel Formulation for Hipec Treatment. Pharm Res. 2012;29:2398–2406. doi: 10.1007/s11095-012-0765-x. [DOI] [PubMed] [Google Scholar]
- 18.Müller RH, Gohla S, Keck CM. State of the art of nanocrystals – Special features, production, nanotoxicology aspects and intracellular delivery. Eur J Pharm Biopharm. 2011;78:1–9. doi: 10.1016/j.ejpb.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 19.Barenholz Y. Doxil® — The first FDA-approved nano-drug: Lessons learned. J Control Release. 2012;160:117–34. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 20.Xiong S, Yu B, Wu J, Li H, Lee RJ. Preparation, therapeutic efficacy and intratumoral localization of targeted daunorubicin liposomes conjugating folate-PEG-CHEMS. Biomed Pharmacother. 2011;65:2–8. doi: 10.1016/j.biopha.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 21.Montero AJ, Adams B, Diaz-Montero CM, Glück S. Nab-paclitaxel in the treatment of metastatic breast cancer: a comprehensive review. Expert Rev Clin Pharmacol. 2011;4:329–34. doi: 10.1586/ecp.11.7. [DOI] [PubMed] [Google Scholar]
- 22.Chan H-K, Kwok PCL. Production methods for nanodrug particles using the bottom-up approach. Adv Drug Deliv Rev. 2011;63:406–16. doi: 10.1016/j.addr.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 23.Lindfors L, Skantze P, Skantze U, Westergren J, Olsson U. Amorphous Drug Nanosuspensions. 3. Particle Dissolution and Crystal Growth. Langmuir. 2007;23:9866–74. doi: 10.1021/la700811b. [DOI] [PubMed] [Google Scholar]
- 24.Thombre AG, Shah JC, Sagawa K, Caldwell WB. In vitro and in vivo characterization of amorphous, nanocrystalline, and crystalline ziprasidone formulations. Int J Pharm. 2012;428:8–17. doi: 10.1016/j.ijpharm.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 25.Shegokar R, Müller RH. Nanocrystals: Industrially feasible multifunctional formulation technology for poorly soluble actives. Int J Pharm. 2010;399:129–39. doi: 10.1016/j.ijpharm.2010.07.044. [DOI] [PubMed] [Google Scholar]
- 26.Hu J, Ng WK, Dong Y, Shen S, Tan RBH. Continuous and scalable process for water-redispersible nanoformulation of poorly aqueous soluble APIs by antisolvent precipitation and spray-drying. Int J Pharm. 2011;404:198–204. doi: 10.1016/j.ijpharm.2010.10.055. [DOI] [PubMed] [Google Scholar]
- 27.de Waard H, Grasmeijer N, Hinrichs WLJ, Eissens AC, Pfaffenbach PPF, Frijlink HW. Preparation of drug nanocrystals by controlled crystallization: Application of a 3-way nozzle to prevent premature crystallization for large scale production. Eur J Pharm Sci. 2009;38:224–9. doi: 10.1016/j.ejps.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 28.de Waard H, Hinrichs WLJ, Frijlink HW. A novel bottom–up process to produce drug nanocrystals: Controlled crystallization during freeze-drying. J Control Release. 2008;128:179–83. doi: 10.1016/j.jconrel.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 29.Merisko-Liversidge E, Liversidge GG. Nanosizing for oral and parenteral drug delivery: A perspective on formulating poorly-water soluble compounds using wet media milling technology. Adv Drug Deliv Rev. 2011;63:427–40. doi: 10.1016/j.addr.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 30.Keck CM, Müller RH. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation. Eur J Pharm Biopharm. 2006;62:3–16. doi: 10.1016/j.ejpb.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 31.Merisko-Liversidge E, Liversidge GG, Cooper ER. Nanosizing: a formulation approach for poorly-water-soluble compounds. Eur J Pharm Biopharm. 2003;18:113–20. doi: 10.1016/s0928-0987(02)00251-8. [DOI] [PubMed] [Google Scholar]
- 32.Juhnke M, Märtin D, John E. Generation of wear during the production of drug nanosuspensions by wet media milling. Eur J Pharm Biopharm. 2012;81:214–22. doi: 10.1016/j.ejpb.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 33.Begat P, Young PM, Edge S, Kaerger JS, Price R. The effect of mechanical processing on surface stability of pharmaceutical powders: Visualization by atomic force microscopy. J Pharm Sci. 2003;92:611–20. doi: 10.1002/jps.10320. [DOI] [PubMed] [Google Scholar]
- 34.Van Eerdenbrugh B, Van den Mooter G, Augustijns P. Top-down production of drug nanocrystals: Nanosuspension stabilization, miniaturization and transformation into solid products. Int J Pharm. 2008;364:64–75. doi: 10.1016/j.ijpharm.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 35.Raghavan SL, Trividic A, Davis AF, Hadgraft J. Crystallization of hydrocortisone acetate: influence of polymers. Int J Pharm. 2001;212:213–21. doi: 10.1016/s0378-5173(00)00610-4. [DOI] [PubMed] [Google Scholar]
- 36.Raghavan SL, Schuessel K, Davis A, Hadgraft J. Formation and stabilisation of triclosan colloidal suspensions using supersaturated systems. Int J Pharm. 2003;261:153–8. doi: 10.1016/s0378-5173(03)00299-0. [DOI] [PubMed] [Google Scholar]
- 37.Dintaman JM, Silverman JA. Inhibition of P-Glycoprotein by D-α-Tocopheryl Polyethylene Glycol 1000 Succinate (TPGS) Pharm Res. 1999;16:1550–6. doi: 10.1023/a:1015000503629. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y, Huang L, Liu F. Paclitaxel Nanocrystals for Overcoming Multidrug Resistance in Cancer. Mol Pharm. 2010;7:863–9. doi: 10.1021/mp100012s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Eerdenbrugh B, Vermant J, Martens JA, Froyen L, Van Humbeeck J, Augustijns P, et al. A screening study of surface stabilization during the production of drug nanocrystals. J Pharm Sci. 2009;98:2091–103. doi: 10.1002/jps.21563. [DOI] [PubMed] [Google Scholar]
- 40.Verma S, Gokhale R, Burgess DJ. A comparative study of top-down and bottom-up approaches for the preparation of micro/nanosuspensions. Int J Pharm. 2009;380:216–22. doi: 10.1016/j.ijpharm.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 41.Gao L, Zhang D, Chen M, Zheng T, Wang S. Preparation and Characterization of an Oridonin Nanosuspension for Solubility and Dissolution Velocity Enhancement. Drug Dev Ind Pharm. 2007;33:1332–9. doi: 10.1080/03639040701741810. [DOI] [PubMed] [Google Scholar]
- 42.Deng J, Huang L, Liu F. Understanding the structure and stability of paclitaxel nanocrystals. Int J Pharm. 2010;390:242–9. doi: 10.1016/j.ijpharm.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng Z, Xu S, Li S. Understanding a relaxation behavior in a nanoparticle suspension for drug delivery applications. Int J Pharm. 2008;351:236–43. doi: 10.1016/j.ijpharm.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 44.Mishra PR, Shaal LA, Müller RH, Keck CM. Production and characterization of Hesperetin nanosuspensions for dermal delivery. Int J Pharm. 2009;371:182–9. doi: 10.1016/j.ijpharm.2008.12.030. [DOI] [PubMed] [Google Scholar]
- 45.Piao H, Kamiya N, Hirata A, Fujii T, Goto M. A Novel Solid-in-oil Nanosuspension for Transdermal Delivery of Diclofenac Sodium. Pharm Res. 2008;25:896–901. doi: 10.1007/s11095-007-9445-7. [DOI] [PubMed] [Google Scholar]
- 46.Piao H, Kamiya N, Watanabe J, Yokoyama H, Hirata A, Fujii T, et al. Oral delivery of diclofenac sodium using a novel solid-in-oil suspension. Int J Pharm. 2006;313:159–62. doi: 10.1016/j.ijpharm.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 47.Adibkia K, Javadzadeh Y, Dastmalchi S, Mohammadi G, Niri FK, Alaei-Beirami M. Naproxen–eudragit® RS100 nanoparticles: Preparation and physicochemical characterization. Colloids Surf B. 2011;83:155–9. doi: 10.1016/j.colsurfb.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 48.Pignatello R, Bucolo C, Ferrara P, Maltese A, Puleo A, Puglisi G. Eudragit RS100® nanosuspensions for the ophthalmic controlled delivery of ibuprofen. Eur J Pharma Sci. 2002;16:53–61. doi: 10.1016/s0928-0987(02)00057-x. [DOI] [PubMed] [Google Scholar]
- 49.Pignatello R, Bucolo C, Puglisi G. Ocular tolerability of Eudragit RS100® and RL100® nanosuspensions as carriers for ophthalmic controlled drug delivery. J Pharm Sci. 2002;91:2636–41. doi: 10.1002/jps.10227. [DOI] [PubMed] [Google Scholar]
- 50.Araújo J, Gonzalez E, Egea MA, Garcia ML, Souto EB. Nanomedicines for ocular NSAIDs: safety on drug delivery. Nanomedicine: NBM. 2009;5:394–401. doi: 10.1016/j.nano.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 51.Kassem MA, Abdel Rahman AA, Ghorab MM, Ahmed MB, Khalil RM. Nanosuspension as an ophthalmic delivery system for certain glucocorticoid drugs. Int J Pharm. 2007;340:126–33. doi: 10.1016/j.ijpharm.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 52.Ali HSM, York P, Ali AMA, Blagden N. Hydrocortisone nanosuspensions for ophthalmic delivery: A comparative study between microfluidic nanoprecipitation and wet milling. J Control Release. 2011;149:175–81. doi: 10.1016/j.jconrel.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 53.Ali HSM, York P, Blagden N. Preparation of hydrocortisone nanosuspension through a bottom-up nanoprecipitation technique using microfluidic reactors. Int J Pharm. 2009;375:107–13. doi: 10.1016/j.ijpharm.2009.03.029. [DOI] [PubMed] [Google Scholar]
- 54.Jacobs C, Müller RH. Production and Characterization of a Budesonide Nanosuspension for Pulmonary Administration. Pharm Res. 2002;19:189–94. doi: 10.1023/a:1014276917363. [DOI] [PubMed] [Google Scholar]
- 55.Kraft WK, Steiger B, Beussink D, Quiring JN, Fitzgerald N, Greenberg HE, et al. The Pharmacokinetics of Nebulized Nanocrystal Budesonide Suspension in Healthy Volunteers. J Clin Pharmacol. 2004;44:67–72. doi: 10.1177/0091270003261490. [DOI] [PubMed] [Google Scholar]
- 56.Zhao R, Hollis CP, Zhang H, Sun L, Gemeinhart RA, Li T. Hybrid Nanocrystals: Achieving Concurrent Therapeutic and Bioimaging Functionalities toward Solid Tumors. Mol Pharm. 2011;8:1985–91. doi: 10.1021/mp200154k. [DOI] [PubMed] [Google Scholar]
- 57.Liu F, Park J-Y, Zhang Y, Conwell C, Liu Y, Bathula SR, et al. Targeted cancer therapy with novel high drug-loading nanocrystals. J Pharm Sci. 2010;99:3542–51. doi: 10.1002/jps.22112. [DOI] [PubMed] [Google Scholar]
- 58.Yao L, Zhao X, Li Q, Zu Y, Fu Y, Zu B, et al. In vitro and in vivo evaluation of camptothecin nanosuspension: A novel formulation with high antitumor efficacy and low toxicity. Int J Pharm. 2012;423:586–8. doi: 10.1016/j.ijpharm.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 59.Wang Y, Liu Z, Zhang D, Gao X, Zhang X, Duan C, et al. Development and in vitro evaluation of deacety mycoepoxydiene nanosuspension. Colloids Surf B. 2011;83:189–97. doi: 10.1016/j.colsurfb.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 60.Gao L, Zhang D, Chen M, Duan C, Dai W, Jia L, et al. Studies on pharmacokinetics and tissue distribution of oridonin nanosuspensions. Int J Pharm. 2008;355:321–7. doi: 10.1016/j.ijpharm.2007.12.016. [DOI] [PubMed] [Google Scholar]
- 61.Lou H, Gao L, Wei X, Zhang Z, Zheng D, Zhang D, et al. Oridonin nanosuspension enhances anti-tumor efficacy in SMMC-7721 cells and H22 tumor bearing mice. Colloids Surf B. 2011;87:319–25. doi: 10.1016/j.colsurfb.2011.05.037. [DOI] [PubMed] [Google Scholar]
- 62.Gao Y, Li Z, Sun M, Li H, Guo C, Cui J, et al. Preparation, characterization, pharmacokinetics, and tissue distribution of curcumin nanosuspension with TPGS as stabilizer. Drug Dev Ind Pharm. 2010;36:1225–34. doi: 10.3109/03639041003695139. [DOI] [PubMed] [Google Scholar]
- 63.Shegokar R, Singh KK. Surface modified nevirapine nanosuspensions for viral reservoir targeting: In vitro and in vivo evaluation. Int J Pharm. 2011;421:341–52. doi: 10.1016/j.ijpharm.2011.09.041. [DOI] [PubMed] [Google Scholar]
- 64.Shubar HM, Dunay IR, Lachenmaier S, Dathe M, Bushrab FN, Mauludin R, et al. The role of apolipoprotein E in uptake of atovaquone into the brain in murine acute and reactivated toxoplasmosis. J Drug Target. 2009;17:257–67. doi: 10.1080/10611860902718680. [DOI] [PubMed] [Google Scholar]
- 65.Shubar HM, Lachenmaier S, Heimesaat MM, Lohman U, Mauludin R, Mueller RH, et al. SDS-coated atovaquone nanosuspensions show improved therapeutic efficacy against experimental acquired and reactivated toxoplasmosis by improving passage of gastrointestinal and blood–brain barriers. J Drug Target. 2011;19:114–24. doi: 10.3109/10611861003733995. [DOI] [PubMed] [Google Scholar]
- 66.Bajaj G, Yeo Y. Drug Delivery Systems for Intraperitoneal Therapy. Pharm Res. 2010;27:735–8. doi: 10.1007/s11095-009-0031-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marchettini P, Stuart OA, Mohamed F, Yoo D, Sugarbaker PH. Docetaxel: pharmacokinetics and tissue levels after intraperitoneal and intravenous administration in a rat model. Cancer Chemoth Pharm. 2002;49:499–503. doi: 10.1007/s00280-002-0439-1. [DOI] [PubMed] [Google Scholar]
- 68.Mohamed F, Stuart OA, Sugarbaker PH. Pharmacokinetics and tissue distribution of intraperitoneal docetaxel with different carrier solutions. J Surg Res. 2003;113:114–20. doi: 10.1016/s0022-4804(03)00162-8. [DOI] [PubMed] [Google Scholar]
- 69.Morgan RJ, Doroshow JH, Synold T, Lim D, Shibata S, Margolin K, et al. Phase I trial of intraperitoneal docetaxel in the treatment of advanced malignancies primarily confined to the peritoneal cavity: Dose-limiting toxicity and pharmacokinetics. Clin Cancer Res. 2003;9:5896–901. [PubMed] [Google Scholar]
- 70.de Bree E, Rosing H, Beijnen JH, Romanos J, Michalakis J, Georgoulias V, et al. Pharmacokinetic study of docetaxel in intraoperative hyperthermic i.p. chemotherapy for ovarian cancer. Anti-Cancer Drug. 2003;14:103–10. doi: 10.1097/00001813-200302000-00003. [DOI] [PubMed] [Google Scholar]
- 71.Hofstra LS, Bos AME, de Vries EGE, van der Zee AGJ, Willemsen ATM, Rosing H, et al. Kinetic modeling and efficacy of intraperitoneal paclitaxel combined with intravenous cyclophosphamide and carboplatin as first-line treatment in ovarian cancer. Gynecol Oncol. 2002;85:517–23. doi: 10.1006/gyno.2002.6665. [DOI] [PubMed] [Google Scholar]
- 72.Witkamp AJ, de Bree E, Van Goethem AR, Zoetmulder FAN. Rationale and techniques of intra-operative hyperthermic intraperitoneal chemotherapy. Cancer Treat Rev. 2001;27:365–74. doi: 10.1053/ctrv.2001.0232. [DOI] [PubMed] [Google Scholar]
- 73.Markman M, Rowinsky E, Hakes T, Reichman B, Jones W, Lewis JL, et al. Phase-I Trial Of Intraperitoneal Taxol - A Gynecologic Oncology Group-Study. J Clin Oncol. 1992;10:1485–91. doi: 10.1200/JCO.1992.10.9.1485. [DOI] [PubMed] [Google Scholar]
- 74.Chambers SK, Chow H-HS, Janicek MF, Cragun JM, Hatch KD, Cui H, et al. Phase I Trial of Intraperitoneal Pemetrexed, Cisplatin, and Paclitaxel in Optimally Debulked Ovarian Cancer. Clin Cancer Res. 2012;18:2668–78. doi: 10.1158/1078-0432.CCR-12-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hasovits C, Clarke S. Pharmacokinetics and pharmacodynamics of intraperitoneal cancer chemotherapeutics. Clin Pharmacokinet. 2012;51:203–24. doi: 10.2165/11598890-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 76.Lu Z, Wang J, Wientjes MG, Au JLS. Intraperitoneal therapy for peritoneal cancer. Future Oncol. 2010;6:1625–41. doi: 10.2217/fon.10.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dedrick RL, Myers CE, Bungay PM, DeVita VT., Jr Pharmacokinetic rationale for peritoneal drug administration in the treatment of ovarian cancer. Cancer Treat Rep. 1978;62:1–11. [PubMed] [Google Scholar]
- 78.Markman M. Intraperitoneal drug delivery of antineoplastics. Drugs. 2001;61:1057–65. doi: 10.2165/00003495-200161080-00003. [DOI] [PubMed] [Google Scholar]
- 79.Nissan A, Stojadinovic A, Garofalo A, Esquivel J, Piso P. Evidence-Based Medicine in the Treatment of Peritoneal Carcinomatosis: Past, Present, and Future. J Surg Oncol. 2009;100:335–44. doi: 10.1002/jso.21323. [DOI] [PubMed] [Google Scholar]
- 80.Zahedi P, Stewart J, De Souza R, Piquette-Miller M, Allen C. An injectable depot system for sustained intraperitoneal chemotherapy of ovarian cancer results in favorable drug distribution at the whole body, peritoneal and intratumoral levels. J Control Release. 2012;158:379–85. doi: 10.1016/j.jconrel.2011.11.025. [DOI] [PubMed] [Google Scholar]
- 81.Markman M. Intraperitoneal antineoplastic drug delivery: rationale and results. Lancet Oncol. 2003;4:277–83. doi: 10.1016/s1470-2045(03)01074-x. [DOI] [PubMed] [Google Scholar]
- 82.Hirano K, Hunt CA. Lymphatic transport of liposome-encapsulated agents: effects of liposome size following intraperitoneal administration. J Pharm Sci. 1985;74:915–21. doi: 10.1002/jps.2600740902. [DOI] [PubMed] [Google Scholar]
- 83.Lu H, Li B, Kang Y, Jiang W, Huang Q, Chen Q, et al. Paclitaxel nanoparticle inhibits growth of ovarian cancer xenografts and enhances lymphatic targeting. Cancer Chemoth Pharm. 2007;59:175–81. doi: 10.1007/s00280-006-0256-z. [DOI] [PubMed] [Google Scholar]
- 84.Yao JH, Elder KR, Guo H, Grant M. Theory and simulation of Ostwald ripening. Phys Rev B. 1993;47:14110–25. doi: 10.1103/physrevb.47.14110. [DOI] [PubMed] [Google Scholar]
- 85.Verma S, Kumar S, Gokhale R, Burgess DJ. Physical stability of nanosuspensions: Investigation of the role of stabilizers on Ostwald ripening. Int J Pharm. 2011;406:145–52. doi: 10.1016/j.ijpharm.2010.12.027. [DOI] [PubMed] [Google Scholar]
- 86.Lindfors L, Skantze P, Skantze U, Rasmusson M, Zackrisson A, Olsson U. Amorphous Drug Nanosuspensions. 1. Inhibition of Ostwald Ripening. Langmuir. 2005;22:906–10. doi: 10.1021/la0523661. [DOI] [PubMed] [Google Scholar]
- 87.Meinders MBJ, van Vliet T. The role of interfacial rheological properties on Ostwald ripening in emulsions. Adv Colloid Interfac. 2004;108–109:119–26. doi: 10.1016/j.cis.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 88.Walstra P. Formation of emulsions. In: Becher P, editor. Encyclopedia of Emulsion Technology: Volume 1 - Basic Theory. New York: Marcel Dekker; 1983. pp. 57–128. [Google Scholar]
- 89.Singare DS, Marella S, Gowthamrajan K, Kulkarni GT, Vooturi R, Rao PS. Optimization of formulation and process variable of nanosuspension: An industrial perspective. Int J Pharm. 2010;402:213–20. doi: 10.1016/j.ijpharm.2010.09.041. [DOI] [PubMed] [Google Scholar]
- 90.Van Eerdenbrugh B, Froyen L, Van Humbeeck J, Martens JA, Augustijns P, Van den Mooter G. Drying of crystalline drug nanosuspensions—The importance of surface hydrophobicity on dissolution behavior upon redispersion. Eur J Pharma Sci. 2008;35:127–35. doi: 10.1016/j.ejps.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 91.Bajaj G, Kim MR, Mohammed SI, Yeo Y. Hyaluronic acid-based hydrogel for regional delivery of paclitaxel to intraperitoneal tumors. J Control Release. 2012;158:386–92. doi: 10.1016/j.jconrel.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dolenc A, Kristl J, Baumgartner S, Planinšek O. Advantages of celecoxib nanosuspension formulation and transformation into tablets. Int J Pharm. 2009;376:204–12. doi: 10.1016/j.ijpharm.2009.04.038. [DOI] [PubMed] [Google Scholar]
- 93.Ambrus R, Kocbek P, Kristl J, Šibanc R, Rajkó R, Szabó-Révész P. Investigation of preparation parameters to improve the dissolution of poorly water-soluble meloxicam. Int J Pharm. 2009;381:153–9. doi: 10.1016/j.ijpharm.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 94.Kakran M, Sahoo NG, Li L, Judeh Z, Wang Y, Chong K, et al. Fabrication of drug nanoparticles by evaporative precipitation of nanosuspension. Int J Pharm. 2010;383:285–92. doi: 10.1016/j.ijpharm.2009.09.030. [DOI] [PubMed] [Google Scholar]
- 95.Xia D, Quan P, Piao H, Piao H, Sun S, Yin Y, et al. Preparation of stable nitrendipine nanosuspensions using the precipitation–ultrasonication method for enhancement of dissolution and oral bioavailability. Eur J Pharm Sci. 2010;40:325–34. doi: 10.1016/j.ejps.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 96.Gao L, Liu G, Wang X, Liu F, Xu Y, Ma J. Preparation of a chemically stable quercetin formulation using nanosuspension technology. Int J Pharm. 2011;404:231–7. doi: 10.1016/j.ijpharm.2010.11.009. [DOI] [PubMed] [Google Scholar]