

Abstract
Many cytotoxic therapies are available to kill cancer cells. Unfortunately, these also inflict significant damage on normal cells. Identifying highly effective cancer treatments that have minimal or no side effects continues to be a major challenge. One of the strategies to minimize damage to normal tissue is to deliver an activating enzyme that localizes only in the tumor and converts a nontoxic prodrug to a cytotoxic agent locally in the tumor. Such strategies have been previously tested but with limited success due in large part to the uncertainty in the delivery and distribution of the enzyme. Imaging the delivery of the enzyme to optimize timing of the prodrug administration to achieve image-guided prodrug therapy would be of immense benefit for this strategy. Here, we have reviewed advances in the incorporation of image guidance in the applications of prodrug enzymes in cancer treatment. These advances demonstrate the feasibility of using clinically translatable imaging in these prodrug enzyme strategies.
Keywords: Prodrug enzymes, Imaging, Cancer
Introduction
The success of chemotherapy in the clinic is limited by insufficient drug concentrations in tumors, systemic toxicity, lack of selectivity for tumor cells compared to normal cells, and the evolution of drug-resistant cancer cells. Several strategies have been developed to improve specific tumor-targeting therapies. One of the most promising is prodrug enzyme therapy where a drug-activating enzyme is targeted or expressed in cancer cells following which a nontoxic prodrug is administered systemically [1]. The enzyme converts the prodrug to an active anticancer drug, achieving high concentrations in the tumor and sparing normal tissue. For such a strategy to work, there are certain requirements. The enzyme should be nonhuman or expressed at very low concentrations in normal tissue and it should have high catalytic activity. The prodrug should be a good substrate for the enzyme but should not be activated in normal tissue. It should be nontoxic and the activated drug should be highly diffusible or actively taken up by adjacent cells for a “bystander cell kill” effect while ideally not leaking out into systemic circulation.
Currently, there are three major categories of enzyme/prodrug strategies: (a) delivery of genes that encode prodrug-activating enzymes into tumor tissue (gene encoding prodrug-activating enzyme therapy, GDEPT, and virus-directed enzyme prodrug therapy, VDEPT), (b) targeted delivery of active enzymes in tumor tissue where the therapeutic enzyme is conjugated with an antibody, small molecular ligand, or peptide that binds to antigens preferentially expressed on the surface of tumor cells or in the tumor vasculature or interstitium (targeting group-directed enzyme/prodrug therapy, TDEPT), and (c) vasculature permeability-dependent enzyme/prodrug therapy (VPDEPT) in which the intratumoral delivery of the enzyme is realized through the higher permeability of tumor vasculature compared with normal vasculature as well as the prolonged circulation lifetime of the macromolecular enzyme [enhanced permeability and retention (EPR) effect] [2].
In TDEPT and VPDEPT, after the clearance of unbound enzyme from normal tissues, the nontoxic prodrug, which is a substrate of the enzyme, is administered. In GDEPT and VDEPT, the injection of the prodrug can be done after confirming the expression of the enzyme in the tumor. The prodrug is converted to the anticancer drug by the enzyme in the tumor, while normal tissues lacking the enzyme are spared from toxicity. Conversion of the prodrug by residual enzyme in normal tissues may lead to toxicity if the prodrug is injected too early, and to low tumor concentrations of the active drug if the prodrug is injected too late, as the enzyme concentration can decrease due to clearance or proteolytic degradation.
Determining the optimal time-window for prodrug injection is therefore critically important for the success of these strategies. The optimum time between enzyme and prodrug injection is usually based on the ratio of ex vivo enzymatic activities between the tumor and normal tissue obtained at selected time points after the injection of the enzyme [3, 4]. There are several disadvantages to this approach. The time point with the highest enzyme activity ratio between tumor and serum is difficult to pinpoint with a few selected time points. Given the variable and heterogeneous nature of tumor vasculature, it is difficult to generalize the time course of enzyme delivery and clearance. These studies also require determining the ex vivo enzymatic activity in different organs at different time points, with the associated costs of time and labor. The ex vivo enzymatic activity determined may not be identical with that in vivo because some enzyme activity may be lost in experimental processing such as homogenizing and extraction. Instrument sensitivity and the quantity of the sample available will also limit accurate measurement of the ex vivo enzyme activity ratio between tumor and normal tissues. Imaging the delivery of a drug-activating enzyme would therefore be ideal to optimize the timing of prodrug delivery. The ability to detect the delivery of the prodrug enzyme within the tumor is critically important because tumor vasculature is typically heterogeneous, and the ability to visualize the prodrug enzyme and the outcome of treatment become especially important in the clinical setting. The purpose of this review is to highlight some of the major prodrug enzyme systems and the applications of multimodality imaging in these prodrug enzyme treatment strategies (summarized in Table 1) with a focus on clinical translation.
Table 1.
Examples of prodrug enzyme and substrate combinations that are compatible with clinically translatable imaging technologies
| Substrates | Enzymes | Imaging technology | Purpose |
|---|---|---|---|
| [18F] FEAU | TK | PET | Imaging |
| [18F]FHBG | TK | PET | Imaging |
| [124I] FIAU/[131I]FIAU | TK | PET/SPECT | Imaging/therapy |
| 5-FC | bCD/yCD | 19F MRS/MRSI | Therapy |
| 5-FU | UPRT | 19F MRS/MRSI | Therapy |
| 3,5-DFBGlu | CPG2 | 19F/13C MRSI | Imaging |
| L-[18F] FAC/L-[18F]FMAC | dCK | PET | Imaging |
5-FC 5-fluorocytosine, 5-FU 5-fluorouracil, TK thymidine kinase, bCD bacterial cytosine deaminase, yCD yeast cytosine deaminase, UPRT uracil phosphoribosyltransferase, CPG2 carboxypeptidase G2, dCK deoxycytidine kinase, PET positron emission tomography, SPECT single-photon emission computed tomography, MRS magnetic resonance spectroscopy, MRSI magnetic resonance spectroscopic imaging
Cytosine deaminase
Cytosine deaminase is one of the most widely researched prodrug enzyme systems. The bacterial and yeast cytosine deaminases (CDs) convert the nontoxic prodrug 5-fluorocytosine (5-FC) to the anticancer drug 5-fluorouracil (5-FU) that is widely used in the treatment of a range of cancers, including colorectal and breast cancer and cancer of the aerodigestive tract [5]. 5-FU is an analog of uracil with a fluorine atom at the C-5 position in place of hydrogen and enters cells through the facilitated transport mechanism of uracil. Intracellularly, it is catalyzed by cellular enzymes to fluoronucleotides (FNucs), such as 2′-deoxy-5-fluorouridine mono-, di-, and triphosphates (FdUMP, FdUDP, and FdUTP), and 5-fluorouridine-5′-mono-, di-, and triphosphates (FUMP, FUDP, and FUTP), which subsequently disrupt RNA synthesis as well as the action of the enzyme thymidylate synthase that is required for DNA synthesis. The rate-limiting enzyme in 5-FU catabolism is dihydropyrimidine dehydrogenase (DPD), which converts 5-FU to dihydrofluorouracil. Since DPD is present at high concentrations in the liver, more than 80% of administered 5-FU is catabolized in the liver providing a strong rationale for synthesizing 5-FU locally within the tumor.
CD has been used extensively in proof-of-principle studies to demonstrate innovative strategies for cancer cell-specific treatments [6–8]. The conversion of 5-FC to 5-FU can be detected noninvasively with 19F magnetic resonance spectroscopy (MRS), providing an additional advantage for establishing enzyme activity in the tissue of interest with noninvasive imaging. While the use of CD in clinical studies has been limited [9, 10], new innovations are likely to lead to the resurgence of its use for cancer cell targeting. Recently Wright et al. [8] have used yeast CD (yCD) to demonstrate a synthetic biology strategy of designing a protein that can activate a therapeutic function in response to a specific cancer marker [8]. In these studies, a hybrid protein was designed that coupled yCD activity to the presence of hypoxia inducible factor-1α [8].
Li et al. [11, 12] have reported on the in vivo detection of bacterial cytosine deaminase (bCD) functionalized with multimodal MR and optical imaging reporters, to allow image-guided delivery of the prodrug. With its noninvasive characteristics and exquisite spatial resolution, magnetic resonance imaging (MRI) is one of the most powerful imaging techniques available in diagnostic imaging and preclinical results can be relatively easily translated to the clinic. However, MRI suffers from less than optimum sensitivity of detection. Optical imaging, on the other hand, is highly sensitive and capable of detecting minute amounts of light-emitting materials, but its image resolution is poor because of the intrinsic absorption and light scattering of heterogeneous tissue and it is not easily translated to the clinic. In preclinical studies, a multimodal strategy incorporating MRI and optical imaging can complement their strengths. Bacterial CD rather than yCD was selected as the therapeutic enzyme due to its high enzymatic stability. Poly-L-lysine (PLL) (Mr=5.6 kD) was selected as a carrier of the imaging reporters because of its extended conformation, which facilitates efficient extravasation into the tumor interstitium. PLL was functionalized with Gd3+-DOTA and rhodamine as previously reported by us, to dynamically monitor the distribution of bCD by either MRI or optical imaging. Rhodamine can also track the enzyme in excised tissue with fluorescence microscopy. The resulting bCD–PLL conjugate (MW>300 kDa) extravasated into the tumor interstitium but not the normal tissues due to the high permeability of tumor vasculature and was easily detected by MRI as well as optical imaging. The use of wild type and vascular endothelial growth factor overexpressing MDA-MB-231 breast cancer xenografts allowed the evaluation of the role of increased vascular permeability in the delivery of the conjugate. Additionally, the conversion of 5-FC to 5-FU was detected noninvasively in vivo with 19F MR spectroscopy.
More recently, this image-guided delivery of bCD has been combined with the delivery of small interfering RNA (siRNA) designed to silence cancer cell-specific targets with the aim of amplifying selective therapeutic targeting of cancer cells while sparing normal tissue, i.e., to improve the treatment efficacy [12]. A nanoplex carrying MRI reporters for in vivo detection and optical reporters for microscopy was used to image the delivery of the siRNA along with the functional prodrug enzyme in breast tumors, to achieve image-guided molecular targeted cancer therapy. As choline kinase-α (Chk-α) is significantly upregulated in aggressive breast cancer cells and a contributing component of this phenotype, Li et al. combined siRNA targeting of Chk-α mRNA with the prodrug enzyme bCD and observed a higher tumor growth delay in a orthotopic xenograft model of breast cancer than with either treatment alone. As shown in Fig. 1a and b, in vivo MRI showed efficient intratumoral nanoplex delivery. siRNA-mediated downregulation of Chk-α (Fig. 1d) and the conversion of 5-FC to 5-FU by bCD (Fig. 1c) were detected noninvasively with 1H magnetic resonance spectroscopic imaging (MRSI) and 19F MRS, respectively. The strategy can be expanded to target multiple pathways with siRNA and to deliver the nanoplex to receptors specific to cancer cells to combine therapy with diagnosis for “theranostic” imaging.
Fig. 1.
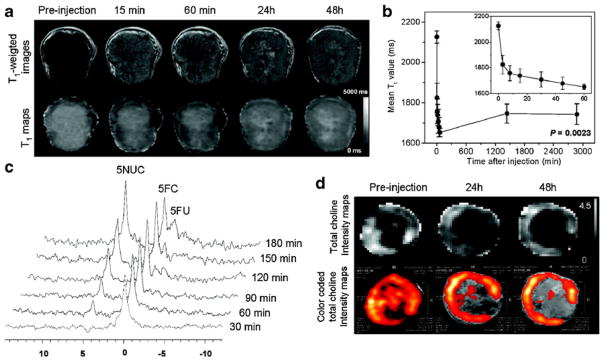
a Representative in vivo T1-weighted MR images (top panel) and quantitative T1 maps (bottom panel) of a tumor (400 mm3) preand post-injection of the nanoplex (300 mg/kg, i.v.). b Time-dependent mean T1 values of tumors (n=4) pre- and post-injection of the nanoplex; a significant decrease of T1 (P<0.005) was observed up to 48 h. Inset panel shows the T1 variation within the first 60 min of injection. c In vivo 19F MRS demonstrated efficient conversion of prodrug 5-FC to 5-FU and its metabolites FNucs by the nanoplex localized in the tumor. 5-FC (200 mg/kg i.v. and 250 mg/kg i.p.) was injected at 24 h after nanoplex injection. d Representative in vivo total choline (tCho) [maps and color-coded tCho intensity maps overlaid on corresponding T1- weighted images of a tumor before and at 24 and 48 h after nanoplex injection (300 mg/kg, i.v.)]. Adapted with permission from [12]
Cytosine deaminase plus uracil phosphoribosyltransferase
Certain tumor cells are not sensitive to or become resistant to 5-FU because of the low efficiency of the conversion of 5-FU to its toxic metabolites. The antitumoral effect of 5-FU can be improved by introducing the gene for uracil phosphoribosyltransferase (UPRT) from bacteria or yeast, which directly converts 5-FU into 5-FUMP [13]. Xing et al. have developed a rat prostate carcinoma cell line that stably expressed a fusion protein containing CD, UPRT, and DsRed fluorescent protein. Red fluorescence detected in cultured cells, as well in ex vivo tumor slices reported on the expression of the fusion protein that also contained the prodrug enzyme. The study showed that simultaneous expression of CD and UPRT significantly increased the sensitivity of rat prostate cancer cells to 5-FC in vitro and improved tumor control in vivo [13]. The conversion of 5-FC into 5-FU, FNuc, α-fluoro-β-ureidopropionic acid, and α-fluoro-β-alanine was monitored in vivo with 19F MRS [13].
Thymidine kinase
Another enzyme/prodrug strategy involves the viral thymidine kinase (TK) enzyme and the prodrug ganciclovir (GCV) that is not toxic by itself but becomes toxic after phosphorylation by the enzyme. Cells expressing viral TK enzyme produce toxic triphosphates leading to cell death. However, human TK cannot phosphorylate and activate the prodrug.
Radiolabeled 5-iodo-2′-fluoro-2′ deoxy-1-beta-D-arabinofuranosyl-uracil (FIAU) has been used as a substrate to image herpes simplex virus type 1 (HSV1)-TK gene expression using clinical imaging techniques such as a clinical gamma camera or a positron emission tomography (PET) system [14].
Yaghoubi et al. demonstrated the feasibility of using a mutant HSV1-TK enzyme (HSV1-sr39TK), which has higher affinity for the PET reporter probe 9-(4-[18F] fluoro-3-hydroxymethylbutyl)guanine ([18F]FHBG), and with which lower doses of GCV are sufficient to inhibit the growth of C6 glioma tumor xenografts [15]. Using [18F]FHBG with PET imaging, it was possible to monitor the expression of HSV1-sr39TK in C6 glioma tumors derived from cells stably transfected with HSV1-sr39TK that were implanted subcutaneously in nude mice. [18F]-Fluorodeoxyglucose ([18F]FDG) was used to assess tumor cell viability and therapeutic efficacy of repeated administration of GCV. Accumulation of [18F]FHBG was observed in vivo in tumors stably expressing HSV1-sr39TK before GCV treatment. After 2 weeks of GCV treatment, significant declines in tumor volumes were observed together with [18F] FHBG and [18F]FDG accumulation [15]. Three weeks after discontinuing treatment, the tumors regrew and the accumulation of [18F]FDG increased significantly. However, tumor [18F]FHBG concentrations remained at background levels. The study demonstrated the feasibility of using [18F]FHBG to detect the tumoral expression of HSV1-sr39TK and the effectiveness of GCV therapy in eradicating HSV1-sr39TK-expressing cells [15]. Schipper et al. have attempted to treat (HSV1)-TK-expressing cells by combining radiotherapy mediated by the trapping of [131I]FIAU, with the prodrug activation of GCV [16]. HSV1-TK-expressing cells accumulated more [131I]FIAU than the parental cell lines, leading to a decrease of their growth. However, a lack of synergy was observed when combining [131I]FIAU with GCV treatment in these cell studies although this does not rule out a synergistic effect in vivo [16].
Imaging of HSV-TK reporter gene expression can be also performed by using a radiopharmaceutical pyrimidine nucleoside analog, such as [18F]FEAU (1-(2′-deoxy-2′-fluoro-β-D-arabinofuranosyl)-5-ethyl-uridine) [17]. It has been shown that [18F]FEAU is better than [124I]FIAU and [18F] FHBG for imaging HSV-TK 1 to 2 h after probe injection in vivo in a rat glioma model. However, for late imaging, [124I] FIAU showed better retention in the HSV-TK-expressing tumor and a greater clearance from the body [17].
Suicide gene therapy can be combined with other therapeutic approaches, such as virotherapy, to achieve a stronger antitumoral effect [18]. Oncolytic adenoviruses expressing TK can be traced by PET imaging, after injection of the appropriate substrate, such as [18F]FEAU. In a preclinical experimental model of pancreatic cancer, the delivery of oncolytic adenovirus was followed noninvasively in vivo with PET imaging, and the combined effects of armed TK oncolytic adenovirus with GCV treatment resulted in an improvement of antitumoral effects, when an appropriate administration protocol was followed [18]. Tseng et al. have described a tumor-specific in vivo transfection of the TK gene using a Sindbis viral vector that can selectively target tumor cells through a laminin receptor [19]. The efficacy of transfection was imaged noninvasively with [18F]FEAU. The study showed the potential of a Sindbis viral vector for GDEPT-based systemic tumor targeting and treatment [19].
Clinical studies with an adenovirus to infect hepatocellular carcinoma in patients to induce HSV1-TK expression in the neoplastic nodules incorporated [18F]FHBG as a tracer for the PET imaging and valganciclovir as a prodrug to induce tumor regression. [18F]FHBG accumulated specifically in tumor nodules following injection of the adenoviral vector encoding HSV1-TK, demonstrating the potential of PET imaging to monitor adenoviral-mediated transgene expression in cancer patients [20]. Further investigations are required to assess the efficacy of the treatment on tumor progression.
Cytosine deaminase combined with thymidine kinase
Jacobs et al. have developed an HSV-1 amplicon vector co-expressing CD as the therapeutic gene and TK as a PET imaging reporter as well as a therapeutic gene, to induce synergistic antitumor activity [21]. Image-guided gene-directed prodrug enzyme therapy was applied to an experimental glioma. MRI was performed on tumor-bearing mice to localize the tumor, while PET images were acquired with [18F]FDG, [11C]MET, and [18F]FLT, which supplies surrogate indications of cellular density, neovascularization, and proliferative activity, respectively. TK gene expression was determined in vivo with [18F]FHBG PET. GCV and 5-FC were injected following transduction of the vector in tumor-bearing mice. Based on volumetric data, out of 22 mice transduced in vivo, 15 responded to the combined therapy. [18F]FLT PET was able to assess tumor response after 1 week of treatment [21]. Image-guided gene-directed pro-drug enzyme therapy will in the future facilitate improvement of protocols for clinical translation.
Serine protease prostate-specific antigen
Prostate cancer-targeted peptide prodrugs have been developed that are activated by the serine protease activity of prostate-specific antigen (PSA) [22]. Thapsigargin is a drug that induces apoptosis in a proliferation-independent manner. It is a potent inhibitor of the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase, a housekeeping enzyme that plays a key role in Ca2+ homeostasis. Inhibition of this ATPase triggers apoptotic death [22]. Normal and malignant prostate epithelial cells synthesize PSA that proteolyzes human seminal proteins semenogelins I and II. After determining the amino acid sequence of the cleavage sites recognized by PSA, a PSA-selective peptide substrate was designed by coupling its C-terminal carboxyl to a primary amine containing analog of thapsigargin. This novel prodrug was selectively activated by PSA within PSA-producing tumors. PSA is a secreted protein, thus cells in the vicinity, such as endothelial and other stromal cells, that do not produce PSA can also be targeted by a local bystander effect [22]. The PSA prodrug HSSKLQ-L12ADT has been designed to release a potent cytotoxic thapsigargin analog (L12ADT) into the tumor microenvironment following hydrolysis by PSA. To enhance delivery and solubility of the peptide, macromolecular carriers, such as N-(2-hydroxypropyl) methacrylamide-based copolymers, can be conjugated to it. Chandran et al. showed that the modified prodrug was toxic in vitro in the presence of active PSA. Moreover, analysis of tumor tissue from treated mice showed release and accumulation of the toxin in prostate tumor tissue [22]. Platforms of PSA inhibitor complexes are being investigated for PET and SPECT imaging of prostate cancer [23] and once available will be useful for PSA-based prodrug enzyme treatments. Other endogeneous enzymes known to be overexpressed by cancer cells such as metalloproteinase [24] could be used in an analogous manner following proteolytic action.
Carboxypeptidase G2
Carboxypeptidase G2 (CPG2) is a zinc-dependent bacterial enzyme that specifically hydrolyzes the amido, carbonyl, or ureido bonds between L-glutamic acid and the corresponding carboxyl-, phenol-, or aniline-substituted aromatic rings [25, 26]. CPG2 has been investigated in cancer therapy, due to its ability to enzymatically convert inactive prodrugs to cytotoxic drugs. The conversion can occur selectively in the tumor by administrating it as an immunoconjugate in which CPG2 is linked to a tumor-specific antibody or by using gene-directed enzyme prodrug therapy [25]. Several prodrugs have been designed to release potent DNA alkylating mustard drugs on CPG2-mediated activation [27]. CPG2 activity can be detected noninvasively in vivo by using 19F MRSI and a reporter probe, such as 3,5-DFBGlu [26]. The conversion of 3,5-DFBGlu to 3,5-DFBA can be measured with 19F MRS because both compounds have distinctly different chemical shifts. As shown in Fig. 2, the activity of the enzyme can be followed in vivo with 19F MRSI in a tumor xenograft over-expressing CPG2 [26]. In the clinic, 19F MRS could provide a biomarker of the enzyme activity in patients undergoing CPG2-based therapy [26]. Jamin et al. have developed 13C-labeled probes to follow the activity of CPG2 with 13C MRS. Hyperpolarization of the probe 3,5-DFBGlu allowed them to measure the conversion of the labeled probe into the resulting cleaved compounds in vitro [28].
Fig. 2.

Noninvasive detection of CPG2 activity in vivo using 19F 3D MRSI. a Serial 19F spectra over time extracted from 19F 3D MRSI datasets originating from a voxel in the middle of a tumor derived from either CPG2-expressing or control WiDr cells, following intravenous injection of 750 mg/kg 3,5-DFBGlu. b Spectral map through the tumor (T) generated from a 19F 3D MRSI dataset acquired 1 h following i.v. injection of 750 mg/kg 3,5-DFBGlu in a mouse bearing a WiDr tumor expressing CPG2. c In vivo 19F pharmacokinetics of 3,5-DFBGlu and its CPG2-mediated conversion in the tumors of mice bearing CPG2- expressing WiDr xenografts. The graph shows the changes in the MR-apparent concentration of 3,5-DFBGlu in xenografts derived from CPG2-expressing (-□-) and control (-△-) WiDr cells and the changes in the concentration of 3,5-DFBA (-■-) in xenografts derived from CPG2 expressing WiDr cells, induced by CPG2 activity following i.v. injection of 3,5-DFBGlu. Data points represent mean±1 SEM; n=6 for CPG2-expressing and n=3 for control tumors. Adapted with permission from [26]
Carboxylesterase
The carboxylesterase enzyme converts the prodrug CPT-11 (Irinotecan) into an active anticancer agent SN-38. Genetically engineered human neuronal stem cells (NSCs) that encode carboxylesterase have been used to target subdural medulloblastomas in an experimental model [29]. Carboxylesterase expressing NSCs (F3.rCE) were labeled with fluorescent magnetic nanoparticles (LEO-Live 675) for in vivo imaging. Figure 3 shows the accumulation of labeled cells in the subdural medulloblastoma site over time. The injection of F3.rCE cells in combination with the prodrug CPT-11 prolonged the survival of tumor-bearing mice, as compared to the mice that received either one or the other alone [29]. With optical imaging, the migratory potential of the F3.rCE cells could be assessed. The study showed the efficacy of therapeutic targeting of subdural medulloblastomas with carboxylesterase enzyme and the prodrug CPT-11 [29].
Fig. 3.

In vivo time-dependent fluorescence imaging of carboxylesterase expressing NSCs (F3.rCE). The mice were randomized into three groups: group A, sham-operated mice with i.v. injection of LEO-Live 675; group B, sham-operated mice with i.v. injection of LEO-Live 675-labeled F3.rCE; group C, subdural medulloblastoma-bearing mice with i.v. injection of LEO-Live 675-labeled F3.rCE cells. Fluorescence images of the mice were taken at the indicated times. In group C, F3.rCE cells migrate to the subdural tumor foci at 2 h and accumulate there over time. Signals from F3.rCE cells have gradually faded in other major organs. Adapted with permission from [29]
Deoxycytidine kinase
As a rate-limiting enzyme in the deoxyribonucleoside salvage pathway, deoxycytidine kinase (dCK), is a critical determinant of therapeutic activity for nucleoside analog prodrugs. PET probes have been synthesized and evaluated in vitro and in vivo to assess dCK activity [30]. dCK phosphorylates and activates several nucleoside analogs that are cytotoxic pro-drugs used in the treatment of hematologic and solid tumors, such as cytarabine, fludarabine, gemcitabine, cladribine, decitabine, and clofarabine. Shu et al. have described several potential PET imaging probes, including 1-(2′-deoxy-2′-[18F]fluoro-beta-L-arabinofuranosyl)cytosine (L-[18F]-FAC) and 1-(2′-deoxy-2′-[18F]fluoro-beta-L-arabinofuranosyl)-5-methylcytosine (L-[18F]FMAC) that accumulate in dCK-positive leukemic tumors [30]. These probes can be used as surrogate markers for cell proliferation, immune activation, treatment stratification, and monitoring, as well as providing a measurement of the metabolic flux through the deoxyribonucleoside salvage pathway [30]. dCK PET imaging probes will be useful in the clinic to stratify and select patients that are likely to benefit from dCK-based prodrug treatments. Likar et al. have described a mutated human dCK reporter gene that could potentially be used for PET imaging during treatment with acycloguanosine-based drugs [31]. Transduced cells showed a high uptake of the pyrimidine-based radiopharmaceutical [18F]FEAU (2′-deoxy-2′-fluoroarabinofuranosylcytosine, 2′-fluoro-2′-deoxyarabinofuranosyl-5-ethyluracil) and displayed suicidal activation after treatment with pyrimidine-based prodrugs, such as cytarabine and gemcitabine [31]. This mutant reporter gene could be used as a suicide gene for prodrug activation of pyrimidine analogs or as a pyrimidine-specific PET reporter gene for imaging patients treated with acycloguanosine-based drugs.
Nitroreductase
Bacterial nitroreductase (NTR) and the prodrug CB1954 is another promising suicide gene/prodrug combination [32]. The nontoxic prodrug CB1954 can be reduced by NTR to its activated cytotoxic form, 5-(aziridin-1-yl)-4-hydroxylamino-2-nitrobenzamide, a DNA inter-strand cross-linking agent. The enzyme activity can be measured in vivo with optical imaging by injecting the probe CytoCy5S, a membrane-permeable quenched substrate that is reduced to its fluorescent form by NTR enzyme [32]. A phase I/II clinical trial with a replication-defective adenoviral vector expressing NTR in combination with CB1984 treatment is currently ongoing in prostate cancer patients [33].
Delivery and targeting
Enzyme prodrug therapies are mainly based on the EPR effect for tumor-selective delivery of the enzyme [12]. However, the efficacy of the therapy can be improved by specifically targeting the enzyme to tumor tissue. Specific targeting will not only improve the delivery to the tumor, but also decrease the deleterious side effects that occur in normal tissues. In vitro experiments combining CD with annexin V to specifically target endothelial cells and breast cancer cells exposing phosphatidylserine (PS) on their surface have been performed [34]. PS is exposed on the surface of vascular endothelial cells within tumor tissue but not on normal endothelium. Moreover, it is exposed on the surface of tumor cells. CD-annexin V fusion protein in combination with 5-FC had significant cytotoxic effects on cells exposing PS on their surface and could be used in vivo for tumor-specific targeting [34]. Tumor-specific targeting can also be achieved by using antibody-directed enzyme prodrug therapy that employs antibody–enzyme conjugates to deliver the enzyme selectively to the tumor [35]. Treatment delivery to the tumor can also be improved by modifying the tumor environment and vasculature [36].
Concluding remarks
There is no dearth of cytotoxic therapies available to kill cancer cells. Unfortunately, most conventional chemotherapies damage normal cells. The basis for prodrug enzyme therapy is to increase the therapeutic index by delivering a drug-activating enzyme to tumor tissue prior to the systemic administration of a nontoxic prodrug. This strategy results in cell killing that is limited to and maximized at the tumor site. Noninvasive imaging can fill an important function, pre-clinically and clinically, in guiding the timing of prodrug administration within the period that the enzyme concentration is high in tumor tissue and low in systemic circulation and normal tissue. To date, most imaging studies in the prodrug field have focused on detecting the activation of the prodrug. Since tumor vasculature is typically heterogeneous, the ability to visualize the prodrug enzyme becomes especially important in the clinical setting. Imaging techniques can be used to detect the delivery of the enzyme to optimize the timing of the prodrug administration in addition to detecting the conversion of the prodrug to its active form. In addition to cancer cells, tumor-associated stromal cells, such as endothelial cells, macrophages, and fibroblasts, are emerging as important functional contributors to tumor progression and dissemination. Potential applications of image-guided prodrug enzyme strategies should in the future include targeting these stromal cells as well as disseminated cancer cells and metastatic lesions. Future development of these technologies can have a major impact in patient therapy by increasing the efficacy of tumor-specific killing while decreasing morbidity.
Acknowledgments
Support from P50 CA103175, P30 CA006973, R01 CA73850, R01 CA82337, R01 CA136576, R01 CA138515, and R01 CA138264 is gratefully acknowledged.
Abbreviations
- bCD
Bacterial cytosine deaminase
- CD
Cytosine deaminase
- Chk
Choline kinase
- dCK
Deoxycytidine kinase
- DPD
Dihydropyrimidine dehydrogenase
- EPR
Enhanced permeability and retention
- FDG
[18F]fluorodeoxyglucose
- 5-FC
5-Fluorocytosine
- FdUMP, FdUDP, and FdUTP
2′-Deoxy-5-fluorouridine mono-, di-, and triphosphates
- [18F]FHBG
9-(4-[18F]fluoro-3-hydroxymethylbutyl)guanine
- FNucs
Fluoronucleotides
- 5-FU
5-Fluorouracil
- FUMP, FUDP, and FUTP
5-Fluorouridine-5′-mono-, di-, and triphosphates
- GCV
Ganciclovir
- GDEPT
Gene encoding prodrug-activating enzyme therapy
- HSV1
Herpes simplex virus type 1
- MRI
Magnetic resonance imaging
- MRS
Magnetic resonance spectroscopy
- MRSI
Magnetic resonance spectroscopic imaging
- PET
Positron emission tomography
- PLL
Poly-L-lysine
- siRNA
Small interfering RNA
- tCho
Total choline
- TDEPT
Targeting group-directed enzyme/prodrug therapy
- TK
Thymidine kinase
- UPRT
Uracil phosphoribosyltransferase
- VDEPT
Virus-directed enzyme prodrug therapy
- VPDEPT
Vasculature permeability-dependent enzyme/prodrug therapy
- yCD
Yeast cytosine deaminase
Contributor Information
Marie-France Penet, JHU ICMIC Program, The Russell H. Morgan Department of Radiology and Radiological Science, The Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA.
Zhihang Chen, JHU ICMIC Program, The Russell H. Morgan Department of Radiology and Radiological Science, The Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA.
Cong Li, JHU ICMIC Program, The Russell H. Morgan Department of Radiology and Radiological Science, The Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA.
Paul T. Winnard, Jr., JHU ICMIC Program, The Russell H. Morgan Department of Radiology and Radiological Science, The Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA
Zaver M. Bhujwalla, Email: zaver@mri.jhu.edu, JHU ICMIC Program, The Russell H. Morgan Department of Radiology and Radiological Science, The Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA. Sidney Kimmel Comprehensive Cancer Center, The Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA. Department of Radiology, Johns Hopkins University School of Medicine, Rm 208C Traylor Bldg., 720 Rutland Avenue, Baltimore, MD 21205, USA
References
- 1.Xu G, et al. Strategies for enzyme/prodrug cancer therapy. Clin Cancer Res. 2001;7(11):3314–24. [PubMed] [Google Scholar]
- 2.Mahato R, et al. Prodrugs for improving tumor targetability and efficiency. Adv Drug Deliv Rev. 2011;63(8):659–70. doi: 10.1016/j.addr.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma SK, et al. Sustained tumor regression of human colorectal cancer xenografts using a multifunctional mannosylated fusion protein in antibody-directed enzyme prodrug therapy. Clin Cancer Res. 2005;11(2 Pt 1):814–25. [PubMed] [Google Scholar]
- 4.Bhatia J, et al. Catalytic activity of an in vivo tumor targeted anti-CEA scFv::carboxypeptidase G2 fusion protein. Int J Cancer. 2000;85(4):571–7. [PubMed] [Google Scholar]
- 5.Longley DB, et al. 5-Fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3(5):330–8. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 6.Aboagye EO, et al. Intratumoral conversion of 5-fluorocytosine to 5-fluorouracil by monoclonal antibody-cytosine deaminase conjugates: noninvasive detection of prodrug activation by magnetic resonance spectroscopy and spectroscopic imaging. Cancer Res. 1998;58(18):4075–8. [PubMed] [Google Scholar]
- 7.Hamstra DA, et al. The use of 19F spectroscopy and diffusion-weighted MRI to evaluate differences in gene-dependent enzyme prodrug therapies. Mol Ther. 2004;10(5):916–28. doi: 10.1016/j.ymthe.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 8.Wright CM, et al. A protein therapeutic modality founded on molecular regulation. Proc Natl Acad Sci U S A. 2011;108 (39):16206–11. doi: 10.1073/pnas.1102803108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crystal RG, et al. Phase I study of direct administration of a replication deficient adenovirus vector containing the E. coli cytosine deaminase gene to metastatic colon carcinoma of the liver in association with the oral administration of the pro-drug 5-fluorocytosine. Hum Gene Ther. 1997;8(8):985–1001. doi: 10.1089/hum.1997.8.8-985. [DOI] [PubMed] [Google Scholar]
- 10.Pandha HS, et al. Genetic prodrug activation therapy for breast cancer: a phase I clinical trial of erbB-2-directed suicide gene expression. J Clin Oncol. 1999;17(7):2180–9. doi: 10.1200/JCO.1999.17.7.2180. [DOI] [PubMed] [Google Scholar]
- 11.Li C, et al. Image-guided enzyme/prodrug cancer therapy. Clin Cancer Res. 2008;14(2):515–22. doi: 10.1158/1078-0432.CCR-07-1837. [DOI] [PubMed] [Google Scholar]
- 12.Li C, et al. Nanoplex delivery of siRNA and prodrug enzyme for multimodality image-guided molecular pathway targeted cancer therapy. ACS Nano. 2010;4(11):6707–16. doi: 10.1021/nn102187v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xing L, et al. Non-invasive molecular and functional imaging of cytosine deaminase and uracil phosphoribosyltransferase fused with red fluorescence protein. Acta Oncol. 2008;47(7):1211–20. doi: 10.1080/02841860802256475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blasberg RG, et al. Herpes simplex virus thymidine kinase as a marker/reporter gene for PET imaging of gene therapy. Q J Nucl Med. 1999;43(2):163–9. [PubMed] [Google Scholar]
- 15.Yaghoubi SS, et al. Imaging progress of herpes simplex virus type 1 thymidine kinase suicide gene therapy in living subjects with positron emission tomography. Cancer Gene Ther. 2005;12(3):329–39. doi: 10.1038/sj.cgt.7700795. [DOI] [PubMed] [Google Scholar]
- 16.Schipper ML, et al. Evaluation of herpes simplex virus 1 thymidine kinase-mediated trapping of (131)I FIAU and prodrug activation of ganciclovir as a synergistic cancer radio/chemotherapy. Mol Imaging Biol. 2007;9(3):110–6. doi: 10.1007/s11307-007-0078-3. [DOI] [PubMed] [Google Scholar]
- 17.Miyagawa T, et al. Imaging of HSV-tk Reporter gene expression: comparison between [18F]FEAU, [18F]FFEAU, and other imaging probes. J Nucl Med. 2008;49(4):637–48. doi: 10.2967/jnumed.107.046227. [DOI] [PubMed] [Google Scholar]
- 18.Abate-Daga D, et al. Oncolytic adenoviruses armed with thymidine kinase can be traced by PET imaging and show potent antitumoural effects by ganciclovir dosing. PLoS One. 2011;6(10):e26142. doi: 10.1371/journal.pone.0026142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tseng JC, et al. Tumor-specific in vivo transfection with HSV-1 thymidine kinase gene using a Sindbis viral vector as a basis for prodrug ganciclovir activation and PET. J Nucl Med. 2006;47 (7):1136–43. [PubMed] [Google Scholar]
- 20.Penuelas I, et al. Positron emission tomography imaging of adenoviral-mediated transgene expression in liver cancer patients. Gastroenterology. 2005;128(7):1787–95. doi: 10.1053/j.gastro.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 21.Jacobs AH, et al. Imaging-guided gene therapy of experimental gliomas. Cancer Res. 2007;67(4):1706–15. doi: 10.1158/0008-5472.CAN-06-2418. [DOI] [PubMed] [Google Scholar]
- 22.Chandran SS, et al. A prostate-specific antigen activated N-(2-hydroxypropyl) methacrylamide copolymer prodrug as dual-targeted therapy for prostate cancer. Mol Cancer Ther. 2007;6 (11):2928–37. doi: 10.1158/1535-7163.MCT-07-0392. [DOI] [PubMed] [Google Scholar]
- 23.LeBeau AM, et al. Optimization of peptide-based inhibitors of prostate-specific antigen (PSA) as targeted imaging agents for prostate cancer. Bioorg Med Chem. 2009;17(14):4888–93. doi: 10.1016/j.bmc.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chuang CH, et al. In vivo positron emission tomography imaging of protease activity by generation of a hydrophobic product from a non-inhibitory protease substrate. Clin Cancer Res. 2011 doi: 10.1158/1078-0432. CCR-11-0608. [DOI] [PubMed] [Google Scholar]
- 25.Rowsell S, et al. Crystal structure of carboxypeptidase G2, a bacterial enzyme with applications in cancer therapy. Structure. 1997;5(3):337–47. doi: 10.1016/s0969-2126(97)00191-3. [DOI] [PubMed] [Google Scholar]
- 26.Jamin Y, et al. Noninvasive detection of carboxypeptidase G2 activity in vivo. NMR Biomed. 2011;24(4):343–50. doi: 10.1002/nbm.1597. [DOI] [PubMed] [Google Scholar]
- 27.Hedley D, et al. Carboxypeptidase-G2-based gene-directed enzyme-prodrug therapy: a new weapon in the GDEPT armoury. Nat Rev Cancer. 2007;7(11):870–9. doi: 10.1038/nrc2247. [DOI] [PubMed] [Google Scholar]
- 28.Jamin Y, et al. Hyperpolarized (13)C magnetic resonance detection of carboxypeptidase G2 activity. Magn Reson Med. 2009;62 (5):1300–4. doi: 10.1002/mrm.22049. [DOI] [PubMed] [Google Scholar]
- 29.Lim SH, et al. Therapeutic targeting of subdural medulloblastomas using human neural stem cells expressing carboxylesterase. Cancer Gene Ther. 2011;18(11):817–24. doi: 10.1038/cgt.2011.52. [DOI] [PubMed] [Google Scholar]
- 30.Shu CJ, et al. Novel PET probes specific for deoxycytidine kinase. J Nucl Med. 2010;51(7):1092–8. doi: 10.2967/jnumed.109.073361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Likar Y, et al. A new pyrimidine-specific reporter gene: a mutated human deoxycytidine kinase suitable for PET during treatment with acycloguanosine-based cytotoxic drugs. J Nucl Med. 2010;51(9):1395–403. doi: 10.2967/jnumed.109.074344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhaumik S, et al. Noninvasive optical imaging of nitroreductase gene-directed enzyme prodrug therapy system in living animals. Gene Therapy. 2011 doi: 10.1038/gt.2011.101. [DOI] [PubMed] [Google Scholar]
- 33.Patel P, et al. A phase I/II clinical trial in localized prostate cancer of an adenovirus expressing nitroreductase with CB1954 [correction of CB1984] Molecular Therapy. 2009;17(7):1292–9. doi: 10.1038/mt.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Rite BD, et al. Annexin V-targeted enzyme prodrug therapy using cytosine deaminase in combination with 5-fluorocytosine. Cancer Lett. 2011;307(1):53–61. doi: 10.1016/j.canlet.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 35.Tietze LF, et al. Prodrugs for targeted tumor therapies: recent developments in ADEPT, GDEPT and PMT. Curr Pharm Des. 2011;17(32):3527–47. doi: 10.2174/138161211798194459. [DOI] [PubMed] [Google Scholar]
- 36.Goel S, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91(3):1071–121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]