

Abstract
Helicases have important roles in nucleic acid metabolism, and their prominence is marked by the discovery of genetic disorders arising from disease-causing mutations. Missense mutations can yield unique insight to molecular functions and basis for disease pathology. XPB or XPD missense mutations lead to Xeroderma pigmentosum, Cockayne’s syndrome, Trichothiodystrophy, or COFS syndrome, suggesting that DNA repair and transcription defects are responsible for clinical heterogeneity. Complex phenotypes are also observed for RECQL4 helicase mutations responsible for Rothmund-Thomson syndrome, Baller-Gerold syndrome, or RAPADILINO. Bloom’s syndrome causing missense mutations are found in the conserved helicase and RecQ C-terminal domain of BLM that interfere with helicase function. Although rare, patient-derived missense mutations in the exonuclease or helicase domain of Werner syndrome protein exist. Characterization of WRN separation-of-function mutants may provide insight to catalytic requirements for suppression of phenotypes associated with the premature aging disorder. Characterized FANCJ missense mutations associated with breast cancer or Fanconi anemia interfere with FANCJ helicase activity required for DNA repair and the replication stress response. For example, a FA patient-derived mutation in the FANCJ Iron-Sulfur domain was shown to uncouple its ATPase and translocase activity from DNA unwinding. Mutations in DDX11 (ChlR1) are responsible for Warsaw Breakage syndrome, a recently discovered autosomal recessive cohesinopathy. Ongoing and future studies will address clinically relevant helicase mutations and polymorphisms, including those that interfere with key protein interactions or exert dominant negative phenotypes (e.g., certain mutant alleles of Twinkle mitochondrial DNA helicase). Chemical rescue may be an approach to restore helicase activity in loss-of-function helicase disorders. Genetic and biochemical analyses of disease-causing missense mutations in human helicase disorders have led to new insights to the molecular defects underlying aberrant cellular and clinical phenotypes.
Keywords: helicase, DNA repair, replication, human genetic disease, genomic stability, mutation
Essential Nature of DNA Helicases Evidenced by Hereditary Helicase Disorders
Helicases are molecular motors that couple the energy of nucleoside triphosphate (NTP) hydrolysis (typically ATP) to the unwinding of polynucleic acid structures by disrupting the noncovalent hydrogen bonds between complementary strands [1, 2]. Helicases can be classified on the basis of their polarity of unwinding double-stranded DNA with respect to the strand that they translocate, either 3′ to 5′ or 5′ to 3′. Helicases have essential roles in virtually all aspects of nucleic acid metabolism, including replication, DNA repair, recombination, transcription, chromosome segregation, and telomere maintenance [2–6]. They can be classified as DNA or RNA unwinding enzymes, although some helicases can function on both DNA and RNA [6]. DNA helicases have been reported to act in a variety of DNA metabolic processes that involve unwinding duplex or alternate DNA structures (e.g., D-loop, Holliday junction (HJ), triplex, G-quadruplex (G4)), stripping proteins bound to either single-stranded or double-stranded DNA, or strand annealing [7–10].
An increasing number of genetic diseases characterized by chromosomal instability are linked to mutations in genes encoding DNA helicase-like proteins. Many of these diseases are very rare, and inherited by autosomal dominant or recessive mutations (Table 1). For example, disease-causing recessive mutations in the BLM and WRN genes of the RecQ helicase family are responsible for Bloom’s syndrome (BS) and Werner syndrome (WS), respectively [11, 12]. Individuals with BS are characterized by proportional small size and display erythema that develops on sun exposed areas of skin. Those with BS are commonly predisposed to cancer, which can encompass a broad spectrum including adult epithelial tumors, leukemias, lymphomas, and rare pediatric tumors [13]. WS is a genetic disorder characterized by premature aging features and the early onset of age-related diseases such as cardiovascular disorders, diabetes mellitus (Type II), osteoporosis, and sarcoma and mesenchymal tumors [14]. Mutations in the RECQL4 gene, encoding a third RecQ family member, can lead to three genetic disorders with overlapping features that display a range in the spectrum of severity which may be influenced by background genetic effects or environmental factors. These are Rothmund–Thomson syndrome (RTS), Baller-Gerold syndrome (BGS), and RAPADILINO [radial hypoplasia/aplasia, patellae hypoplasia/aplasia and cleft or highly arched palate, diarrhoea and dislocated joints, little size (height at least 2 S.D. smaller than the average height) and limb malformation, nose slender and normal intelligence] syndrome [15]. Individuals with RTS are stunted in growth, and characterized by photosensitivity with poikiloderma, early graying and hair loss, juvenile cataracts and osteogenic sarcomas. It should be noted there are cases of RTS in which no RECQL4 mutation has been found. BGS is characterized by radial aplasia/hypoplasia and craniosynostosis, but not the poikiloderma typical of RTS patients [16]. In addition to WRN, BLM and RECQL4, two other human RecQ helicases are RECQL1 (RECQ1) and RECQL5 (RECQ5), which are not yet linked to a disease, but it seems likely that these helicases will also play a role in cancer predisposition or a hereditary disorder characterized by chromosomal instability [17].
Table 1.
DNA Helicases, Clinical Symptoms, and Salient Features
| Helicase | Clinical Symptoms | Salient Features |
|---|---|---|
| WRN | Werner syndrome (WS) | Premature aging Early onset of age-related diseases (e.g., cardiovascular disorders, diabetes mellitus (type II), osteoporosis, sarcoma and mesenchymal tumors) |
| BLM | Bloom syndrome (BS) | An erythema that develops on sun exposed areas of skin Broad spectrum of cancers in early life |
| RECQL4 | Rothmund–Thomson syndrome (RTS) | Stunted in growth, photosensitivity with poikiloderma, early graying and hair loss, juvenile cataracts and osteogenic sarcomas |
| Baller-Gerold syndrome (BGS) | BGS is characterized by radial aplasia/hypoplasia and craniosynostosis, but not the poikiloderma typical of RTS patients | |
| RAPADILINO syndrome | Radial hypoplasia/aplasia, patellae hypoplasia/aplasia and cleft or highly arched palate, diarrhoea and dislocated joints, little size (height at least 2 S.D. smaller than the average height) and limb malformation, nose slender and normal intelligence] | |
| FANCJ | Fanconi Anemia (FA) | Congenital abnormalities, progressive bone marrow failure, and cancer accompanied by chromosomal instability and hypersensitivity to agents that induce DNA ICLs |
| XPD | Xeroderma pigmentosum (XP) | Severe photosensitivity, leading to hyper pigmentation, premature skin aging, and predisposition to skin cancers |
| XP combined with Cockayne’s syndrome (CS) | Photosensitivity, neurological/developmental abnormalities, and skin cancer neurological and developmental abnormalities in addition to UV sensitivity, but have no predisposition to skin cancer | |
| Trichothiodystrophy (TTD) | Brittle hair and nails ichtyosis, postnatal growth failure, and impaired sexual development | |
| Cerebro-oculo-facial-skeletal (COFS) syndrome | Progressive neurological degeneration, calcifications, cataracts, microcornea, optic atrophy, progressive joint contractures, and growth failure | |
| XPB | Xeroderma pigmentosum (XP) | Multiple clinical disorders including XP/CS, XP with neurological abnormalities, and TTD |
| DDX11 | Warsaw Breakage syndrome (WABS) | Cellular features of both FA and Roberts syndrome, characterized by sister chromatid cohesion defects |
Another family of DNA helicases with a linkage to human diseases that have acquired a great deal of interest is the so-called Iron-Sulfur (Fe-S) cluster helicases, named after a conserved metal binding domain in the helicase core [18, 19]. Mutations in the Fe-S cluster helicase XPD, a key factor of the TFIIH complex implicated in basal transcription and nucleotide excision repair (NER), can give rise to multiple rare autosomal recessive disorders: xeroderma pigmentosum (XP), XP combined with Cockayne’s syndrome (CS), Trichothiodystrophy (TTD), and Cerebro-oculo-facial-skeletal (COFS) syndrome (for a recent review, see [20]). Photosensitivity, neurological/developmental abnormalities, and skin cancer can be used to distinguish between XP, TTD, and CS; however, related or overlapping clinical features can arise from XPD mutations. Recessive mutations in XPB, which encodes a second motor ATPase helicase-like protein in the TFIIH complex, give rise to multiple clinical disorders including XP/CS, XP with neurological abnormalities, and TTD.
Homozygous recessive mutations in the FANCJ gene encoding the FANCJ Fe-S cluster DNA helicase (also called BACH1 or BRIP1), and at least fourteen other genes, are responsible for Fanconi anemia (FA), a disorder characterized by progressive bone marrow failure, chromosomal instability, and cancer. In addition, many FA individuals have congenital abnormalities such as commonly observed radial ray abnormalities. Moreover, in severe cases there are developmental abnormalities that can affect many organ systems [21, 22]. Cells from FA individuals display hypersensitivity to agents that induce DNA interstrand cross-links (ICLs). Notably, FANCJ mutations have also been associated with breast cancer [23]. The latest Fe-S cluster helicase implicated in a genetic disease is DDX11 (also known as ChlR1), in which homozygous recessive mutations are linked to Warsaw Breakage syndrome (WABS), a unique disease with cellular features of both FA (hypersensitivity to DNA cross-linking agents) and Roberts syndrome, a cohesinopathy disorder in which cells from affected individuals display sister chromatid cohesion defects [24].
Some clinical syndromes characterized by linkage to mutations in genes encoding DNA helicase-like proteins (including the motor ATPases CSB and FANCM) are also linked to mutations in other DNA repair genes (Table 2). A review by DiGiovanna and Kraemer (2012) [20] provides an excellent perspective on the relationships among XP, CS, TTD and related clinical disorders with molecular defects in DNA helicase-like proteins (XPB, XPD, CSB) or gene products involved in steps such as DNA damage recognition (e.g., XPA), incision (e.g., XPF, XPG), or DNA synthesis (e.g., POLH). A complex spectrum of genotype/phenotype relationships exist for these diseases with significant heterogeneity in clinical presentation in many cases. The genetic linkage of the XP/CS/TTD diseases to helicase-like genes as well as other DNA repair genes emphasizes the important role of helicase proteins in the basic biology underlying the DNA repair defects in the genetic disorders. A similar case can be made for the DNA repair disorder FA that is linked to recessive mutations in genes encoding DNA helicase-like proteins (FANCJ, FANCM) as well as other DNA repair proteins involved in damage signaling (e.g., FANCA) or promote homologous recombination (HR) (e.g., FANCD1, FANCN)[25] (Table 2).
Table 2.
Clinical Syndromes with Genetic Defects in Helicase-like Proteins or Other DNA Repair Factors
| DNA Repair Disorder | Helicase Gene Mutation | Other DNA Repair Gene Mutation |
|---|---|---|
| XP | XPD | XPC, DDB2, XPV (POLH), XPF, XPG, XPA, XPE |
| CS | CSB | CSA |
| TTD | XPB, XPD | GTF2H6, C7orf11 |
| COFS | XPD, CSB | XPG |
| XP/CS | XPB, XPD, CSB | XPG |
| UV-sensitive syndrome | CSB | CSA |
| FA | FANCJ (BACH1), FANCM | FANCA, FANCB, FANCC, FANCD1 (BRCA2), FANCD2, FANCE, FANCF, FANCG (XRCC9), FANCI, FANCL (PHF9), FANCN (PALB2), FANCO (RAD51C), FANCP (SLX4) |
Besides the nuclear DNA helicases, missense mutations of the Twinkle mitochondrial DNA helicase co-segregate with a number of diseases with mitochondrial defects including adult-onset progressive external ophthalmoplegia, hepatocerebral mitochondrial DNA depletion syndrome, and infantile-onset spinocerebellar ataxia [26]. Twinkle is required for replication of human mitochondrial DNA, and mutations in the C10orf2 gene encoding Twinkle helicase can lead to mitochondrial deletions in post-mitotic tissues.
Primary Sequence Arrangement and Structural Architecture of DNA Helicases
DNA helicases are classified according to their amino acid sequence homology into two large families, superfamilies 1 and 2 (SF1 and SF2) and four small families [2]. Conserved clusters of amino acids, the so-called helicase motifs, constitute the helicase core domain. Structural and biochemical analyses have provided insight to how the helicase core domain is organized to bind nucleotide and DNA, and couple the binding/hydrolysis of NTP to unwind double-stranded DNA. The crystal structures of SF1 helicases UvrD [27] and PcrA [28], and SF2 helicases, including RecG [29], NS3 [30], RecQ [31], RECQL1 (RECQ1) [32], VASA [33], and Hel308 [34, 35], revealed that there are two universal RecA folds, which are essential for nucleic acid binding, NTP binding and hydrolysis, and coupling of NTP hydrolysis to nucleic acid unwinding. The conserved helicase motifs reside at the interface between the two RecA-like domains. These seven signature helicase motifs are designated motif I (or Walker A), Ia, II (or Walker B), III, IV, V and VI. In addition, there are other less conserved, family-typical motifs, including a Q motif (also known as motif 0) before motif I [2, 36]. Generally speaking, motifs I, II, VI, and Q are important for ATP binding/hydrolysis, and motifs Ia, IV, and V are primarily responsible for DNA binding. Motif III is also involved in DNA binding, and is important for coupling ATP hydrolysis to DNA unwinding [6, 27].
Structural elements and accessory domains outside the RecA core domains are important for the specialized functions of each enzyme, thereby permitting helicases to be involved in diverse aspects of RNA and DNA metabolism. For example, the crystal structure of human RECQ1 bound to DNA substrate revealed a prominent β-hairpin that acts as a pin and abuts the end of the duplex DNA to promote strand separation [32]. Other DNA helicases such as bacterial UvrD [27] and archaeal Hel308 [34] also possess a conserved β hairpin to act as an unwinding element. The hairpin is located within the helicase domain and can display various orientations toward the DNA substrate, depending on the helicase. The structure of the isolated Winged helix (WH) found in the conserved RecQ C-terminal (RQC) region from Werner syndrome helicase-nuclease (WRN) in complex with DNA identifies a novel mode of DNA interaction [37], which may be conserved in other members of the RecQ family. However, the structural rearrangement of the WH domain upon binding the DNA substrate may be a distinguishing feature of the enzymatic mechanism that differs among RecQ helicases [38]. Other auxiliary domains in certain SF2 helicases exist, such as the helicase-and-RNaseD-like-C-terminal (HRDC) domain, which is believed to be important for recognition of specific DNA structures and protein interactions [39].
Recent studies of the SF2 XPD helicase revealed two unique domains (Arch and Fe-S) that separate this family of DNA helicases from others. A conserved metal binding domain between the Walker A and B boxes in the XPD protein family of SF2 DNA helicases was first recognized by the White lab [40]. Recently solved crystal structures of archaeal XPD confirmed the existence of a novel Fe-S domain and revealed the architecture of the conserved helicase motifs [41–43]. It is thought that the intimate association of the Fe-S and Arch domains with conserved motifs of the helicase core domain are responsible for conformational changes induced by nucleotide binding and hydrolysis which facilitate the catalytic separation of complementary strands. The Fe-S domain was proposed to form a wedge shape with the nearby Arch motif to separate the DNA duplex as the enzyme translocates in an ATP-dependent manner. Biochemical studies of the purified recombinant XPD protein and mutated XPD proteins with amino acid substitutions experimentally introduced by site-directed mutagenesis suggest that the Fe-S cluster has dual functions to stabilize elements of protein secondary structure and target the helicase to the junction of single-stranded DNA and double-stranded DNA [44].
The first structure of XPD from Thermoplasma acidophilium (ta) in complex with a short DNA fragment suggested a path of the translocated DNA strand through the taXPD protein [45]. Structural, mutational and biochemical analyses of taXPD suggest that Fe-S domain helicases achieve a polarity of translocation opposite to that of 3′ to 5′ helicases by conformational changes within the motor domain rather than binding single-stranded DNA with an opposite orientation. This was further supported by mutational analysis of taXPD and biochemical experiments to map intimate contacts between domains of the helicase protein and its DNA substrate using a partial duplex DNA molecule that has an artificial protease chemically incorporated into the DNA backbone that causes proteolytic cleavage of the protein at sites of contact [46].
The emerging wealth of information gleaned from structural and biochemical analyses of purified recombinant helicase proteins has provided strong incentive for researchers to investigate the molecular pathology of disease-causing mutations in DNA helicases. In the following section, we will provide specific examples of disease-causing missense mutations, and in some cases the genetic and biochemical analyses of the molecular and cellular defects associated with them.
Molecular Insights from Studying Missense Mutations in Human Helicase Genes
For many DNA repair disorders, studies of both unicellular and multicellular organisms with defined genetic mutations have led to new insights to the molecular roles and pathways of the proteins that are biochemically defective. Promoter or splicing mutations that disrupt the normal transcriptional or translational control of a gene, or nonsense mutations that encode truncated proteins can provide valuable information for genetic diseases. Missense mutations which result in single amino acid substitutions can also be informative and yield unique insight to the molecular functions and mechanisms of a cellular protein such as a DNA metabolic enzyme. In some cases, separation-of-function mutants that eliminate one protein function but not another have been enlightening because they help to dissect helicase enzyme mechanism or molecular requirements for pathway function in vivo.
A class of missense mutations useful for studying the role of helicases in cellular DNA metabolism are those that inactivate DNA unwinding and exert a dominant-negative phenotype in vivo [47]. A very typical type of dominant-negative helicase mutation is one that results in catalytic inactivation of ATP hydrolysis and DNA unwinding. However, care should be taken in the interpretation of experimental data from experiments that assess the effect of expressing a helicase variant with an ATPase-inactivating mutation in a cell-based system because the level of expression may not be physiological. Overexpression of a catalytically inactive helicase protein may have consequences that are different from normal expression of the mutant allele as would be found in a heterozygous individual.
Cellular expression of a mutant DNA helicase protein compromised in its ATPase and/or DNA unwinding activity but able to stably bind DNA can result in a static protein-DNA complex that perturbs processes such as replication, DNA repair, or transcription. Alternatively, or in addition, the mutant dead helicase protein may retain its ability to bind other proteins and highjack critical DNA repair factors. A third possibility arises because a number of helicases can form either homo-oligomers or hetero-oligomers. The mixed mutant/normal helicase multimer may be defective in its catalytic DNA unwinding function or fail to function properly in vivo.
A major challenge in the field is to understand how naturally occurring disease-causing mutations exert their phenotypic consequences. This requires a systematic investigation of the molecular defects caused by missense mutations and characterization of the biological pathways that fail to operate properly when a defective helicase protein is expressed. We will expound upon this topic discussing some clinically relevant missense mutations in human SF2 DNA helicases belonging to the RecQ and Fe-S families. In addition to these helicases, the Twinkle mitochondrial DNA helicase is highly relevant for consideration as several genetic disorders mentioned earlier are linked to mutations (including ones that are missense) in the human C10orf2 mitochondrial helicase gene. Some of the disease-causing mutations in the Twinkle mitochondrial DNA helicase are dominant. It was reported that overexpression of disease-causing Twinkle mutant proteins in normal cells or transgenic mice led to the accumulation of mitochondrial DNA replication intermediates and mitochondrial DNA depletion [48–50], consistent with the idea that dysfunctional Twinkle helicase can cause mitochondrial replication forks to stall. The reduction in Twinkle helicase activity for those variants that were examined correlated to the extent of mitochondrial DNA depletion and accumulation of replication intermediates [48]. A recently published research article by Longley, Copeland and colleagues describes the biochemical characterization of twenty disease variants of the human mitochondrial DNA helicase which showed that the mutations are quite heterogeneous in terms of their molecular effects on ATPase and helicase function as well as effects in some cases on protein stability [51]. Interestingly, all twenty mutant Twinkle variants retained at least partial helicase activity, leading the authors to propose that these defects are consistent with the delayed presentation of mitochondrial diseases associated with the c10orf2 mutations. The fact that only missense mutations in Twinkle have been identified suggests that it is an essential gene, consistent with its requirement for mitochondrial DNA replication [26].
Disease-Causing Mutations in Fe-S Helicase Disorders
SF2 DNA helicases including XPD, DDX11, and FANCJ that contain a conserved Fe-S motif between helicase motifs IA and II are implicated in genomic instability disorders. Human RTEL-1 also belongs to this Fe-S cluster family of DNA helicases, and RTEL-1 has been suggested to play a role in maintenance of telomere length and genome stability in mice and human [52–54]. The roles of Fe-S DNA helicases mutated in hereditary disorders in diverse nucleic acid metabolic pathways such as transcription and NER (XPD), ICL repair (FANCJ), and sister chromatid cohesion (DDX11) makes them an attractive group of proteins to study the molecular basis of diseases affected by alterations in genome homeostasis.
FANCJ Missense Mutations Associated with Breast Cancer or Genetically Linked to Fanconi Anemia
Quite recently, an excellent review from the Cantor lab was published that provides a nice perspective on the physiological consequences of FANCJ mutations [23]. Here, we will limit our discussion to some relevant points about the location of missense mutations in the FANCJ protein and their predicted or demonstrated biochemical effects on FANCJ catalytic function or protein interaction. FANCJ (BACH1) was originally discovered by its interaction with the tumor suppressor BRCA1, and the first FANCJ mutations identified were in early-onset breast cancer patients or probands from breast cancer families [55]. Subsequent studies have also identified FANCJ mutations in breast cancer patients [56, 57]. However, other studies suggest that only very rare germ-line variants of FANCJ contribute to breast cancer development, suggesting low to moderate penetrancy and requirement for additional mutated alleles in order for certain FANCJ alleles to be clinically relevant [23]. Nonetheless, the probability that FANCJ serves as a tumor suppressor was bolstered by the discovery from three groups that mutations in FANCJ are genetically linked to FA (Complementation Group J), a disease characterized by a strong predisposition to cancer [58–60].
The clinical spectrum of FANCJ missense mutations genetically linked to FA or associated with breast cancer suggests that the majority reside in the N-terminal portion of the protein where the helicase core domain is located (Figure 1). Two of these reside within (R173C) or very near (V193I) the nuclear localization sequence (NLS) of FANCJ and near the MLH1 protein interaction domain required for normal resistance to DNA cross-linking agents [61]. The FANCJ-Q944E mutation identified in a breast cancer patient is positioned near the BRCA1 binding domain located in the C-terminal region of FANCJ. To our knowledge, it remains to be shown the biochemical effects of the R173C, V193I, or Q944E mutations on the catalytic function or protein interactions of FANCJ. Given the relatively close linear proximity of residue Q944 to S990, the site for FANCJ phosphorylation required for the physical interaction between FANCJ and BRCA1 [62], it will be of interest to determine if BRCA1 binding to FANCJ is affected by the Q944E mutation. Such a FANCJ missense mutation may influence the molecular mechanism employed for DNA repair, as it was recently shown that disruption of the FANCJ-BRCA1 interaction blocks DNA repair by HR and promotes polη–dependent bypass [63].
Figure 1. Clinically relevant missense mutations in FANCJ helicase responsible for Fanconi Anemia complementation group J or associated with breast cancer.
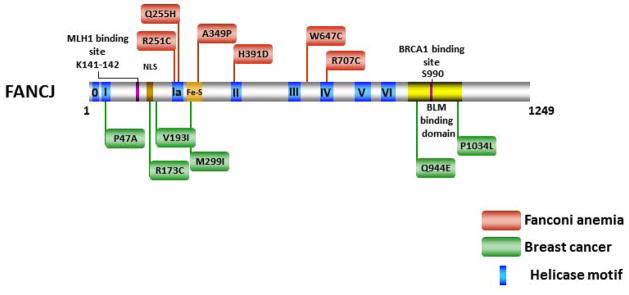
See text for details. For a comprehensive listing of FANCJ mutations, see The Rockefeller University-Fanconi anemia mutation database www.rockefeller.edu/fanconi/mutate; also see [23]. FANCJ protein interaction sites for MLH1 [61] and BRCA1 [62] are indicated as well as region of FANCJ shown to interact with BLM helicase [138]. Nuclear localization sequence (NLS) is also designated. Note in the figure the spatial relationships between FA disease-linked or breast cancer associated mutations in FANCJ and domains responsible for helicase activity, protein interactions, or the NLS.
The direct interaction of FANCJ with BRCA1 suggested the helicase was involved in fundamental processes of DNA metabolism that would be important for genomic stability and tumor suppression [55]. This notion was further bolstered by the observation of Cantor et al. that two patients with early onset breast cancer were found to harbor germline mutations P47A (motif I) and M299I (Fe-S cluster) of FANCJ [55]. Biochemical analysis of these purified recombinant proteins demonstrated an abolishment of ATPase and helicase activity for the P47A mutant, whereas the M299I mutant showed increased significantly elevated ATPase activity [64]. The elevated motor ATPase of the M299I variant enabled the helicase to unwind a damaged DNA substrate in a more proficient manner [65]. The distinct biochemical effects exerted by the two missense mutations (P47A, M299I) associated with breast cancer suggest that dysfunctionally down-regulated or up-regulated FANCJ ATP-driven protein displacement [66] or helicase activity may be deleterious to its tumor suppressor function. Two other FANCJ missense mutations found in breast cancer patients, R173C and V193I, both reside between motif I and Ia (Fig. 1), but they have not yet been biochemically characterized. The close proximity of FANCJ lysines 141 and 142 required for the FANCJ-MLH1 interaction and normal resistance to a DNA cross-linking agent [61] raises the possibility that mutations in this region may have clinical significance. Interestingly, the R173C mutation resides within the nuclear localization sequence (NLS) of FANCJ, raising the possibility that the mutant proteins fails to localize properly to the nucleus where it performs its DNA metabolic function.
Nearly all of the FANCJ missense mutations genetically linked to FA are located in the conserved helicase motifs of the protein, supporting the idea that FANCJ ATPase and/or helicase function is essential for its role in the FA pathway. This premise is consistent with cellular genetic complementation studies in human cells which show that cross-link resistance is dependent on catalytically active FANCJ protein [61]. Two FANCJ disease-causing missense mutations (R251C and Q255H) reside very close to one another in helicase motif Ia, consistent with a predicted role of the motif in DNA binding; however, the biochemical defects of the corresponding mutant proteins remain to be elucidated. Structural and biochemical analysis of an archaeal XPD protein suggests that motif Ia interacts with DNA [45]. Western blot analysis of extracts from FANCJ patient cells harboring the FANCJ-Q255H allele, as well as FANCJ-W647C between motifs III and IV and FANCJ-R707C in motif IV suggested that these FANCJ mutant proteins are unstable [58]. From the XPD crystal structure [45], motif IV is believed to have a role in DNA binding, suggesting that any residual FANCJ-R707C protein expressed would be defective in helicase function. One FANCJ disease-causing missense mutation (H391D) located in motif II (Walker A box) is likely to be defective in ATP hydrolysis, but this remains to be formally shown. Of all these FANCJ mutant alleles, none have been evaluated for their ability to complement the hypersensitivity of an FA-J cell line to DNA ICL agents such as mitomycin C (MMC) or diepoxybutane.
One of the FANCJ missense mutations responsible for FA is an alanine-to-proline mutation at residue 349 [59]. The Ala349 residue resides immediately adjacent to a highly conserved cysteine of the predicted Fe-S domain of FANCJ (Fig. 2A). Inheritance of the paternal FANCJ-A349P missense allele and a maternal truncating FANCJ-R798X allele resulted in phenotypic abnormalities, including intrauterine growth failure and death as a stillborn fetus with a gestational age of 22 weeks [59]. Biochemical analysis of purified recombinant FANCJ-A349P protein demonstrated that it was defective in coupling ATP-dependent DNA translocase activity to unwinding duplex DNA (Fig. 2B) or displacing proteins bound to DNA [67]. From a genetic standpoint, expression of a recombinant Green Fluorescent Protein (GFP)-tagged FANCJ-A349P protein in patient-derived FANCJ null cells failed to rescue sensitivity to a DNA cross-linking agent or the G4 binding drug telomestatin whereas expression of the normal GFP-tagged FANCJ recombinant protein did, indicating that the A349P allele was unable to perform its role in ICL repair or resolution of G4 DNA [67]. In addition, expression of the FANCJ-A349P allele in a normal background at a level approximately equivalent to that of endogenous FANCJ exerted a negative effect on resistance to DNA cross-linkers or telomestatin, suggesting that expression of the FANCJ-A349P protein is deleterious to normal cellular DNA metabolism. To our knowledge, there is no information pertaining to the phenotypes of the cells from the parent who transmitted the FANCJ-A349P allele. From these biochemical and cellular studies of the FANCJ-A349P protein, it was suggested that the ability of FANCJ to couple ATP hydrolysis and DNA translocase activity to higher order functions such as unwinding structured nucleic acids or stripping protein from DNA is essential for its biological roles.
Figure 2. Disease-causing FANCJ-A349P Fe-S domain mutation uncouples DNA translocation from helicase activity.

Panel A, FANCJ protein with the conserved helicase core domain and position of the Fe-S cluster. The conserved helicase motifs are indicated by yellow boxes, and the protein interaction domains for MLH1 and BRCA1 are shown by aqua green and blue boxes, respectively. The expanded Fe-S domain shows the locations for conserved cysteine residues in orange, and the A349P missense mutation of a FANCJ patient in bold. Panel B, Biochemical analysis of the purified recombinant FANCJ-A349P protein demonstrated that the amino acid substitution in the Fe-S domain uncouples ATPase and single-stranded DNA translocase activity from its duplex DNA unwinding function. See text and reference [67] for details.
DDX11 Mutations Genetically Linked to Warsaw Breakage Syndrome
Mutations in DDX11 (also named ChlR1) are genetically linked to the autosomal recessive chromosomal instability disorder WABS [24]. Until very recently, only one individual in the world has been reported as having WABS [24]. This person was described as having severe microcephaly, pre- and postnatal growth retardation, and abnormal skin pigmentation. The person with WABS had compound heterozygous mutations: a splice-site mutation in intron 22 of the maternal allele (IVS22+2T>C) and a 3 bp deletion in exon 26 of the paternal allele (c.2689_2691del). Thus, cells of the described genotype would only express the K897del mutant protein, suggesting that the protein is incompletely functional or nonfunctional. Biochemical analysis of the DDX11-K897del protein confirmed that it was devoid of catalytic activity [68].
The K897del mutation results in a single amino acid deletion of a lysine residue very near the C-terminus of the protein. The endogenous DDX11-K897del protein was hardly detected by immunoblot analysis of lysates from fibroblasts or lymphoblasts of the affected individual despite normal levels of the acetyltransferases ESCO1, ESCO2, and tubulin [24]. Cells from the patient exhibit chromosomal instability characterized by sister chromatid cohesion defects, chromosomal breakage, and hyper-sensitivity to MMC or the topoisomerase inhibitor camptothecin. Based on the cellular phenotypes, it was suggested that WABS is a unique disease with cellular features of both FA and the cohesinopathy disorder Roberts syndrome. The sister chromatid cohesion defect of the cells from the person with WABS is consistent with studies of the yeast ChlR1 homolog in which mutants also showed cohesion defects [69]. A homozygous deletion in the chl-1 gene in C. elegans resulting in the introduction of several missense codons and a premature stop codon resulted in worms that have germ-line abnormalities and are sterile [70]. The germ cells of the chl-1 mutant displayed chromosomal instability. In mice, the absence of Ddx11 resulted in embryonic lethality [71]. Cells isolated from Ddx11-/-embryos displayed chromosome missegregation, decreased chromosome cohesion, and increased aneuploidy. Depletion of human ChlR1 by RNA interference resulted in abnormal sister chromatid cohesion and a prometaphase delay leading to mitotic failure [72].
Greater insight to WABS will likely be gained from more biochemical and genetic studies, including the identification and characterization of other mutant DDX11 alleles. Quite recently, a novel homozygous mutation (c.788G>A [p.R263Q]) in DDX11 in three affected siblings with severe intellectual disability and many of the congenital abnormalities reported in the WABS original case was identified [73] (Fig. 3). This mutation affects a highly conserved arginine residue in the Fe-S domain found in a number of SF2 helicases including FANCJ, XPD, and RTEL [10]. A mutation of the homologous arginine residue in XPD (R112H) which causes TTD (Fig. 3) results in loss of helicase activity, defective nucleotide excision repair, and reduction in TFIIH, indicating the importance of the conserved Fe-S domain residue [74, 75]. Lymphocytes from the WABS individuals with the DDX11-R263Q mutant allele showed elevated MMC-induced chromosomal breakage and sister chromatid cohesion defects [73], consistent with findings from the first WABS case reported. Biochemical analysis of the purified recombinant DDX11-R263Q protein demonstrated that it was defective in DNA binding, ATP hydrolysis, and helicase activity [73]. Thus, the involvement of DDX11 in WABS was confirmed by the characterization of the mutation p.R263Q in DDX11. Moreover, the molecular significance of the conserved Fe-S domain in this conserved group of helicases for interaction with the DNA substrate and catalytic activity was confirmed by this most recent study. The discovery and characterization of disease-causing missense mutations in the Fe-S cluster of DNA helicases DDX11 [73], FANCJ [67], and XPD [74, 75] that occur in WABS, FA, and TTD, respectively, emphasize the clinical relevance of this particular domain.
Figure 3. Pathogenic mutation of homologous residue in Fe-S domain of DDX11 and XPD.

Amino acid alignment showing start of Fe-S cluster in human DDX11, XPD, FANCJ, and RTEL1 helicase proteins. Mutation of conserved arginine (R263 in DDX11, and R112 in XPD (red)), residing four residues upstream of highly conserved cysteine (green), is responsible for WABS and TTD, respectively. Distinguishing cellular phenotypes and biochemical effects of the DDX11 and XPD mutations linked to WABS and TTD, respectively, are noted. See references [73–75] and text for details.
XPD and XPB Missense Mutations Genetically Linked to Xeroderma Pigmentosum, Cockayne’s Syndrome, Trichothiodystrophy, and COFS Syndrome
Missense mutations in the XPD gene are linked to four hereditary diseases: XP, XP combined with CS, TTD, and COFS syndrome with some cases of partially overlapping clinical features [20] (Fig. 4). XP is characterized by a severe photosensitivity, leading to hyper-pigmentation, premature skin aging, and predisposition to skin cancers. A limited number of persons with XPD mutations exhibit a combination of XP and CS (XP/CS), and display neurological and developmental abnormalities in addition to UV sensitivity, but have no predisposition to skin cancer. Individuals with TTD develop symptoms such as brittle hair and nails ichthyosis, postnatal growth failure, and gonadal abnormalities. XPD mutations are also linked to COFS syndrome which is characterized by progressive neurological degeneration, calcifications, cataracts, microcornea, optic atrophy, progressive joint contractures, and growth failure [76].
Figure 4. Clinically relevant missense mutations in XPD helicase responsible for Xeroderma pigmentosum, Xeroderma pigmentosum combined with Cockayne syndrome, Trichothiodystrophy, or COFS Syndrome.
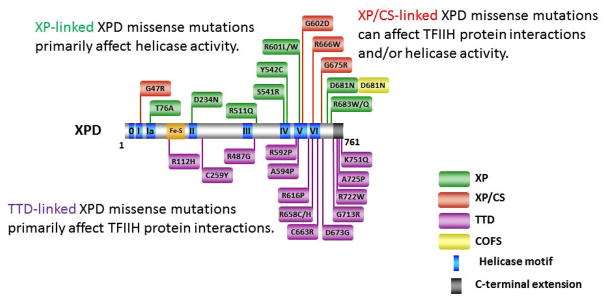
For discussion of XPD mutations and genetic heterogeneity, see [20]. Note in the figure that some of the XPD mutations linked to TTD reside outside the helicase core domain in the C-terminal extension, whereas all XP- or XP/CS-linked mutations reside within or very near the helicase core domain. XPD-D681N mutation is associated with XP and COFS syndrome.
XPD is a 5′ to 3′ SF2 family DNA helicase that is an integral subunit of the TFIIH complex required for NER and basal transcription [77]. NER (that is defective in XP) corrects UV radiation-induced photoproducts (e.g., cyclobutane pyrimidine dimers) and helix-distorting lesions induced by chemical carcinogens or certain DNA damaging agents. These bulky lesions are removed by the coordinate action of the TFIIH complex and other proteins including DNA damage recognition factors, Replication Protein A (RPA), structure-specific nucleases, DNA polymerase, proliferating cell nuclear antigen (PCNA), and ligase [78]. Within the TFIIH complex, two DNA helicases XPD and XPB with opposite translocation polarities are responsible for unwinding the duplex DNA in the vicinity of the lesion to create a bubble containing the lesion, a prerequisite for proper removal of the damaged fragment [77].
Underlying deficiencies in DNA repair and/or basal transcription are responsible for XPD-related diseases [77]. Because TFIIH is part of the basal transcription machinery as opposed to machinery in transcription activation, disease-causing mutations in XPD responsible for XP/CS or TTD are likely to reflect a general impairment of transcription. Ongoing efforts have been aimed at establishing clear genotype-phenotype relationships. Structural and mutational analyses have provided important insights to the molecular basis for disease-causing mutations in XPD leading to XP, XP/CS, or TTD [41–43]. For simplicity, we will highlight some take-home messages and discuss a few unique XPD alleles of interest. Disease-causing missense mutations in XPD responsible for XP are located mainly in the helicase core domain. Examples include T76A (motif Ia), D234N (motif II), S541R (motif IV), Y542C (motif IV), and R601L or R601W (motif V) (Fig. 4). All of these missense mutations either seriously impair ATPase/helicase activity or completely inactivate catalytic function. The XPD mutations linked to XP do not affect basal transcription [75], consistent with the idea that XPD ATPase/helicase activity is required for NER, but not for transcription. The fact that biallelic mutations of the type that generate premature protein translation in the XPD protein and are therefore overtly deleterious have not been described in humans is consistent with the belief that XPD plays a structural role in TFIIH, which plays a fundamental role in basal transcription. The essentiality of XPD is evidenced by the finding that disruption of both alleles of the XPD gene in mice (deletion of helicase motifs IV-VI) resulted in embryonic lethality at the two-cell stage [79].
Many XP individuals are compound heterozygous, and it is informative to characterize the contribution of both alleles to the disease phenotype. Ueda et al. reported that the clinical heterogeneity of compound heterozygous XP individuals with a XPD-R683W allele was attributed to the nature of the second XPD allele which differentially affected molecular steps of transcription [80]. The clinical heterogeneity was evident because two brothers had XP with cancer and neurodegeneration, another patient had numerous skin cancers and progressive neurodegeneration, and two XP siblings from a third family had neither skin cancer nor neurodegeneration. All these XP individuals had in common the XPD-R683W allele, but the second XPD allele was different. It should be noted that the XPD-R683W protein was shown to be defective in its interaction with the p44 subunit of TFIIH, attesting to the importance of this shared mutation [80]. The nature of the second XPD allele (Q452X, I455del, or 199insPP) resulted in distinguishable effects on basal transcription activity of the TFIIH complex despite the fact that immunoprecipitated TFIIIH with any of these three alleles lacked helicase activity. XPD/Q452X interfered with transactivation, XPD/I455del impaired RNA polymerase II phosphorylation, and XPD/199insPP inhibited kinase activity of the cdk7 subunit of TFIIH. The clinical heterogeneity in the XP individuals in terms of neurodegeneration can likely be explained on a molecular basis by the differential effects on transcription resulting from the XPD allele combinations. However, genetic background in some cases is likely to play a role. In this scenario, it could be that variants of the other components of TFIIH and/or environmental factors contribute to the clinical heterogeneity. Nonetheless, the suggestion that both XPD alleles contributed to the phenotype of these compound heterozygous XP individuals is an important lesson that may apply to other helicase disorders characterized by clinical heterogeneity. For example, the inheritance of the previously mentioned FANCJ-A349P missense allele (Fig. 2) and a truncating R798X allele resulted in severe phenotypic abnormalities including intrauterine growth failure and gestational death [59]. Although not certain, the dominant-negative nature of the FANCJ-A349P allele [67] may play a role in the severity or phenotypic presentation of disease.
XPD mutations responsible for XP combined with CS also reside within or near the conserved helicase motifs as well (Fig. 4). It is generally believed that transcription defects associated with certain missense mutations in XPD that lead to XP/CS are due to defective protein interactions with other factors of the TFIIH complex. The partial transcription defect exerted by the motif I XPD-G47R mutation [75] is likely to be responsible for the CS phenotype associated with this particular mutation, and may be relevant for other XPD mutations responsible for XP/CS.
Unlike the XPD mutations implicated in XP or XP/CS which are only found in or very near the helicase core domain, XPD mutations responsible for TTD can also be found in the C-terminal region of the protein (Fig. 4). The C-terminus of XPD is important for interaction with other proteins (e.g., p44 subunit of TFIIH), which can be critical for optimal XPD helicase activity and/or stability of the TFIIH complex (for review, see [81]). Such mutations in XPD reduce DNA repair activity and basal transcription [82]. Several XPD missense mutations linked to TTD that have been examined inhibit basal transcription, suggesting a molecular defect distinct from that of XP. Interestingly, the XPD-R616P mutation very near helicase motif V abolished transcription in a reconstituted in vitro system, and impaired p44 binding, but did not affect helicase activity [75]. This result is consistent with the idea that defective XPD protein interaction contributes to TTD. Another mutation found in TTD is XPD-R112H, which replaces a highly conserved arginine in the Fe-S cluster of the helicase core domain. In addition to TTD, this patient also displays a mild phenotype characteristic of XP [83]. It has been proposed that the combined helicase inactivation and interference with TFIIH protein interactions that leads to partial inhibition of transcription is responsible for the complex phenotype associated with the R112H allele.
Compound heterozygous XPD mutations identified in an individual with COFS syndrome were a R616W mutation, found in an XP patient, and a unique D681N mutation residing on the C-terminal side of helicase motif VI [76]. The aspartic acid (D681) is highly conserved in all known XPD genes. UV survival assays performed with diploid fibroblast cultures derived from skin biopsy of the individual with COFS syndrome demonstrated UV sensitivity comparable to that of cells from a XP-A patient with severe XP, leading the authors to conclude that UV-sensitive COFS syndrome is an additional disease state of NER with mutations in the DNA repair genes CSB and XPG already linked to COFS syndrome. It remains puzzling how the missense mutation XPD-D681N can result in distinct clinical phenotypes of COFS syndrome versus XP. To our knowledge, the molecular defect(s) of the XPD-D681N protein has not been determined. The range of XPD missense mutations resulting in heterogeneity in clinical phenotype prompt continued interest to characterize the molecular and cellular defects of XPD variants, such as the D681N allele linked to COFS syndrome, in order to gain a better understanding of disease relevant genotype-phenotype relationships. The clinical spectrum of XPD missense mutations suggests that the site of mutation may determine the disease phenotype. Thus, XPD-associated phenotypes and diseases arise from allelic series in which different alleles can affect the function of the gene in different ways. However, genetic background and environmental effects may also contribute to phenotype. This subject requires further study.
The XPB gene encodes a DNA helicase with opposite polarity to that of XPD that is also found in the TFIIH complex, and XPB mutations can lead to clinical disorders with overlapping phenotypes including XP/CS, XP with neurological abnormalities, and TTD [20]. In contrast to XPD where helicase activity is indispensable for NER, only XPB ATPase is necessary for the pathway to remove bulky lesions and UV photoproducts. XPB ATPase activity is also necessary for DNA opening for transcription to occur, but its helicase activity is important for promoter escape during transcription (for review, see [84]). Only limited information has been acquired from the clinical spectrum of XPB mutations due to the very small number of patients mutated in the XPB gene. This may reflect the essential nature of XPB for survival, consistent with its paramount importance in basal transcription. The phenotypic heterogeneity of XPB may be attributed to the nature of the mutations. Missense mutations which only partially affect XPB biochemical and cellular function could result in mild XP/CS, whereas severe XP/CS may be a consequence of a hypo-morphic allele resulting in a XPB protein-truncating mutation combined with a XPB missense mutation [85]. A disease-causing mutation in XPB resulting in a single amino acid substitution (T119P) in the N-terminus of the protein prior to the conserved helicase motifs was identified in a patient with mild versions of both TTD and photosensitivity [86]. Cellular studies from the Sarasin lab demonstrated that the XPB-T119P allele is associated with moderately defective DNA repair [87]. This is in contrast to XPB-F99S allele (also located in the N-terminus) of an XP/CS patient which is profoundly reduced in its DNA repair function [87]. Interestingly, both the T119P and F99S mutations in XPB do not impair TFIIH helicase activity [84]; however, the F99S substitution was shown to impair the interaction of XPB with p52, one of the subunits of the TFIIH complex [82]. Although less is known about XPB compared to XPD, the studies of TFIIH have helped to dissect the complex heterogeneity of XPB and XPD DNA repair-transcription syndromes.
Disease-Causing Mutations in RecQ Helicase Disorders
Double strand breaks (DSB) that arise during the processing of ICLs, at broken replication forks, or that are directly induced by ionizing radiation (IR) or certain chemical agents represent a highly toxic form of DNA damage that leads to genomic instability; consequently, several prominent pathways of DSB repair exist to rejoin broken chromosome ends [88, 89]. Correction of DSBs during S or G2 phases can occur by HR, which is a high fidelity repair mechanism to rejoin broken DNA ends. Another major pathway of DSB repair is nonhomologous end-joining (NHEJ), which can take place anytime during the cell cycle, but is a highly error-prone process. RecQ helicases are generally believed to have important roles in HR repair of DSBs and telomere maintenance as well as during occasions of replication stress to ensure normal fork progression or restart broken replication forks [9, 90]. In the absence of normal RecQ function, cells are prone to accumulate chromosomal instability. Therefore, the function of RecQ helicases is of broad interest in the field, and even more so because of the genetic linkage of human diseases to autosomal recessive mutations in RecQ helicase genes.
WRN Missense Mutations Genetically Linked to Werner Syndrome
WS is a genetic disorder characterized by premature aging, genomic instability, sensitivity to DNA damaging agents, aberrant recombination, and replication defects. The WRN gene encodes a RecQ 3′ to 5′ DNA helicase and 3′ to 5′ exonuclease that is proposed to play a role in regulation of recombination events, primarily HR [3, 90]. The mutational spectrum in WS suggests that inactivation of both the helicase and exonuclease functions of WRN is the predominant genetic mechanism underlying WS [91]. It seems probable that WRN has a specialized function when the replication fork encounters a blocking lesion or alternate DNA structure. Stalled replication forks or DSBs are dealt with by a myriad of proteins and is dependent upon the lesion’s chemical nature, cell cycle phase, cell type, etc. A key early step of HR repair is DSB end resection which enables a single-stranded DNA overhang to be generated that can invade and base pair with a complementary strand of homologous duplex and begin the HR process. Since DSB resection occurs at a critically early stage, it is of great interest to understand the proteins (e.g., nucleases, DNA binding and signaling proteins, recombinases, helicases) and steps involved.
How WRN and likely other RecQ helicases facilitate accurate processing of DSB is still not well understood. Results from a plasmid end-joining assay using WS mutant cells transfected with WRN site-directed mutants suggested that WRN exonuclease and helicase activities are both required for optimal recombinational repair [92]. The dual importance of balanced and concerted action of WRN helicase and exonuclease activities are further supported by other cellular and biochemical studies [93, 94]. However, relating the findings from experimental studies to the molecular pathology and clinical manifestation of WS is still a daunting challenge. Despite the progress that has been made, the relative importance of WRN helicase or exonuclease activities to suppress the disease phenotypes of WS remains poorly understood. Unfortunately, the clinical spectrum of disease mutations has not provided much insight; however, a few recently identified WRN patient mutations (discussed below) are provocative and should be studied in experimental systems.
Since the original discovery of the WRN gene and its linkage to the premature aging disorder WS in 1996 [95], the majority of disease-causing WRN mutations identified result in truncated proteins. As the NLS in WRN protein was mapped to a region near the extreme C-terminus [96], the spectrum of WS clinical mutations were rather uninformative because the truncated mutant WRN proteins would fail to localize to the nucleus, where WRN presumably performs its essential functions in DNA replication, repair, and the maintenance of genomic stability. However, more recently, five disease-causing WRN missense mutations have been identified (Fig. 5). Two missense mutations reside in the N-terminal exonuclease domain of WRN and have been shown to result in WRN protein instability [97]. Two were found in the WRN helicase domain, one in the Walker A box (motif I) (G574R)[91] just three amino acids away from the invariant lysine residue implicated in nucleotide binding and the other (R637W) [98] very near helicase motif V. Although not yet biochemically characterized, these two helicase core domain mutants would be predicted to interfere with normal ATPase and helicase function.
Figure 5. Clinically relevant missense mutations in WRN helicase-nuclease responsible for Werner syndrome.
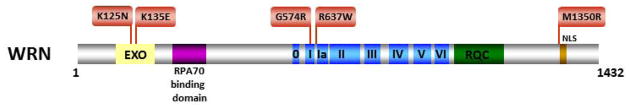
See text for details. For a comprehensive listing of WRN mutations, see The International Registry of Werner Syndrome www.wernersyndrome.org; also see [91]. The high affinity RPA70 interaction domain of WRN [139] is indicated. WRN exonuclease domain (EXO) and NLS are also shown. See text for details.
The R637W mutation was the first example of a missense allele in a WS patient [98]. The second allele in this WS patient resulted in a Val 1082 frameshift mutation. He was diagnosed definitively as having WS at the age of 48 according to the International Registry scoring system with typical skin and pigmentary alterations, cataracts, aged hair, osteoporosis, and cancer. It will be interesting to ascertain the effect of the WRN-R637W mutation on biochemical functions. The R637W mutation may affect protein stability or abrogate both helicase and exonuclease functions. In addition, the study of disease-causing separation-of-function mutations would likely provide insight to the catalytic requirements to suppress cellular phenotypes associated with WS.
The fifth missense mutation, M1350R, resides immediately N-terminal to the NLS of WRN [91] (Fig. 5). Given the fact that of the approximately sixty WRN disease mutations reported, the majority result in truncated WRN protein fragments that lack the C-terminal NLS, it will be of interest to determine if the WRN-M1350R mutation affects the ability of the WRN protein to effectively localize to nuclei where it is believed to have its function. For the WRN alleles G574R, R637W, and M1350R, it is possible that promoter, enhancer, or intron mutations linked to these changes contribute to disease phenotype. Alternatively, a mutation outside the WRN gene that segregates with the WRN allele may be responsible for disease phenotype. To test this possibility, investigators will have to transfect the mutant alleles into WRN-negative cells and demonstrate that the allele is incapable of complementing the WS phenotype. Quite recently, centenarian genome wide association studies suggested that specific variants of the WRN gene determine progeria and accelerated aging [99]. Thus genetic signatures may have a useful predictive value for exceptional longevity.
BLM Helicase Missense Mutations Genetically Linked to Bloom’s Syndrome
The hallmark of BS cells is an elevated rate of sister chromatid exchange (SCE) [100]. BS is inherited by homozygous recessive mutations in the BLM gene, which encodes a protein that belongs to the RecQ family of DNA helicases [101]. BLM is believed to function in HR repair to maintain genomic stability. There are two explanations for the role of BLM in SCE suppression: 1) a BLM protein complex containing topoisomerase IIIα, RPA, RMI, and RMI2 dissolves double HJ structures which may form during recombination by a mechanism in which BLM helicase activity working in concert with topoisomerase cleavage/ligation is required for the double HJ dissolution reaction [102, 103]; 2) BLM dismantles D-loops [104] and channels the repair pathway towards synthesis-dependent strand annealing by promoting DNA synthesis [105]. Recent results suggest that BLM is believed to have additional roles (early and late) in HR repair of DSBs as well [106, 107]. BLM may also play a role in the resolution of DNA structure(s) that arise at converging replication forks, which present an attractive native substrate for the BLM complex. BLM is required for the resolution of DNA linkages that arise from common fragile sites [108]. The mapping of BLM-associated ultra-fine DNA bridges to fragile-site DNA suggests that the inefficient resolution of DNA linkages at fragile sites in BS cells is responsible for the chromosomal instability at these loci.
Some studies have addressed the biological importance of BLM catalytic activity in cell-based systems. An SV40-transformed BS fibroblast cell line transfected with a construct encoding a catalytically inactive BLM protein with a Walker A box mutation (K695T) expressed the mutant BLM protein poorly, and the transfected cell line showed genomic instability characteristic of BS cells [109]. BLM-K695T could be expressed in hTERT-transformed normal human fibroblasts, resulting in sensitization to DNA damaging agents; however, expression of the helicase-dead BLM protein did not exert a dominant-negative effect on the formation of foci that contained the DNA damage response proteins BRCA1 and NBS1, suggesting that BLM may play a structural role independent of its helicase activity in assembling BRCA1 and/or NBS1 protein complexes [110].
BLM helicase activity is likely to be required for at least some of its important biological functions because inspection of the disease-causing BLM missense mutations reveals that all thirteen identified to date reside in conserved motifs of the helicase core or in the highly conserved RQC domain that resides immediately after the helicase core domain [111] (Fig. 6). Furthermore, all of the disease-causing helicase domain BLM mutants that have been biochemically characterized show defects in ATP or DNA binding, which resulted in low or no detectable helicase activity. This is logical because the conserved helicase motifs, as discussed above, have been implicated in essential functions necessary for coupling the motor ATPase to strand separation. For example, three disease-causing BLM missense mutations residing within or very near motif IV (BLM-C878R, BLM-G891E, BLM-C901Y) all affect DNA binding [112], a finding that is consistent with structural and mutational analysis data of other DNA helicases.
Figure 6. Clinically relevant missense mutations in BLM helicase responsible for Bloom’s syndrome.
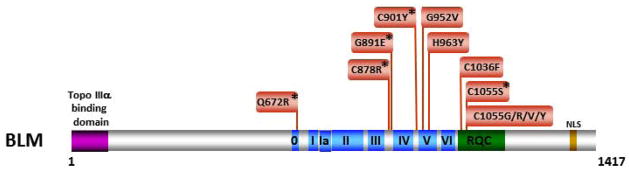
See text for details. For a comprehensive listing of BLM mutations, see The Bloom’s Syndrome Registry www.med.cornell.edu/bsr/; also see [111]. Region of BLM shown to interact with Topoisomerase 3α [140] is shown. Asterisk indicates disease-causing BLM mutations which have been shown to significantly impair or abolish BLM helicase activity.
Of particular interest to us is a disease-causing BLM helicase core domain missense mutation in the conserved Q motif, designated BLM-Q672R [101, 113]. The Q motif in RNA helicases is suggested to have a role in either nucleotide binding [114, 115] and/or the interaction with single-stranded RNA [116]. Structural data suggests a role of the Q motif in nucleotide binding for certain DNA helicases [31, 41, 42, 45, 117]; however, it is not entirely clear if the Q motif may facilitate or orchestrate other essential functions of the helicase, such as coupling ATP hydrolysis to DNA unwinding, protein interactions, or other unpredicted functions. The BLM-Q672R mutation was shown to abolish helicase activity and severely diminish ATPase activity of the purified recombinant protein; however, the BLM-Q672R protein retained its normal DNA binding but was defective in ATP binding, leading the authors to propose that residue Q672 is involved in ATP binding [112]. It may be of interest to determine the effect of the Q672R mutation on BLM oligomerization since replacement of this highly conserved residue in the FANCJ helicase perturbed its ability to dimerize and abolished its helicase function [118]. Although purified recombinant BLM protein was shown to form oligomeric ring-like structures by electron microscopy [119], the functional importance of BLM assembly state is debated because several studies have shown that a BLM protein fragment consisting of the helicase domain and C-terminal region is active as a helicase in its monomeric form [120–122]. It is unknown if a higher-order multimer of BLM is important for its physiological function. Nonetheless, expression of BLM-Q672R in BS cells failed to correct the high rate of SCE in BS cells [109], demonstrating that the molecular defect of the mutant protein displayed phenotypic consequences.
In addition to the helicase core domain, BLM shares with many RecQ helicases a conserved RQC region consisting of the Zn2+ binding and Winged Helix (WH) domains. Amino acid substitutions of two cysteine residues that are sites of disease-causing BS missense mutations (C1036 and C1055) [101, 123] (Fig. 6) were found to interfere with Zn2+ binding [124]. Interestingly, five different amino acids substitutions (S, G, R, V, and Y) resulting from disease-causing mutations in BLM occur at C1055, suggesting that this particular locus in the BLM Zn2+ domain may be a hotspot for mutation and that the cysteine residue is highly important for BLM structure and function. Of all the BLM RQC domain mutants, the BLM-C1055S mutant protein is the only one that has been biochemically characterized, and it was shown to lack ATPase and helicase activity [109, 113]. Microinjection of the BLM-C1055S mutant protein into BS fibroblasts failed to rescue the p53-mediated apoptosis defect [125]. When purified recombinant BLM-C1055S protein was microinjected into normal cells, there was a defect in its localization to promyelocytic leukemia (PML) bodies and decreased p53-mediated apoptosis compared to cells microinjected with normal recombinant BLM protein. The localization of BLM to DNA repair foci outside of PML bodies upon replication stress suggests that BLM has roles outside PML bodies [126], which may contribute to the observed differences in the effects of microinjected normal versus BLM-C1055S proteins on p53-mediated apoptosis.
RECQL4 Missense Mutations Genetically Linked to Rothmund-Thomson Syndrome, Baller-Gerold Syndrome, or RAPADILINO
The RECQL4 helicase presents a rather unusual case for the RecQ helicase family because three genetic disorders with overlapping features and a range of severity can be attributed to mutations in the RECQL4 gene: RTS, RAPADILINO, and BGS [15, 127]. Unlike the BLM helicase, disease-causing missense mutations for all three RECQL4 diseases have been identified in regions outside the conserved helicase domain shared by the other RecQ helicases (Fig. 7). Of the seven known RTS missense mutations, four reside in the C-terminal region, one in the N-terminal region, and two in the helicase core domain. Notably, the disease-causing RTS mutation Q253H resides in the N-terminus where the conserved SLD2 protein interaction domain implicated in chromosomal DNA replication can also be found [128]. Only two RAPADILINO missense mutations have been reported, one in the N-terminal region and one in the helicase core domain. For BGS, a single patient missense mutation is known that resides in the C-terminal region. Interestingly, the C-terminal R1021W mutation was genetically linked to both BGS and RTS. The effects of the R1021W mutation, as well as all of the other missense mutations in the RECQL4 gene, on the biochemical functions of the RECQL4 helicase protein have not yet been determined. However, a commonly found RECQL4 mutation linked to RAPADILINO leading to a 44 amino acid deletion was recently characterized for its effects on biochemical functions of the protein and found to severely reduce its ATPase activity and abolish its helicase activity [129]. Further studies to establish genotype-phenotype relationships for RECQL4 are warranted.
Figure 7. Clinically relevant missense mutations in RECQL4 helicase responsible for Rothmund-Thomson syndrome, Baller-Gerold syndrome, or RAPADILINO.
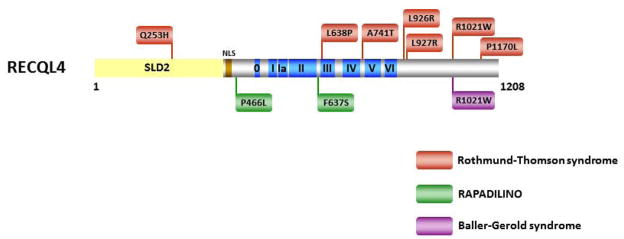
See text for details. For comprehensive listing of RECQL4 mutations, see [15]. RECQL4-R1021W is associated with both RTS and BGS. Region of RECQL4 shown to interact with SLD2 [128] is shown.
Summary and Perspective
In this review, we have largely focused our discussion on helicase disease variants characterized by single amino acid substitutions. We believe that such mutants can be highly valuable for combined biochemical, molecular and genetic studies, particularly in those cases where the intact mutant protein is expressed in vivo and remains stable. In this scenario, unique molecular aspects of helicase mechanism can be revealed as well as new insights to pathway function and relevance. From a clinical view, helicase missense mutations can provide a window to human hereditary diseases and illuminate new insights to pathways important for maintenance of genomic stability.
The analysis of helicase mutations should also include the study of polymorphic variants, even if they are not yet associated with cancer or a genetic disorder. For example, the Rockefeller University – Fanconi anemia mutation database lists an extensive collection of variants of the human FA genes, including FANCJ. It will be of great interest to ascertain the potential genetic deficiencies and molecular defects associated with these variants. The emergence of FANCJ as a breast cancer tumor suppressor places a new emphasis on the molecular characterization of FANCJ variants [23]. Numerous studies have examined the association of single nucleotide polymorphisms in XPD with various types of cancers; however, a causal relationship between XPD polymorphisms, DNA repair capacity and cancer risk has been brought into question [130]. Clearly more work is necessary to clarify if clear associations between polymorphisms in DNA repair helicases (such as XPD or FANCJ) and clinical phenotypes exist. This has become a growing field of interest in clinical practice and potential application in personalized medicine. Studies of DNA repair gene polymorphisms have also led to new insights in basic research. For example, studies of WRN polymorphic variants have helped to provide a better understanding of WRN helicase function and potential implications for human health [131–133].
While this review largely focused its discussion on the probability that disease-based helicase domain missense mutations are likely to exert a negative effect on catalytic function which would impair its cellular role in a homozygous recessive inherited disease, it is also possible that certain catalytic inactivating mutations may exert a dominant-negative effect, such as the FANCJ-A349P mutant allele residing in the conserved Fe-S domain (Fig. 2) [67]. The potential clinical significance of the FANCJ helicase-inactivating mutation in heterozygote carriers remains to be determined. In addition, a helicase missense mutation that affects a critical protein interaction (such as an XPD mutation leading to TTD) could also result in aberrant function. Thus, as mentioned previously, the effect of the FANCJ-Q944E mutation identified in a breast cancer patient on the protein’s ability to interact with the tumor suppressor BRCA1 and/or catalytically unwind structured DNA molecules will be of interest.
An emerging field of chemical biology is the prospect of pharmacologically rescuing misfolded mutant proteins using small molecules [134]. For example, screening small molecule libraries for compounds that rescue mutant p53 function has attracted interest [135]. As a field, chemical rescue is in its infancy, but it is provocative to consider this strategy for the helicase disease missense variants. Missense mutations in DNA helicases such as FANCJ, XPD, DDX11, BLM, RECQL4, and WRN are genetically linked to chromosomal instability disorders characterized by an early incidence of aging phenotypes and/or cancer. It is hypothesized that cellular phenotype(s) associated with a helicase missense mutant can be reversed by a compound that restores helicase catalytic function in a DNA repair pathway. An a priori argument can be made that it is more likely that the small molecule approach to restoring helicase function would have greater probability for success in the case of a missense mutation as opposed to a truncating or deletion mutation that is likely to have a greater distorting effect on helicase structure.
How might chemical rescue restore helicase function? A small molecule that allosterically binds to a helicase protein (e.g., FANCJ-A349P (67)) and induces a subtle conformational change in the ATPase/helicase core domain that enables efficient coupling of ATP hydrolysis to DNA helicase activity may restore its function in the cellular response to ICL-inducing agents. A small molecule that binds to a helicase mutant protein (e.g., FANCJ-Q25A (118)) in such a manner to restore its oligomerization by remolding its dimerization interface may increase its DNA unwinding efficiency. Chemical rescue of a defective helicase protein characterized by a disease-causing missense mutation that only partially reduces DNA binding affinity, nucleotide hydrolysis, or thermal stability but leaves some intact helicase activity (e.g., C10orf2 encoding the mitochondrial DNA helicase Twinkle [51]) may be achievable in the future. Virtual screening of chemical libraries for small molecules that bind to mutant helicase proteins and induce conformational changes to restore function may move to the forefront as we gain greater insight to structure-activity relationships from high resolution structures of DNA helicase proteins. This approach may provide a paradigm for therapeutic intervention by modulation of other key DNA repair proteins required for a robust DNA damage response.
Although to our knowledge there has been no published data describing chemical rescue of a misfolded DNA repair protein, we reported a small molecule inhibitor of the WRN helicase that effectively impaired WRN function in the cellular response to DNA damage or replication stress [136]. Future studies that will address the use of helicase-specific inhibitors in cancer therapy [137] may also include chemical rescue as an approach to restore helicase activity in loss-of-function helicase disorders.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute on Aging and the Fanconi Anemia Research Fund (RMB). We wish to thank Dr. Junko Oshima (University of Washington) for helpful discussion.
Footnotes
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lohman TM, Tomko EJ, Wu CG. Non-hexameric DNA helicases and translocases: mechanisms and regulation. Nat Rev Mol Cell Biol. 2008;9:391–401. doi: 10.1038/nrm2394. [DOI] [PubMed] [Google Scholar]
- 2.Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 3.Brosh RM, Jr, Bohr VA. Human premature aging, DNA repair and RecQ helicases. Nucleic Acids Res. 2007;35:7527–7544. doi: 10.1093/nar/gkm1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lohman TM, Bjornson KP. Mechanisms of helicase-catalyzed DNA unwinding. Annu Rev Biochem. 1996;65:169–214. doi: 10.1146/annurev.bi.65.070196.001125. [DOI] [PubMed] [Google Scholar]
- 5.Patel SS, Donmez I. Mechanisms of helicases. J Biol Chem. 2006;281:18625–18628. doi: 10.1074/jbc.R600008200. [DOI] [PubMed] [Google Scholar]
- 6.Pyle AM. Translocation and unwinding mechanisms of RNA and DNA helicases. Annu Rev Biophys. 2008;37:317–336. doi: 10.1146/annurev.biophys.37.032807.125908. [DOI] [PubMed] [Google Scholar]
- 7.Bernstein KA, Gangloff S, Rothstein R. The RecQ DNA helicases in DNA repair. Annu Rev Genet. 2010;44:393–417. doi: 10.1146/annurev-genet-102209-163602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dillingham MS. Superfamily I helicases as modular components of DNA-processing machines. Biochem Soc Trans. 2011;39:413–423. doi: 10.1042/BST0390413. [DOI] [PubMed] [Google Scholar]
- 9.Singh DK, Ghosh AK, Croteau DL, Bohr VA. RecQ helicases in DNA double strand break repair and telomere maintenance. Mutat Res. 2011 doi: 10.1016/j.mrfmmm.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu Y, Suhasini AN, Brosh RM., Jr Welcome the Family of FANCJ-like helicases to the block of genome stability maintenance proteins. Cell Mol Life Sci. 2008;66:1209–1222. doi: 10.1007/s00018-008-8580-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu WK, Hickson ID. RecQ helicases: multifunctional genome caretakers. Nat Rev Cancer. 2009;9:644–654. doi: 10.1038/nrc2682. [DOI] [PubMed] [Google Scholar]
- 12.Harrigan JA, Bohr VA. Human diseases deficient in RecQ helicases. Biochimie. 2003;85:1185–1193. doi: 10.1016/j.biochi.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 13.German J. Bloom’s syndrome. XX. The first 100 cancers. Cancer Genet Cytogenet. 1997;93:100–106. doi: 10.1016/s0165-4608(96)00336-6. [DOI] [PubMed] [Google Scholar]
- 14.Martin GM. Genetics and aging; the Werner syndrome as a segmental progeroid syndrome. Adv Exp Med Biol. 1985;190:161–170. doi: 10.1007/978-1-4684-7853-2_5. [DOI] [PubMed] [Google Scholar]
- 15.Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet J Rare Dis. 2010;5:2. doi: 10.1186/1750-1172-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Maldergem L, Siitonen HA, Jalkh N, Chouery E, De Roy M, Delague V, Muenke M, Jabs EW, Cai J, Wang LL, Plon SE, Fourneau C, Kestila M, Gillerot Y, Megarbane A, Verloes A. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. J Med Genet. 2006;43:148–152. doi: 10.1136/jmg.2005.031781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma S, Doherty KM, Brosh RM., Jr Mechanisms of RecQ helicases in pathways of DNA metabolism and maintenance of genomic stability. Biochem J. 2006;398:319–337. doi: 10.1042/BJ20060450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.White MF. Structure, function and evolution of the XPD family of iron-sulfur-containing 5′-->3′ DNA helicases. Biochem Soc Trans. 2009;37:547–551. doi: 10.1042/BST0370547. [DOI] [PubMed] [Google Scholar]
- 19.White MF, Dillingham MS. Iron-sulphur clusters in nucleic acid processing enzymes. Curr Opin Struct Biol. 2011;22:94–100. doi: 10.1016/j.sbi.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 20.Digiovanna JJ, Kraemer KH. Shining a light on Xeroderma Pigmentosum. J Invest Dermatol. 2012;132:785–796. doi: 10.1038/jid.2011.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crossan GP, Patel KJ. The Fanconi anaemia pathway orchestrates incisions at sites of crosslinked DNA. J Pathol. 2012;226:326–337. doi: 10.1002/path.3002. [DOI] [PubMed] [Google Scholar]
- 22.Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer. 2011;11:467–480. doi: 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cantor SB, Guillemette S. Hereditary breast cancer and the BRCA1-associated FANCJ/BACH1/BRIP1. Future Oncol. 2011;7:253–261. doi: 10.2217/fon.10.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van der LP, Chrzanowska KH, Godthelp BC, Rooimans MA, Oostra AB, Stumm M, Zdzienicka MZ, Joenje H, De Winter JP. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am J Hum Genet. 2010;86:262–266. doi: 10.1016/j.ajhg.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kee Y, D’Andrea AD. Molecular pathogenesis and clinical management of Fanconi anemia. J Clin Invest. 2012;122:3799–3806. doi: 10.1172/JCI58321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Copeland WC. Defects in mitochondrial DNA replication and human disease. Crit Rev Biochem Mol Biol. 2012;47:64–74. doi: 10.3109/10409238.2011.632763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JY, Yang W. UvrD helicase unwinds DNA one base pair at a time by a two-part power stroke. Cell. 2006;127:1349–1360. doi: 10.1016/j.cell.2006.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Velankar SS, Soultanas P, Dillingham MS, Subramanya HS, Wigley DB. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell. 1999;97:75–84. doi: 10.1016/s0092-8674(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 29.Singleton MR, Scaife S, Wigley DB. Structural analysis of DNA replication fork reversal by RecG. Cell. 2001;107:79–89. doi: 10.1016/s0092-8674(01)00501-3. [DOI] [PubMed] [Google Scholar]
- 30.Yao N, Hesson T, Cable M, Hong Z, Kwong AD, Le HV, Weber PC. Structure of the hepatitis C virus RNA helicase domain. Nat Struct Biol. 1997;4:463–467. doi: 10.1038/nsb0697-463. [DOI] [PubMed] [Google Scholar]
- 31.Bernstein DA, Zittel MC, Keck JL. High-resolution structure of the E.coli RecQ helicase catalytic core. EMBO J. 2003;22:4910–4921. doi: 10.1093/emboj/cdg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pike AC, Shrestha B, Popuri V, Burgess-Brown N, Muzzolini L, Costantini S, Vindigni A, Gileadi O. Structure of the human RECQ1 helicase reveals a putative strand-separation pin. Proc Natl Acad Sci U S A. 2009;106:1039–1044. doi: 10.1073/pnas.0806908106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sengoku T, Nureki O, Nakamura A, Kobayashi S, Yokoyama S. Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell. 2006;125:287–300. doi: 10.1016/j.cell.2006.01.054. [DOI] [PubMed] [Google Scholar]
- 34.Buttner K, Nehring S, Hopfner KP. Structural basis for DNA duplex separation by a superfamily-2 helicase. Nat Struct Mol Biol. 2007;14:647–652. doi: 10.1038/nsmb1246. [DOI] [PubMed] [Google Scholar]
- 35.Richards JD, Johnson KA, Liu H, McRobbie AM, McMahon S, Oke M, Carter L, Naismith JH, White MF. Structure of the DNA repair helicase hel308 reveals DNA binding and autoinhibitory domains. J Biol Chem. 2008;283:5118–5126. doi: 10.1074/jbc.M707548200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fairman-Williams ME, Guenther UP, Jankowsky E. SF1 and SF2 helicases: family matters. Curr Opin Struct Biol. 2010;20:313–324. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kitano K, Kim SY, Hakoshima T. Structural basis for DNA strand separation by the unconventional winged-helix domain of RecQ helicase WRN. Structure. 2010;18:177–187. doi: 10.1016/j.str.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 38.Woodman IL, Bolt EL. Winged helix domains with unknown function in Hel308 and related helicases. Biochem Soc Trans. 2011;39:140–144. doi: 10.1042/BST0390140. [DOI] [PubMed] [Google Scholar]
- 39.Vindigni A, Marino F, Gileadi O. Probing the structural basis of RecQ helicase function. Biophys Chem. 2010;149:67–77. doi: 10.1016/j.bpc.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 40.Rudolf J, Makrantoni V, Ingledew WJ, Stark MJ, White MF. The DNA repair helicases XPD and FancJ have essential Iron-Sulfur domains. Mol Cell. 2006;23:801–808. doi: 10.1016/j.molcel.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 41.Fan L, Fuss JO, Cheng QJ, Arvai AS, Hammel M, Roberts VA, Cooper PK, Tainer JA. XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell. 2008;133:789–800. doi: 10.1016/j.cell.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu H, Rudolf J, Johnson KA, McMahon SA, Oke M, Carter L, McRobbie AM, Brown SE, Naismith JH, White MF. Structure of the DNA repair helicase XPD. Cell. 2008;133:801–812. doi: 10.1016/j.cell.2008.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wolski SC, Kuper J, Hanzelmann P, Truglio JJ, Croteau DL, Van Houten B, Kisker C. Crystal structure of the FeS cluster-containing nucleotide excision repair helicase XPD. PLoS Biol. 2008;6:e149. doi: 10.1371/journal.pbio.0060149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pugh RA, Honda M, Leesley H, Thomas A, Lin Y, Nilges MJ, Cann IK, Spies M. The iron-containing domain is essential in Rad3 helicases for coupling of ATP hydrolysis to DNA translocation and for targeting the helicase to the single-stranded DNA-double-stranded DNA junction. J Biol Chem. 2008;283:1732–1743. doi: 10.1074/jbc.M707064200. [DOI] [PubMed] [Google Scholar]
- 45.Kuper J, Wolski SC, Michels G, Kisker C. Functional and structural studies of the nucleotide excision repair helicase XPD suggest a polarity for DNA translocation. EMBO J. 2011;31:494–502. doi: 10.1038/emboj.2011.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pugh RA, Wu CG, Spies M. Regulation of translocation polarity by helicase domain 1 in SF2B helicases. EMBO J. 2011;31:503–514. doi: 10.1038/emboj.2011.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu Y, Brosh RM., Jr Helicase-inactivating mutations as a basis for dominant negative phenotypes. Cell Cycle. 2010;9:4080–4090. doi: 10.4161/cc.9.20.13667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goffart S, Cooper HM, Tyynismaa H, Wanrooij S, Suomalainen A, Spelbrink JN. Twinkle mutations associated with autosomal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling. Hum Mol Genet. 2009;18:328–340. doi: 10.1093/hmg/ddn359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tyynismaa H, Mjosund KP, Wanrooij S, Lappalainen I, Ylikallio E, Jalanko A, Spelbrink JN, Paetau A, Suomalainen A. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci U S A. 2005;102:17687–17692. doi: 10.1073/pnas.0505551102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wanrooij S, Goffart S, Pohjoismaki JL, Yasukawa T, Spelbrink JN. Expression of catalytic mutants of the mtDNA helicase Twinkle and polymerase POLG causes distinct replication stalling phenotypes. Nucleic Acids Res. 2007;35:3238–3251. doi: 10.1093/nar/gkm215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Longley MJ, Humble MM, Sharief FS, Copeland WC. Disease variants of the human mitochondrial DNA helicase encoded by C10orf2 differentially alter protein stability, nucleotide hydrolysis, and helicase activity. J Biol Chem. 2010;285:29690–29702. doi: 10.1074/jbc.M110.151795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Uringa EJ, Lisaingo K, Pickett HA, Brind’Amour J, Rohde JH, Zelensky A, Essers J, Lansdorp PM. RTEL1 contributes to DNA replication and repair and telomere maintenance. Mol Biol Cell. 2012;23:2782–2792. doi: 10.1091/mbc.E12-03-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell. 2012;149:795–806. doi: 10.1016/j.cell.2012.03.030. [DOI] [PubMed] [Google Scholar]
- 54.Youds JL, Mets DG, McIlwraith MJ, Martin JS, Ward JD, ONeil NJ, Rose AM, West SC, Meyer BJ, Boulton SJ. RTEL-1 enforces meiotic crossover interference and homeostasis. Science. 2010;327:1254–1258. doi: 10.1126/science.1183112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA, Livingston DM. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105:149–160. doi: 10.1016/s0092-8674(01)00304-x. [DOI] [PubMed] [Google Scholar]
- 56.De Nicolo A, Tancredi M, Lombardi G, Flemma CC, Barbuti S, Di Cristafono C, Sobhian B, Bevilacqua G, Drapkin R, Caligo MA. A novel breast cancer-associated BRIP1 (FANCJ/BACH1) germ-line mutation impairs protein stability and function. Clin Cancer Res. 2008;14:4672–4680. doi: 10.1158/1078-0432.CCR-08-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, North B, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR, Rahman N. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006;38:1239–1241. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- 58.Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, Arwert F, Mathew CG, Zdzienicka MZ, Hiom K, De Winter JP, Joenje H. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group. J, Nat Genet. 2005;37:934–935. doi: 10.1038/ng1625. [DOI] [PubMed] [Google Scholar]
- 59.Levran O, Attwooll C, Henry RT, Milton KL, Neveling K, Rio P, Batish SD, Kalb R, Velleuer E, Barral S, Ott J, Petrini J, Schindler D, Hanenberg H, Auerbach AD. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat Genet. 2005;37:931–933. doi: 10.1038/ng1624. [DOI] [PubMed] [Google Scholar]
- 60.Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, Cantor SB. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell. 2005;8:255–265. doi: 10.1016/j.ccr.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 61.Peng M, Litman R, Xie J, Sharma S, Brosh RM, Jr, Cantor SB. The FANCJ/MutLalpha interaction is required for correction of the cross-link response in FA-J cells. EMBO J. 2007;26:3238–3249. doi: 10.1038/sj.emboj.7601754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302:639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 63.Xie J, Litman R, Wang S, Peng M, Guillemette S, Rooney T, Cantor SB. Targeting the FANCJ-BRCA1 interaction promotes a switch from recombination to poleta-dependent bypass. Oncogene. 2010;29:2499–2508. doi: 10.1038/onc.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cantor S, Drapkin R, Zhang F, Lin Y, Han J, Pamidi S, Livingston DM. The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc Natl Acad Sci U S A. 2004;101:2357–2362. doi: 10.1073/pnas.0308717101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gupta R, Sharma S, Doherty KM, Sommers JA, Cantor SB, Brosh RM., Jr Inhibition of BACH1 (FANCJ) helicase by backbone discontinuity is overcome by increased motor ATPase or length of loading strand. Nucleic Acids Res. 2006;34:6673–6683. doi: 10.1093/nar/gkl964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sommers JA, Rawtani N, Gupta R, Bugreev DV, Mazin AV, Cantor SB, Brosh RM., Jr FANCJ uses its motor ATPase to disrupt protein-DNA complexes, unwind triplexes, and inhibit Rad51 strand exchange. J Biol Chem. 2009;284:7505–7517. doi: 10.1074/jbc.M809019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu Y, Sommers JA, Suhasini AN, Leonard T, Deakyne JS, Mazin AV, Shin-Ya K, Kitao H, Brosh RM., Jr Fanconi anemia Group J mutation abolishes its DNA repair function by uncoupling DNA translocation from helicase activity or disruption of protein-DNA complexes. Blood. 2010;116:3780–3791. doi: 10.1182/blood-2009-11-256016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu Y, Sommers JA, Khan I, De Winter JP, Brosh RM., Jr Biochemical characterization of Warsaw Breakage syndrome helicase. J Biol Chem. 2012;287:1007–1021. doi: 10.1074/jbc.M111.276022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Skibbens RV. Chl1p, a DNA helicase-like protein in budding yeast, functions in sister-chromatid cohesion. Genetics. 2004;166:33–42. doi: 10.1534/genetics.166.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chung G, O’Neil NJ, Rose AM. CHL-1 provides an essential function affecting cell proliferation and chromosome stability in Caenorhabditis elegans. DNA Repair (Amst) 2011;10:1174–1182. doi: 10.1016/j.dnarep.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 71.Inoue A, Li T, Roby SK, Valentine MB, Inoue M, Boyd K, Kidd VJ, Lahti JM. Loss of ChlR1 helicase in mouse causes lethality due to the accumulation of aneuploid cells generated by cohesion defects and placental malformation. Cell Cycle. 2007;6:1646–1654. doi: 10.4161/cc.6.13.4411. [DOI] [PubMed] [Google Scholar]
- 72.Parish JL, Rosa J, Wang X, Lahti JM, Doxsey SJ, Androphy EJ. The DNA helicase ChlR1 is required for sister chromatid cohesion in mammalian cells. J Cell Sci. 2006;119:4857–4865. doi: 10.1242/jcs.03262. [DOI] [PubMed] [Google Scholar]
- 73.Capo-chichi JM, Bharti SK, Sommers JA, Yammine T, Chouery E, Patry L, Rouleau GA, Samuels ME, Hamden FF, Michaud JL, Brosh RM, Jr, Megarbane A, Kibar Z. Identification and biochemical characterization of a novel mutation in DDX11 causing Warsaw Breakage Syndrome. Human Mutation. doi: 10.1002/humu.22226. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Botta E, Nardo T, Lehmann AR, Egly JM, Pedrini AM, Stefanini M. Reduced level of the repair/transcription factor TFIIH in trichothiodystrophy. Hum Mol Genet. 2002;11:2919–2928. doi: 10.1093/hmg/11.23.2919. [DOI] [PubMed] [Google Scholar]
- 75.Dubaele S, Proietti De Santis L, Bienstock RJ, Keriel A, Stefanini M, Van Houten B, Egly JM. Basal transcription defect discriminates between xeroderma pigmentosum and trichothiodystrophy in XPD patients. Mol Cell. 2003;11:1635–1646. doi: 10.1016/s1097-2765(03)00182-5. [DOI] [PubMed] [Google Scholar]
- 76.Graham JM, Jr, Anyane-Yeboa K, Raams A, Appeldoorn E, Kleijer WJ, Garritsen VH, Busch D, Edersheim TG, Jaspers NG. Cerebro-oculo-facio-skeletal syndrome with a nucleotide excision-repair defect and a mutated XPD gene, with prenatal diagnosis in a triplet pregnancy. Am J Hum Genet. 2001;69:291–300. doi: 10.1086/321295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Egly JM, Coin F. A history of TFIIH: Two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair (Amst) 2011;10:714–721. doi: 10.1016/j.dnarep.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 78.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 79.de Boer J, Donker I, de Wit J, Hoeijmakers JH, Weeda G. Disruption of the mouse xeroderma pigmentosum group D DNA repair/basal transcription gene results in preimplantation lethality. Cancer Res. 1998;58:89–94. [PubMed] [Google Scholar]
- 80.Ueda T, Compe E, Catez P, Kraemer KH, Egly JM. Both XPD alleles contribute to the phenotype of compound heterozygote xeroderma pigmentosum patients. J Exp Med. 2009;206:3031–3046. doi: 10.1084/jem.20091892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lehmann AR. XPD structure reveals its secrets. DNA Repair (Amst) 2008;7:1912–1915. doi: 10.1016/j.dnarep.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 82.Coin F, Oksenych V, Egly JM. Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Mol Cell. 2007;26:245–256. doi: 10.1016/j.molcel.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 83.Broughton BC, Berneburg M, Fawcett H, Taylor EM, Arlett CF, Nardo T, Stefanini M, Menefee E, Price VH, Queille S, Sarasin A, Bohnert E, Krutmann J, Davidson R, Kraemer KH, Lehmann AR. Two individuals with features of both xeroderma pigmentosum and trichothiodystrophy highlight the complexity of the clinical outcomes of mutations in the XPD gene. Hum Mol Genet. 2001;10:2539–2547. doi: 10.1093/hmg/10.22.2539. [DOI] [PubMed] [Google Scholar]
- 84.Oksenych V, Coin F. The long unwinding road: XPB and XPD helicases in damaged DNA opening. Cell Cycle. 2010;9:90–96. doi: 10.4161/cc.9.1.10267. [DOI] [PubMed] [Google Scholar]
- 85.Oh KS, Khan SG, Jaspers NG, Raams A, Ueda T, Lehmann A, Friedmann PS, Emmert S, Gratchev A, Lachlan K, Lucassan A, Baker CC, Kraemer KH. Phenotypic heterogeneity in the XPB DNA helicase gene (ERCC3): xeroderma pigmentosum without and with Cockayne syndrome. Hum Mutat. 2006;27:1092–1103. doi: 10.1002/humu.20392. [DOI] [PubMed] [Google Scholar]
- 86.Weeda G, Eveno E, Donker I, Vermeulen W, Chevallier-Lagente O, Taieb A, Stary A, Hoeijmakers JH, Mezzina M, Sarasin A. A mutation in the XPB/ERCC3 DNA repair transcription gene, associated with trichothiodystrophy. Am J Hum Genet. 1997;60:320–329. [PMC free article] [PubMed] [Google Scholar]
- 87.Riou L, Zeng L, Chevallier-Lagente O, Stary A, Nikaido O, Taieb A, Weeda G, Mezzina M, Sarasin A. The relative expression of mutated XPB genes results in xeroderma pigmentosum/Cockayne’s syndrome or trichothiodystrophy cellular phenotypes. Hum Mol Genet. 1999;8:1125–1133. doi: 10.1093/hmg/8.6.1125. [DOI] [PubMed] [Google Scholar]
- 88.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 89.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 90.Monnat RJ., Jr Human RECQ helicases: roles in DNA metabolism, mutagenesis and cancer biology. Semin Cancer Biol. 2010;20:329–339. doi: 10.1016/j.semcancer.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Friedrich K, Lee L, Leistritz DF, Nurnberg G, Saha B, Hisama FM, Eyman DK, Lessel D, Nurnberg P, Li C, Garcia FV, Kets CM, Schmidtke J, Cruz VT, Van den Akker PC, Boak J, Peter D, Compoginis G, Cefle K, Ozturk S, Lopez N, Wessel T, Poot M, Ippel PF, Groff-Kellermann B, Hoehn H, Martin GM, Kubisch C, Oshima J. WRN mutations in Werner syndrome patients: genomic rearrangements, unusual intronic mutations and ethnic-specific alterations. Hum Genet. 2010;128:103–111. doi: 10.1007/s00439-010-0832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Swanson C, Saintigny Y, Emond MJ, Monnat RJ., Jr The Werner syndrome protein has separable recombination and survival functions. DNA Repair (Amst) 2004;3:475–482. doi: 10.1016/j.dnarep.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 93.Chen L, Huang S, Lee L, Davalos A, Schiestl RH, Campisi J, Oshima J. WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging Cell. 2003;2:191–199. doi: 10.1046/j.1474-9728.2003.00052.x. [DOI] [PubMed] [Google Scholar]
- 94.Opresko PL, Otterlei M, Graakjaer J, Bruheim P, Dawut L, Kolvraa S, May A, Seidman MM, Bohr VA. The Werner Syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol Cell. 2004;14:763–774. doi: 10.1016/j.molcel.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 95.Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, Martin GM, Mulligan J, Schellenberg GD. Positional cloning of the Werner’s syndrome gene. Science. 1996;272:258–262. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- 96.Suzuki T, Shiratori M, Furuichi Y, Matsumoto T. Diverged nuclear localization of Werner helicase in human and mouse cells. Oncogene. 2001;20:2551–2558. doi: 10.1038/sj.onc.1204344. [DOI] [PubMed] [Google Scholar]
- 97.Huang S, Lee L, Hanson NB, Lenaerts C, Hoehn H, Poot M, Rubin CD, Chen DF, Yang CC, Juch H, Dorn T, Spiegel R, Oral EA, Abid M, Battisti C, Lucci-Cordisco E, Neri G, Steed EH, Kidd A, Isley W, Showalter D, Vittone JL, Konstantinow A, Ring J, Meyer P, Wenger SL, von Herbay A, Wollina U, Schuelke M, Huizenga CR, Leistritz DF, Martin GM, Mian IS, Oshima J. The spectrum of WRN mutations in Werner syndrome patients. Hum Mutat. 2006;27:558–567. doi: 10.1002/humu.20337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Uhrhammer NA, Lafarge L, Dos SL, Domaszewska A, Lange M, Yang Y, Aractingi S, Bessis D, Bignon YJ. Werner syndrome and mutations of the WRN and LMNA genes in France. Hum Mutat. 2006;27:718–719. doi: 10.1002/humu.9435. [DOI] [PubMed] [Google Scholar]
- 99.Sebastiani P, Solovieff N, Dewan AT, Walsh KM, Puca A, Hartley SW, Melista E, Andersen S, Dworkis DA, Wilk JB, Myers RH, Steinberg MH, Montano M, Baldwin CT, Hoh J, Perls TT. Genetic signatures of exceptional longevity in humans. PLoS ONE. 2012;7:e29848. doi: 10.1371/journal.pone.0029848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chaganti RS, Schonberg S, German J. A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proc Natl Acad Sci U S A. 1974;71:4508–4512. doi: 10.1073/pnas.71.11.4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–666. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- 102.Bachrati CZ, Hickson ID. Dissolution of double Holliday junctions by the concerted action of BLM and topoisomerase IIIalpha. Methods Mol Biol. 2009;582:91–102. doi: 10.1007/978-1-60761-340-4_8. [DOI] [PubMed] [Google Scholar]
- 103.Wu L, Hickson ID. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–874. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- 104.Bachrati CZ, Borts RH, Hickson ID. Mobile D-loops are a preferred substrate for the Bloom’s syndrome helicase. Nucleic Acids Res. 2006;34:2269–2279. doi: 10.1093/nar/gkl258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Adams MD, McVey M, Sekelsky JJ. Drosophila BLM in double-strand break repair by synthesis-dependent strand annealing. Science. 2003;299:265–267. doi: 10.1126/science.1077198. [DOI] [PubMed] [Google Scholar]
- 106.Chu WK, Hanada K, Kanaar R, Hickson ID. BLM has early and late functions in homologous recombination repair in mouse embryonic stem cells. Oncogene. 2010;29:4705–4714. doi: 10.1038/onc.2010.214. [DOI] [PubMed] [Google Scholar]
- 107.Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011;25:350–362. doi: 10.1101/gad.2003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 2009;11:753–760. doi: 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- 109.Neff NF, Ellis NA, Ye TZ, Noonan J, Huang K, Sanz M, Proytcheva M. The DNA helicase activity of BLM is necessary for the correction of the genomic instability of Bloom syndrome cells. Mol Biol Cell. 1999;10:665–676. doi: 10.1091/mbc.10.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Davalos AR, Campisi J. Bloom syndrome cells undergo p53-dependent apoptosis and delayed assembly of BRCA1 and NBS1 repair complexes at stalled replication forks. J Cell Biol. 2003;162:1197–1209. doi: 10.1083/jcb.200304016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.German J, Sanz MM, Ciocci S, Ye TZ, Ellis NA. Syndrome-causing mutations of the BLM gene in persons in the Bloom’s Syndrome Registry. Hum Mutat. 2007;28:743–753. doi: 10.1002/humu.20501. [DOI] [PubMed] [Google Scholar]
- 112.Guo RB, Rigolet P, Ren H, Zhang B, Zhang XD, Dou SX, Wang PY, Amor-Gueret M, Xi XG. Structural and functional analyses of disease-causing missense mutations in Bloom syndrome protein. Nucleic Acids Res. 2007;35:6297–6310. doi: 10.1093/nar/gkm536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bahr A, De Graeve F, Kedinger C, Chatton B. Point mutations causing Bloom’s syndrome abolish ATPase and DNA helicase activities of the BLM protein. Oncogene. 1998;17:2565–2571. doi: 10.1038/sj.onc.1202389. [DOI] [PubMed] [Google Scholar]
- 114.Tanner NK, Cordin O, Banroques J, Doere M, Linder P. The Q motif: a newly identified motif in DEAD box helicases may regulate ATP binding and hydrolysis. Mol Cell. 2003;11:127–138. doi: 10.1016/s1097-2765(03)00006-6. [DOI] [PubMed] [Google Scholar]
- 115.Tanner NK. The newly identified Q motif of DEAD box helicases is involved in adenine recognition. Cell Cycle. 2003;2:18–19. doi: 10.4161/cc.2.1.296. [DOI] [PubMed] [Google Scholar]
- 116.Cordin O, Tanner NK, Doere M, Linder P, Banroques J. The newly discovered Q motif of DEAD-box RNA helicases regulates RNA-binding and helicase activity. EMBO J. 2004;23:2478–2487. doi: 10.1038/sj.emboj.7600272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Theis K, Chen PJ, Skorvaga M, Van Houten B, Kisker C. Crystal structure of UvrB, a DNA helicase adapted for nucleotide excision repair. EMBO J. 1999;18:6899–6907. doi: 10.1093/emboj/18.24.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wu Y, Sommers JA, Loiland JA, Kitao H, Kuper J, Kisker C, Brosh RM., Jr The Q motif of FANCJ DNA helicase regulates its dimerization, DNA binding, and DNA repair function. J Biol Chem. 2012;287:21699–21716. doi: 10.1074/jbc.M112.351338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Karow JK, Newman RH, Freemont PS, Hickson ID. Oligomeric ring structure of the Bloom’s syndrome helicase. Curr Biol. 1999;9:597–600. doi: 10.1016/s0960-9822(99)80264-4. [DOI] [PubMed] [Google Scholar]
- 120.Janscak P, Garcia PL, Hamburger F, Makuta Y, Shiraishi K, Imai Y, Ikeda H, Bickle TA. Characterization and mutational analysis of the RecQ core of the Bloom syndrome protein. J Mol Biol. 2003;330:29–42. doi: 10.1016/s0022-2836(03)00534-5. [DOI] [PubMed] [Google Scholar]
- 121.Xu YN, Bazeille N, Ding XY, Lu XM, Wang PY, Bugnard E, Grondin V, Dou SX, Xi XG. Multimeric BLM is dissociated upon ATP hydrolysis and functions as monomers in resolving DNA structures. Nucleic Acids Res. 2012;40:9802–9814. doi: 10.1093/nar/gks728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yang Y, Dou SX, Xu YN, Bazeille N, Wang PY, Rigolet P, Xu HQ, Xi XG. Kinetic mechanism of DNA unwinding by the BLM helicase core and molecular basis for its low processivity. Biochemistry. 2010;49:656–668. doi: 10.1021/bi901459c. [DOI] [PubMed] [Google Scholar]
- 123.Foucault F, Vaury C, Barakat A, Thibout D, Planchon P, Jaulin C, Praz F, Amor-Gueret M. Characterization of a new BLM mutation associated with a topoisomerase II alpha defect in a patient with Bloom’s syndrome. Hum Mol Genet. 1997;6:1427–1434. doi: 10.1093/hmg/6.9.1427. [DOI] [PubMed] [Google Scholar]
- 124.Guo RB, Rigolet P, Zargarian L, Fermandjian S, Xi XG. Structural and functional characterizations reveal the importance of a zinc binding domain in Bloom’s syndrome helicase. Nucleic Acids Res. 2005;33:3109–3124. doi: 10.1093/nar/gki619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang XW, Tseng A, Ellis NA, Spillare EA, Linke SP, Robles AI, Seker H, Yang Q, Hu P, Beresten S, Bemmels NA, Garfield S, Harris CC. Functional interaction of p53 and BLM DNA helicase in apoptosis. J Biol Chem. 2001;276:32948–32955. doi: 10.1074/jbc.M103298200. [DOI] [PubMed] [Google Scholar]
- 126.Rao VA, Fan AM, Meng L, Doe CF, North PS, Hickson ID, Pommier Y. Phosphorylation of BLM, dissociation from topoisomerase IIIalpha, and colocalization with gamma-H2AX after topoisomerase I-induced replication damage. Mol Cell Biol. 2005;25:8925–8937. doi: 10.1128/MCB.25.20.8925-8937.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Capp C, Wu J, Hsieh TS. RecQ4: the second replicative helicase? Crit Rev Biochem Mol Biol. 2010;45:233–242. doi: 10.3109/10409231003786086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sangrithi MN, Bernal JA, Madine M, Philpott A, Lee J, Dunphy WG, Venkitaraman AR. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell. 2005;121:887–898. doi: 10.1016/j.cell.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 129.Croteau DL, Rossi ML, Ross J, Dawut L, Dunn C, Kulikowicz T, Bohr VA. RAPADILINO RECQL4 mutant protein lacks helicase and ATPase activity. Biochim Biophys Acta. 2012;1822:1727–1734. doi: 10.1016/j.bbadis.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Clarkson SG, Wood RD. Polymorphisms in the human XPD (ERCC2) gene, DNA repair capacity and cancer susceptibility: an appraisal. DNA Repair (Amst) 2005;4:1068–1074. doi: 10.1016/j.dnarep.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 131.Bohr VA, Metter EJ, Harrigan JA, von Kobbe C, Liu JL, Gray MD, Majumdar A, Wilson DM, III, Seidman MM. Werner syndrome protein 1367 variants and disposition towards coronary artery disease in Caucasian patients. Mech Ageing Dev. 2004;125:491–496. doi: 10.1016/j.mad.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 132.Hsu JJ, Kamath-Loeb AS, Glick E, Wallden B, Swisshelm K, Rubin BP, Loeb LA. Werner syndrome gene variants in human sarcomas. Mol Carcinog. 2010;49:166–174. doi: 10.1002/mc.20586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kamath-Loeb AS, Welcsh P, Waite M, Adman ET, Loeb LA. The enzymatic activities of the Werner syndrome protein are disabled by the amino acid polymorphism R834C. J Biol Chem. 2004;279:55499–55505. doi: 10.1074/jbc.M407128200. [DOI] [PubMed] [Google Scholar]
- 134.Janovick JA, Park BS, Conn PM. Therapeutic rescue of misfolded mutants: validation of primary high throughput screens for identification of pharmacoperone drugs. PLoS ONE. 2011;6:e22784. doi: 10.1371/journal.pone.0022784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bullock AN, Fersht AR. Rescuing the function of mutant p53. Nat Rev Cancer. 2001;1:68–76. doi: 10.1038/35094077. [DOI] [PubMed] [Google Scholar]
- 136.Aggarwal M, Sommers JA, Shoemaker RH, Brosh RM., Jr Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc Natl Acad Sci U S A. 2011;108:1525–1530. doi: 10.1073/pnas.1006423108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Aggarwal M, Brosh RM., Jr Hitting the bull’s eye: novel directed cancer therapy through helicase-targeted synthetic lethality. J Cell Biochem. 2009;106:758–763. doi: 10.1002/jcb.22048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Suhasini AN, Rawtani NA, Wu Y, Sommers JA, Sharma S, Mosedale G, North PS, Cantor SB, Hickson ID, Brosh RM., Jr Interaction between the helicases genetically linked to Fanconi anemia group J and Bloom’s syndrome. EMBO J. 2011;30:692–705. doi: 10.1038/emboj.2010.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Doherty KM, Sommers JA, Gray MD, Lee JW, von Kobbe C, Thoma NH, Kureekattil RP, Kenny MK, Brosh RM., Jr Physical and functional mapping of the RPA interaction domain of the Werner and Bloom syndrome helicases. J Biol Chem. 2005;280:29494–29505. doi: 10.1074/jbc.M500653200. [DOI] [PubMed] [Google Scholar]
- 140.Hu P, Beresten SF, van Brabant AJ, Ye TZ, Pandolfi PP, Johnson FB, Guarente L, Ellis NA. Evidence for BLM and Topoisomerase IIIalpha interaction in genomic stability. Hum Mol Genet. 2001;10:1287–1298. doi: 10.1093/hmg/10.12.1287. [DOI] [PubMed] [Google Scholar]