

Abstract
The Uba6 (E1)-Use1 (E2) ubiquitin transfer cascade is a poorly understood alternative arm of the ubiquitin proteasome system (UPS) required for mouse embryonic development, independent of the canonical Uba1-E2-E3 pathway. Loss of neuronal Uba6 during embryonic development results in altered patterning of neurons in the hippocampus and the amygdala, decreased dendritic spine density, and numerous behavioral disorders. The levels of the E3 ubiquitin ligase Ube3a (E6-AP) and Shank3, both linked with dendritic spine function, are elevated in the amygdala of Uba6-deficient mice, while levels of the Ube3a substrate Arc are reduced. Uba6 and Use1 promote proteasomal turnover of Ube3a in mouse embryo fibroblasts (MEFs) and catalyze Ube3a ubiquitylation in vitro. These activities occur in parallel with an independent pathway involving Uba1-UbcH7, but in a spatially distinct manner in MEFs. These data reveal an unanticipated role for Uba6 in neuronal development, spine architecture, mouse behavior, and turnover of Ube3a.
The ubiquitin proteasome system (UPS) regulates signaling functions in eukaryotes, and provides a mechanism for controlling protein homeostasis and developmental decisions. In this pathway, ubiquitin is transferred to targets via an E1-E2-E3 cascade, wherein ubiquitin-charged E2 enzymes function together with E3 ubiquitin ligases to bring about substrate ubiquitylation (Ye and Rape, 2009). Historically, the canonical E1 ubiquitin activating enzyme Uba1 was thought to reside at the apex of the entire UPS by catalyzing ATP-dependent charging of all ubiquitin E2s, which subsequently function with individual E3s to promote substrate ubiquitylation (Schulman and Harper, 2009). However, we and others previously identified a related E1-like enzyme in vertebrates, Uba6, which constitutes an independent arm of the ubiquitin system and co-exists with Uba1 in all cell types and tissues examined (Chiu et al., 2007; Jin et al., 2007; Pelzer et al., 2007). Uba6 is required to activate the E2 Use1 (Uba6-specific E2, also called Ube2z) in vitro and in tissue culture cells, and while it is capable of activating a subset of canonical ubiquitin E2s in vitro, the high levels of Uba1 in most cells is thought to obviate the need for Uba6 to promote canonical ubiquitin transfer (Jin et al., 2007). Uba6 is required for the viability of mouse embryo fibroblasts (MEFs) (Lee et al., 2011) and mice lacking Uba6 die ∼E5.5 of embryonic development for unknown reasons (Chiu et al., 2007), indicating a unique role for this pathway during embryogenesis. To date, only a single class of E3s – the UBR and RING domain containing E3s involved in the N-end rule pathway - has been shown to function with Uba6-Use1 (Lee et al., 2011). Uba6-Use1-Ubr1/2 promotes turnover of regulators of G-protein signaling Rgs4 and Rgs5 in the cytoplasm (Lee et al., 2011). This pathway occurs in parallel with a Uba1-Rad6-Ubr1/2 pathway in the nucleus and cytoplasm (Lee et al., 2011). UBR1 is mutated in Johanson-Blizzard syndrome (Zenker et al., 2005), characterized by mental retardation and developmental malformations, although the relevant pathways remain poorly defined.
While our understanding of the biochemistry and mechanisms of the UPS is advanced, our understanding of the physiological roles in metazoan biology, for even the best-studied UPS components, is still rudimentary. Moreover, in cases where a biological function is known, the mechanisms and targets are often not understood. However, recent work in the mammalian nervous system is starting to close this gap. The canonical UPS controls multiple aspects of brain development and function, including dendrite and axon length determination as well as dendritic spine architecture, and dynamics. (Bingol and Schuman, 2006; Bingol and Sheng, 2011; Hegde, 2010; Hung et al., 2010; Kuczera et al., 2011; Mabb and Ehlers, 2010; Pak and Sheng, 2003; Yang et al., 2010). Several E3s have been linked to neuronal development, including the RING-finger based E3s - the Anaphase Promoting Complex, SCFβ-TRCP, OBSL1-CUL7-FBW8, and the TRIM3 ubiquitin ligase – and in some cases, specific targets have been identified (Ang et al., 2008; Hung et al., 2010; Yang et al., 2010); Litterman et al., 2011).
Given widespread expression of Uba6 in the mammalian brain together with the known role of the canonical UPS in neuronal function, we sought to determine whether the non-canonical system also controls neuronal development and function. We generated mice in which Uba6 was deleted during embryonic development via Nestin-Cre, referred to subsequently as Uba6NKO mice. These mice display defects in the morphology and abundance of neurons specifically in the CA3 region of the hippocampus and in the amygdala, with evidence of decreased dendritic spine density, and a requirement for Uba6 for hippocampal neuron survival in culture. Moreover, Uba6NKO mice display numerous behavioral phenotypes including social interaction defects. Unexpectedly, analysis of several proteins known to regulate dendritic spine function revealed elevated levels of the ubiquitin ligase Ube3a and the post-synaptic density protein Shank3 specifically in the amygdala. Ube3a (also referred to as E6-AP), the founding member of HECT-domain ubiquitin ligases (Huibregtse et al., 1995; Scheffner et al., 1993), is mutated in Angelman's Syndrome (Dindot et al., 2008; Yashiro et al., 2009; Mabb et al., 2011), and has been linked with autism spectrum disorders (ASD) through both gain of function and loss of function mechanisms (Smith et al., 2011). Interestingly, we found that the increase in Ube3a levels seen in Uba6NKO animals correlates with a decrease in the abundance of the previously characterized Ube3a substrate Arc (Greer et al., 2010). Arc is a synaptic protein that promotes internalization of AMPA-type glutamate receptors, and control of its levels via Ube3a is important for spine function. Consistent with a direct role for Uba6-Use1 in Ube3a stability, mouse embryo fibroblasts (MEFs) lacking either Uba6 or Use1 display elevated Ube3a levels and half-life, and human USE1 can ubiquitylate UBE3A in vitro. The control of Ube3a levels by Uba6-Use1 in MEFs occurs in parallel with an independent system involving the canonical Uba1-UbcH7 (also called Ube2l) cascade, wherein the Uba6-Use1 pathway functions exclusively in the cytoplasm and the Uba1-UbcH7 pathway functions in the nucleus and to a lesser extent in the cytoplasm. These data indicate that alterations of the Uba6 pathway in the brain lead to defects in neuronal structure and function likely through affects on multiple targets, and identify Ube3a as a new substrate of the Uba6-Use1 system in MEFs.
Results
Reduced Organismal Size and Post-natal Lethality in Mice Lacking Neuronal Uba6
To examine the role of Uba6 in brain function, we generated mice in which exons 16 and 17 encoding the essential adenylation domain were flanked by lox sites, allowing deletion with Cre recombinase driven by the Nestin promoter on a fully C57B6 background (Figure 1A). Nestin expression occurs in the neuroectoderm at or before embryonic day 9 (including both glia and neuronal cells) (Tsien et al., 1996). Deletion of Uba6 in the brains of Nestin-Cre+/-;Uba6Flox/Flox mutant mice (referred to hereafter as Uba6NKO) was confirmed by western blotting of extracts from the cortex, hippocampus, and amygdala at 3 months of age, with no change in Use1 levels (Figure 1B).
Figure 1. Generation and characterization of Uba6 brain-specific knockout mice.
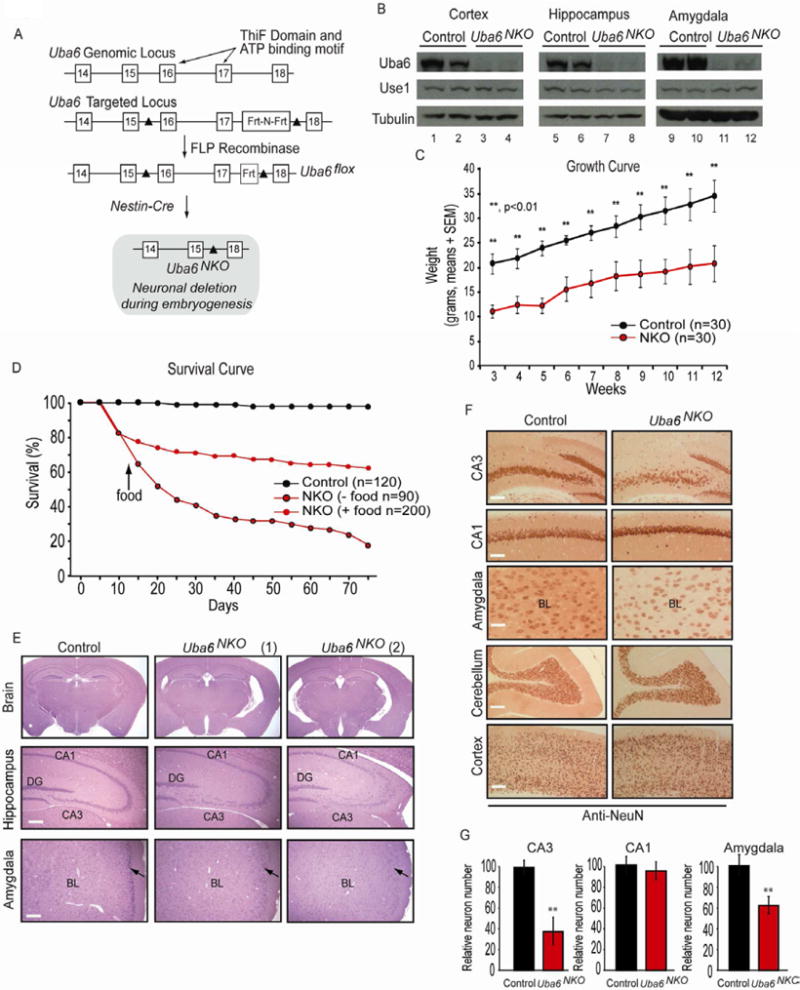
(A) Strategy used to generate a conditional Uba6 allele. The targeted Uba6Flox allele was generated in ES cells using a targeting construct with an Frt-neo-Frt-loxP cassette. The NEO gene was removed in vivo by crossing to FLPe mice. The Uba6Flox allele includes two loxP sites flanking exons 16 and 17, which encode the ThiF domain and the ATP binding site. Uba6NKO mice were generated using heterozygous Nestin-CRE, which generates one loxP site preceding exon 18.
(B) Western blot of Uba6 and Use1 in cerebellar cortex, hippocampus and amygdala extracts of control (Uba6Flox/Flox) and Uba6NKO mice. Tubulin was used as a loading control.
(C-D) Growth and survival of Uba6NKO mice. (C) Littermate control (n=30) and Uba6NKO (n=30) mice were weighed weekly beginning at 3 weeks of age. (D) Mice {Control (n=120), Uba6NKO (n=90) with food placed conventionally in the cage, and Uba6NKO (n=200) with food placed in the bottom of the cage} were followed up to 75 day of age, and survival determined. Each bar represents the mean ± SEM and asterisks denote the level of statistical significance (Student t test) between the Control and Uba6NKO mice. **P < 0.01.
(E) H & E staining of coronal brain sections with enlargements of the hippocampus and the amygdala regions from Uba6NKO and Control mice (3 months of age). Two Uba6NKO animals were examined, as indicated. All Scale bars (white lines): 500 μm. Arrowhead identifies piriform cortex.
(F) Anti-NeuN staining of coronal sections of the hippocampus (CA1 and CA3 regions), cortex, cerebellum, and the amygdala from Uba6NKO and Control mice (3 months of age). All Scale bars (white lines): 250 μm.
(G) Quantification of NeuN-positive neurons in the CA1 and CA3 regions of the hippocampus and in the amygdala of Uba6NKO mice determined using Metamorph Software. Control (n=8), Uba6NKO (n=8), (mean ± SEM). **P < 0.01. See also Figure S1.
Uba6NKO mice were born at Mendelian ratios (Figure S1A), but showed a significant (∼50%) reduction in weight at birth, which persists over time (Figure 1C). Animals are born with similarly reduced body length and at 11 weeks of age have a body length ∼80% of heterozygous litter mates (Figure S1B). Fifty percent of Uba6NKO pups died between P5 and P21, and an additional 25% died before reaching 2 months of age (Figure 1D). Full histological analysis of Uba6NKO animals at death revealed no obvious changes in vital organs, other than the specific effects seen in the brain described below. However, we noted the absence of stomach contents in several animals at death. We found that by making food more accessible, we could substantially rescue the early lethality, from 30% viability at 2 months of age without food access to 70% viability with food access (Figure 1D). These data indicate a requirement for Uba6 in the brain during mouse embryonic development in order to achieve proper body size and post-natal viability.
Morphological Defects in the Hippocampus and Amygdala in Uba6NKO Mice
Histological analyses of brain sections from 3 month old Uba6NKO mice revealed morphological abnormalities in the brain that were limited specifically to the hippocampus and amygdala, although the brain size overall was 11% smaller for Uba6NKO mice (Figure S1B). Hematoxylin and Eosin (H&E) staining (Figure 1E and Figure S1C-F) and immunohistochemistry with neuronal-specific anti-NeuN antibodies (Figure 1F) on coronal brain sections from Uba6NKO mice revealed a 65% reduction in the number of neurons in the CA3 region with little evidence for altered neuronal content in CA1, dentate gyrus, cortex, or cerebellar regions (Figure 1E-G, and Figure S1C-F), with no obvious change in GFAP-positive glial cell populations (not shown). Neurons that remained in the CA3 region were significantly scattered when compared with Uba6Flox/Flox mice (Figure 1E, F, and Figure S1C-F). This was particularly striking in the posterior aspect of the hippocampus (Figure S1C-F). Defects in the architecture of neurons in the CA3 region were seen as early as post-natal day 5 (P5), although they were not as dramatic as that seen in older mice (Figure S1G). We also noted alterations in neuronal architecture in the amygdala, with a 40% reduction in neuronal number in the basolateral amygdala and reduced neuronal density in the piriform cortex (Figure 1E-G). The mechanism underlying these developmental defects in CA3 and the amygdala are unknown, although we did not observe an increase in the abundance of apoptotic cells in the brain at either E14.5 or post-natal day 5 (Figure S1H and I).
Behavioral Characterization of Uba6NKO Mice
Given the roles of the hippocampus and amygdala in cognition and behavior, we performed a battery of behavioral tests to assess learning/memory performance, as well as social and emotional behaviors in a cohort of female mice at 3 months of age (Silverman et al., 2010). We first assessed basic exploratory behaviors, locomotor activity, and responses to a novel environment using an open field test (Figure 2A, B). Control Uba6Flox/Flox mice progressively reduced their activity during the 60-minute test (Figure 2A), indicating habituation to a novel environment. In contrast, Uba6NKO mice displayed hyperactivity and a complete lack of habituation (Figure 2A), suggesting profound deficits in this simple form of memory (Thompson and Spencer, 1966). Moreover, Uba6NKO mice spent significantly less time than control mice in the field center, preferring instead to traverse the periphery (thigmotaxis) (Figure 2B, and Supplemental Movie 1), consistent with a profound anxiety-like phenotype in Uba6NKO mice. When tested for motor coordination (rotarod), Uba6NKO mice performed significantly better than control mice, despite having reduced grip strength (Figure S2A, B). Moreover, Uba6NKO mice displayed constitutive piloerection without an alteration in body temperature (Figure S2C), suggesting an involuntary anxiety response independent of their environment.
Figure 2. Behavioral analysis ofUba6NKOanimals reveals hyperactivity.
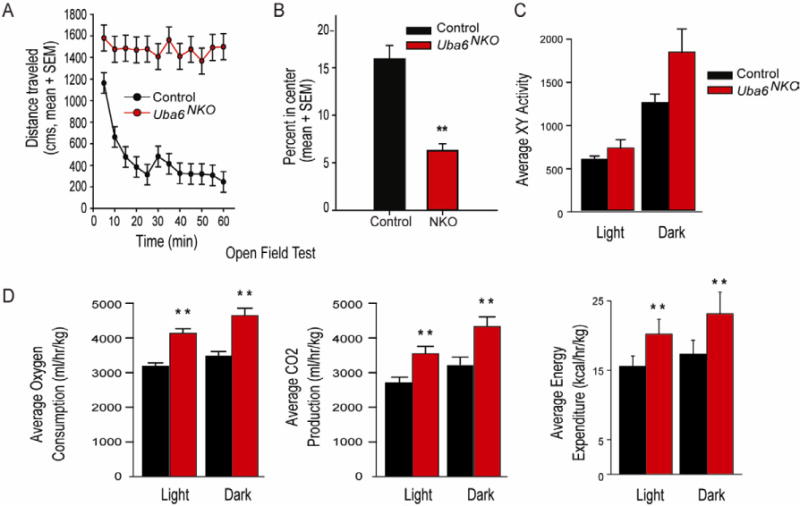
(A-B) Mice (female) were exposed to an open field arena for 60 min and distance traveled (A) and the time spent in the center (B) was recorded (mean ± SEM). Asterisks denote level of statistical significance (Student t test) between the control and Uba6NKO. **P < 0.01. Control (n=8) and Uba6NKO (n=6).
(C-D) Energy homeostasis analysis of control (Uba6Flox/Flox; n=6) and Uba6NKO (n=6) male mice analyzed for 48 h. (C) XY activity, (D) oxygen consumption, carbon dioxide production, and energy expenditure. All values are mean ± SEM. For complete data set, see Table S1. See also Figure S2, S3, and Supplemental Movie 1.
Uba6NKO Mice Display Increased Metabolic Activity Consistent with Hyperactivity
Excessive locomotor activity in the open field test suggests a level of hyperactivity in immediate response to an unfamiliar environment. We therefore directly measured home cage activity as well as metabolic rates over a 48 h period. Uba6NKO mice showed a trend toward increased X-Y activity that was pronounced in the dark phase (47% increased activity, p = 0.08) (Figure 2C, Table S1). Additionally, the Uba6NKO mice were hypermetabolic compared to controls: Uba6NKO mice consumed 32% more O2 (p = 0.003) and produced 33% more CO2 (p = 0.005) than control mice, and therefore have higher energy expenditure (Figure 2D, Table S1). Importantly, the respiratory exchange ratio (RER) remained constant throughout the diurnal cycle, indicating that both control and Uba6NKO mice had similar preferences for burning fats and carbohydrates (Table S1). These data illustrate a general shift towards increased metabolic activity in Uba6NKO mice.
Uba6NKO Mice Display Social Behavior Abnormalities
We next investigated whether Uba6NKO mice display altered social behaviors. As visual inspection of Uba6NKO mice suggested unusual home-cage nesting behavior, we measured nest building over time. Control mice created a nest within a 90 min period upon introduction of nesting material (Figure 3A), while Uba6NKO mice display a dramatic aversion to nesting, with no evidence of nest building or interaction with nesting material after 48 h (Figure 3A and S2D).
Figure 3. Uba6NKOmice display social interaction deficits.
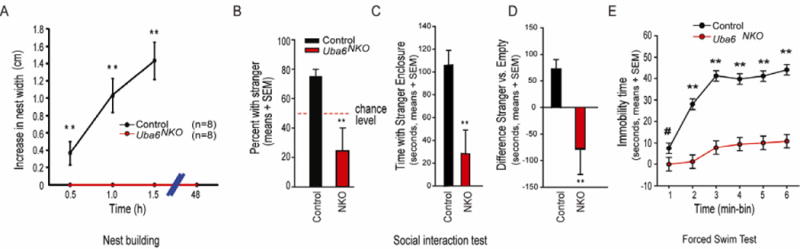
(A) Uba6NKO mice (female) exhibit impaired nest-building behavior. Mice were habituated in an empty cage for 15 min, and a 5 × 5 cm square of pressed cotton was placed in a random cage corner. The width and height of the nest built by the mouse, with subtraction of the original width and height of the cotton pad, was measured at 30, 60, 90 min and 48 h. All values are mean ± SEM. **P < 0.01, ***P < 0.001. Control (n=8) and Uba6NKO (n=8).
(B-D) Uba6NKO mice (female) exhibit social interaction defects, as determined by percentage of total interaction time with stranger in three-chamber social interaction test (B) and the time with the stranger enclosure (C-D). Dashed line in (B) represents chance-level performance (i.e. 50%) when mice equally explore the chamber containing a stranger mouse and the opposite chamber. Error bars are mean ± SEM and the statistical significance between control (n=8) and Uba6NKO (n=6) mice is **P < 0.01.
(E) Forced swim test. Uba6NKO (n=6) mice displayed significantly less immobility time than Control (n=8) mice (all female). All values are mean ± SEM. **P < 0.01. Also see Figure S2 and S3.
To measure social interactions, Uba6NKO and control Uba6Flox/Flox mice were subjected to a three-chamber social–interaction test. After habituation, mice are allowed to choose between a chamber containing a caged age-match stranger mouse or a chamber containing an empty cage. As illustrated in Figure 3B-D and Supplemental Movie 2, when faced with the choice to explore either chamber, control mice spent ∼75% of their time interacting with the stranger mouse, as expected. In contrast, Uba6NKO mice spent only ∼25% of the time interacting with the stranger mouse (Figure 3B-D), Supplemental Movie 2), preferring the empty chamber. Thus, Uba6NKO mice display defects in social interactions. We also employed the forced swim test in order to examine defects in behavioral despair. Uba6NKO mice spent 5-fold less time immobilized than did control Uba6Flox/Flox mice over a 5 min test period (Figure 3E). These data indicate severe social interaction deficits and emotional disturbances in Uba6NKO mice.
Uba6NKO Mice Display Defects in Learning and Memory
Defects in CA3 organization led us to examine whether Uba6NKO mice display learning and memory defects using the fear conditioning test. During training, age-matched Uba6NKO and control Uba6Flox/Flox mice demonstrated comparable low levels of freezing prior to shock application (Figure 4A). However, Uba6NKO mice demonstrated significantly reduced freezing in response to shock compared with control mice, indicating an altered response to an aversive stimulus. When re-exposed to the conditioning chamber (i.e. context) 24h later, Uba6NKO mice displayed a 5-fold reduction in freezing compared to control mice (Figure 4B).
Figure 4. Uba6NKOmice display enhanced anxiety phenotypes and a decrease in dendritic spine density in the CA3 region of the hippocampus and the amygdala.
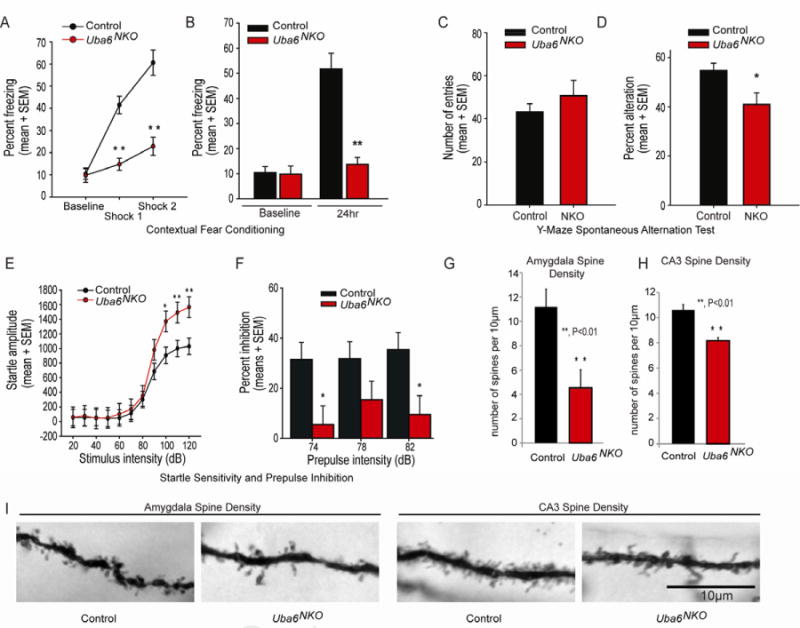
(A) Measurement of contextual fear conditioning expressed as percentage of total time spent freezing over each 1-min time bin. Mice were introduced to the novel conditioning chamber at t = 0, and a 2-second duration electric footshock was delivered at t = 2 min and t = 4 min. **P < 0.01. Control (n=8) and Uba6NKO (n=6).
(B) Retention of the contextual fear memory. The bars on the right represent the time spent freezing in response to the conditioned context 24 h following initial acquisition, in the absence of footshock. For comparison, baseline-freezing levels (recorded during the first two minutes of exposure to the novel context during training) are shown on the left. Error bars represents mean ± SEM and asterisks denote level of statistical significance (Student t test) between the control and Uba6NKO female mice. **P < 0.01. Control (n=8) and Uba6NKO (n=6).
(C-D) The average number of arm entries does not significantly differ between groups during the 6-min Y-maze session (C) while the average percent of spontaneous alternation is significantly decreased in Uba6NKO mice when compared with control and Uba6NKO mice (D). Error bars are mean ± SEM. *P < 0.05. Control (n=12) and Uba6NKO (n=9). Also see Figure S2 and Supplemental Movie 2.
(E-F) Uba6NKO mice exhibit impaired startle responses. Startle responses to eight presentations of the following sound pulses (presented in pseudorandom order): no stimulus, 20, 40, 60, 80, 100, and 120 dB pulse. For each pulse, the mean startle amplitudes were averaged (E). Pre-pulse inhibition measurements were performed in mice randomly exposed to five trial types: pulse alone (120 dB), three different prepulse/pulse pairings [prepulses at 74, 78, or 82 dB, followed by a 120 dB pulse with a 150-ms onset-onset interval], and no stimulus (F). Error bars are means ± SEMs (*, P < 0.05; and **, P < 0.01). Control (n=8) and Uba6NKO (n=6), all female).
(G-I) Reduced dendritic spines in neurons from amygdala (G) and the CA3 (H). The number of mushroom-shaped spines were determined per 10 μm in WT (n=4) and Uba6NKO mice (n=4). **P < 0.01. Error bars represent mean ± SEM. Seventy-five 10 μm sections were analyzed per mouse. Representative images are shown in (I).
As the impairment in contextual fear conditioning was confounded by the altered shock response, we submitted Uba6NKO mice to a Y-maze arena test, a distinct hippocampal-dependent task, which relies only on the spontaneous tendency of rodents to alternate arm entries. Arm entries were comparable between Uba6NKO and control mice (Figure 4C). However, the extent of spontaneous alternation was significantly reduced in Uba6NKO mice (Figure 4D), consistent with working memory performance impairment in Uba6 NKO mice.
As Uba6NKO mice displayed behavioral alterations suggestive of impaired emotional responses, we investigated their emotional responses using the startle sensitivity and pre-pulse inhibition test and the forced swim test. These models have been shown to be sensitive to amygdala and/or hippocampal dysfunction (Silverman et al., 2010). Uba6NKO mice demonstrated an exaggerated response to acoustic stimuli (Figure 4E) and subsequently impaired pre-pulse inhibition (Figure 4F). Thus, Uba6NKO mice display defects in learning and memory.
Reduced Dendritic Spine Density in Uba6NKO Mice
Given defects in CA3 and amygdala organization, as well as defects in behavior associated these regions of the brain of Uba6NKO mice, we employed Golgi staining to examine dendritic spine density. In the CA3 region, a 15% reduction in spine number was observed (p<0.01) (Figure 4H, I) while in the amygdala, a 45% reduction in spine number was found (p <0.01) (Figure 4G, I). The magnitude of loss in spine density is comparable to or greater than that seen previously in neurons from Shank3-deficient mice (Peca et al., 2011a). Thus, defects in dendritic spine number may contribute to the behavioral defects seen in Uba6NKO mice.
Increased Ube3a and Shank3 Levels in Uba6-deficient Amygdala
Given the role of Uba6-Use1 in controlling protein abundance and the defects seen in spine number, we surveyed a series of proteins previously linked to spine development or function for changes in abundance in the amygdala of Uba6NKO mice. No change in the abundance of Neuroligin3, Psd95, Gkap, Shank1, or Shank2 was observed in Uba6NKO amygdala extracts when compared with control Uba6Flox/Flox extracts (Fig 5A, B). Surprisingly, a marked increase in the abundance of Angelman's Syndrome protein and HECT-domain ubiquitin ligase Ube3a (average of 2.6-fold) as well as the PSD protein Shank3 (average of 4.2-fold) was observed in the amygdala of Uba6NKO (Figure 5A, B). UBE3A transcription has been shown to be activity dependent (Greer et al., 2010). Therefore, to rule out transcriptional effects, we examined UBE3A mRNA by qPCR and found no changes in mRNA abundance in the amygdala (Figure 5B), indicating that the increased levels of Ube3a are not due to altered transcription. This increase in Ube3a abundance was validated with amygdala extracts from 6 independent Uba6NKO mice (data not shown). Likewise, qPCR revealed no change in Shank3 transcription in Uba6NKO mice (Figure 5C), indicating a post-transcriptional effect.
Figure 5. Altered expression of Ube3a and Shank3 in the amygdala ofUba6NKOmice.
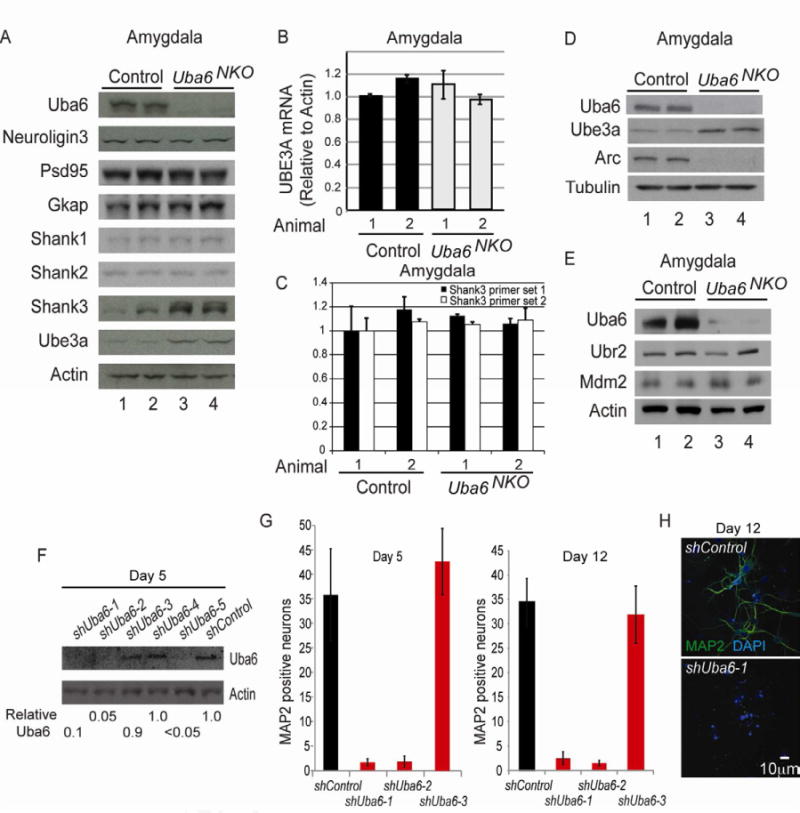
(A) Immunoblotting of amygdala extracts from Uba6NKO mice at 3 months of age with the indicated antibodies. Tubulin was used as controls. Extracts from two mice of each genotype were employed.
(B,C) qPCR analysis of Ube3a and Shank3 mRNA from the amygdala of two control and Uba6NKO mice. Data are mean ± SEM, with n=3 independent experiments. (D,E) Increased abundance of Ube3a in Uba6NKO mice correlates with loss of Arc expression in the amygdala in two animals (D), but without alterations in Ubr2 or Mdm2 (E). (F-H) Uba6 is required for hippocampal neuron viability in dissociated cultures in vitro. Hippocampal cells from E16.5 embryos were depleted with shRNAs targeting Uba6 for 5 or 12 days and the presence of Uba6 at day 5 (F) or the presence of neurons at day 5 (G) or 12 (G,H) determined using anti-MAP2 antibodies. For quantification, the number of neurons in 12 random 360,000 μm2 fields were determined from duplicate (5 h) or triplicate (12 h) experiments. Error bars in panel G are means of ± SEMs (n=3 independent experiments).
Previous studies indicate a role for Ube3a in the control of Arc levels in neurons, presumably through the canonical Uba1 pathway upstream of Ube3a (Greer et al., 2010). Arc is a synaptic protein that controls the internalization of AMPA type glutamate receptors via the endocytic pathway (Chowdhury et al., 2006; Rial Verde et al., 2006; Shepherd et al., 2006). Remarkably, loss of Uba6 in the amygdala correlates with a dramatic reduction in the steady-state abundance of Arc (Figure 5D), as expected if Ube3a is the E3 acting upon Arc. Previous work has also implicated Mdm2 in the control of Arc in cardiomyocytes (Foo et al., 2007), but we did not see a consistent alteration in the levels of either Mdm2 or the Uba6-Use1-dependent E3 Ubr2 in Uba6NKO amygdala extracts (Figure 5E).
Uba6-Use1 Controls Ube3a Stability in MEFs
The increase in Ube3a levels could reflect either a direct role for Uba6 in Ube3a turnover or an indirect effect on Ube3a stability through developmental defects. We initially attempted to directly test this model in cultured primary hippocampal neurons using RNAi. First, E16.5 hippocampal neurons were infected with 5 independent lentiviral vectors targeting Uba6 in order to identify the most efficacious hairpins for Uba6 depletion at day 5 post infection (Figure 5F). We then infected two potent shRNAs (shUba6-1 and shUba6-2) as well as an ineffective hairpin (shUba6-3) into E16.5 hippocampal neurons and assessed neuronal viability at 5 and 12 days post-infection. Unexpectedly, we found that Uba6 expression is required for viability of hippocampal neurons in culture. A dramatic reduction in the number of surviving neurons as determined by MAP2 staining was observed at day 5 and day 12 post-infection with shUba6-1 and shUba6-2, while the ineffective shRNA (shUba6-3) had no effect on neuronal viability when compared with a control shRNA (Figure 5F-H). The survival of glial cells in the culture was unaffected. Rare neurons remaining at day 5 and 12 post-infection were devoid of projections. Thus, the rapid loss of neurons and projections precluded an analysis of Uba6 activity and Ube3a abundance or spine density in this setting.
We therefore examined the abundance and stability of Ube3a in MEFs with or without Uba6 or Use1. Uba6Flox/Flox MEFs were incubated with or without adenoviral-Cre, and after 48 hours, the abundance of Ube3a was examined by immunoblotting. Ube3a abundance was increased by 3.7-fold in the absence of Uba6 (Figure 6A, lane 2), without an increase in Uba3a mRNA (Figure 6B). In MEFs, Ube3a has an apparent half-life of 8-12 hours in a cycloheximide (CHX) chase experiment (Figure 6C, lanes 1-5), consistent with previous reports in tissue culture cells (Cooper et al., 2004). In contrast, UBA6 deletion resulted in dramatic stabilization of Ube3a (Figure 6H, lanes 6-10), indicating that Uba6 is required turnover of Ube3a in MEFs.
Figure 6. The Uba6-Use1 pathway promotes Ube3a turnover in MEFs.
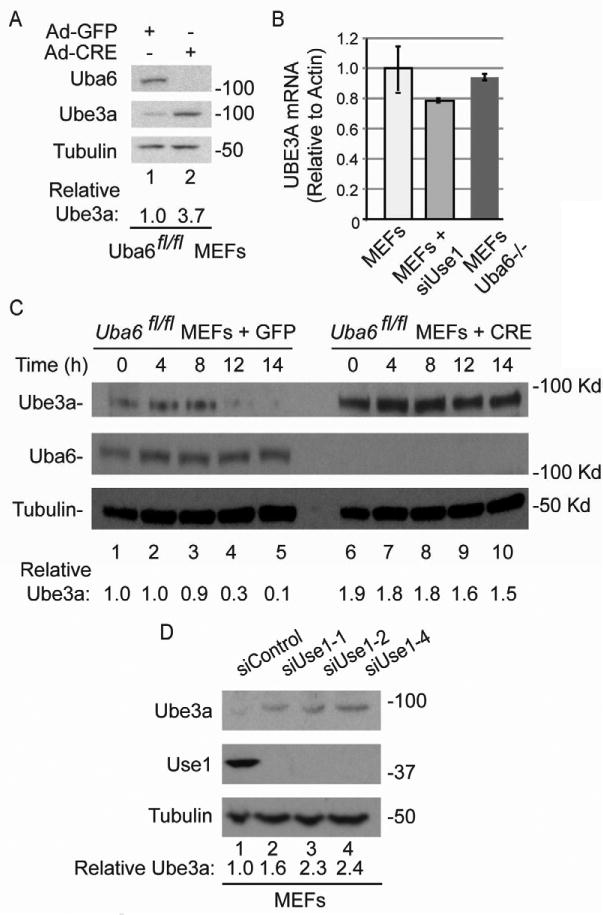
(A) Uba6Flox/Flox MEFs were incubated with Adenoviral-GFP (as control) or with Adenoviral-CRE and after 48h, cell extracts were analyzed by immunoblotting using the indicated antibodies. (B) qPCR analysis of Ube3a mRNA from Control MEFs, MEFs depleted of Use1 by RNAi, and Uba6Flox/Flox f MEFS in which Uba6 was deleted with Adenoviral-CRE. Error bars are mean ± SEM, with n=3 independent experiments.
(C) Uba6Flox/Flox MEFs were incubated with Adenoviral-GFP (as control) or with Adenoviral-CRE and after 48h, cells were treated with cycloheximide (CHX). Cell extracts harvested at the indicated times were analyzed by immunoblotting using the indicated antibodies.
(D) MEFs were transfected with either control siRNA or 3 individual siRNAs targeting Use1. After 72 h, cell extracts were analyzed by immunoblotting using the indicated antibodies.
The only E2 known to be specifically activated by Uba6 is Use1 (Jin et al., 2007). Depletion of Use1 with 3 independent siRNAs resulted in a 1.6-2.4-fold increase in the abundance of Ube3a (Figure 6D, lanes 1-4), independent of its mRNA (Figure 6B). Thus, Use1, like its activating enzyme Uba6, is required to maintain the levels of Ube3a in cultured MEFs. We were unable to detect endogenous expression of Shank3 or Arc in MEFs, precluding an analysis of any regulatory connection between these proteins and Uba6-Use1 in this system.
USE1 Ubiquitinates UBE3a in vitro via the HECT Domain
UBE3A, a HECT-domain ubiquitin ligase, employs UBCH7 as an E2 for charging of its active site cysteine and subsequent transfer to its substrates (Huang et al., 1999; Kumar et al., 1997; Cooper et al., 2004). We examined whether the human UBA6-USE1 cascade might be capable of transferring ubiquitin to lysine residues in human UBE3A, and if so, whether this required the active site cysteine residue of Ube3a. USE1 specifically promotes K48-dependent poly-ubiquitylation on its known substrate RGS4 in vitro (Lee et al., 2011), and would therefore be expected to promote turnover of UBE3A via the proteasome via K48-dependent ubiquitylation. We found that incubation of HA-UBE3A purified from HEK293 cells with bacterially-produced USE1 in the presence of recombinant UBA6 led to poly-ubiquitylation of HA-UBE3A but this reaction did not occur when the catalytic cysteine residue of HA-UBE3A (C840) was replaced by alanine (Figure 7A, lanes 5 and 7). Very large conjugates primarily retained in the stacking gel were produced, consistent with highly processive ubiquitin chain elongation. In addition, the active site residue of USE1 (C188) was required for UBE3A ubiquitylation, as its replacement by alanine abolished UBE3A ubiquitylation (data not shown). Poly-ubiquitylation occurred with wild-type ubiquitin and ubiquitin whose only lysine residue is a position 48 (Figure 7B lanes 7 and 8), but did not occur with lysine-free ubiquitin nor with K63-only ubiquitin (Figure 7B lanes 6 and 9). Both lysine-free ubiquitin and lysine-63 only ubiquitin produced a major product migrating at the position of mono-ubiquitylated UBE3A (Figure 7B, lanes 6 and 9). While this is consistent with a single major site of ubiquitylation, we cannot exclude the possibility that ubiquitylation occurs stochastically on distinct but single lysine residues.
Figure 7. Uba6-Use1 and Uba1-UbcH7 pathways ubiquitylate Ube3ain vitroand spatially control Ube3a abundance in MEFs.
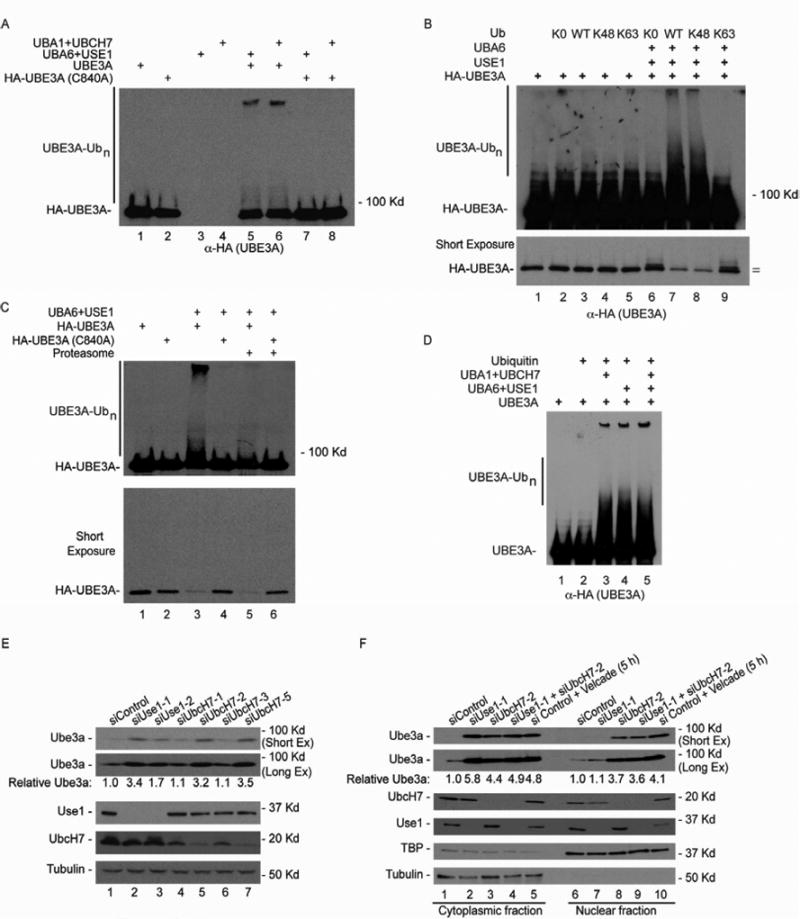
(A) HA-UBE3A (purified from 293T cells) with or without mutation of its active site cysteine residue (C840A) was incubated with ATP, ubiquitin, and UBA6-USE1 or UBA1-UBCH7.
(B) USE1 promotes K48-linked ubiquitylation of UBE3A. The “=” indicates the positions of HA-UBE3A and its mono-ubiquitin conjugate migrating more slowly.
(C) Ubiquitylated UBE3A is degraded by the 26S proteasome. The indicated reaction mixtures were treated with of proteasomes (360 nM) in which associated ubiquitin specific proteases were inhibited with ubiquitin-vinyl-sulfone.
(D) Patterns of UBE3A ubiquitylation in vitro are indistinguishable with UBA6-USE1, UBA1-UBCH7, or both pathways together.
(E) Use1 and UbcH7 independently function to control Ube3a abundance in MEFs. The indicated siRNAs were transfected into MEFs and the levels of the indicated proteins accessed after 72 hours using western blotting. Tubulin as used as a loading control.
(F) Spatially distinct control of Ube3a in MEFs via Uba6-Use1 and Uba1-UbcH7 pathways. As in (E), except that cytoplasmic and nuclear fractions were isolated and analyzed separately by immunoblotting.
Given that Use1 depletion in MEFs leads to stabilization of Ube3a, the expectation is that ubiquitylation of UBE3A in vitro would produce a substrate for the proteasome. Indeed, addition of purified human proteasomes to UBE3A ubiquitylation reactions led to a loss on UBE3A-polyubiquitylated conjugates, indicating that the conjugates formed by USE1 are sufficient to promote proteasomal degradation in vitro (Figure 7C, lanes 3 and 5). As expected, UBE3A levels were not decreased with the catalytically inactive form of UBE3A (C380A) was used (lane 6).
Uba6-Use1 and Uba1-UbcH7 function in parallel to control Ube3a abundance in MEFs
A previous study demonstrated that UBE3A functions with the canonical K48-specific E2 enzyme UBCH7 to both autoubiquitylate itself and to ubiquitylate its targets in vitro (Cooper et al., 2004). This finding raised the question of whether there was any relationship between the canonical and non-canonical pathways for regulation of UBE3A, or if the two pathways functioned in parallel in vivo. Consistent with previous work (Cooper et al., 2004), we initially found that addition of UBCH7 to UBE3A resulted in UBA3A ubiquitylation in vitro and this required the catalytic cysteine in UBA3A (Figure 7A, lanes 6 and 8). Addition of UBA1-UBCH7 and UBA6-USE1 together did not alter the pattern of reaction products (Figure 7D compare lanes 3-5) Thus, both UBA6-USE1 and UBA1-UBCH7 have the capacity to conjugate degradative signals on UBE3A. This scenario is reminiscent of that seen with N-end rule substrates Rgs4 and Rgs5, where both Uba6-Use1 and Uba1-Ube2a/b control substrate turnover in a spatially defined manner (Lee et al., 2011). To examine whether the two pathways can both contribute to Ube3a turnover, we transfected MEFs with 4 independent siRNAs targeting UbcH7, two of which resulted in significant depletion (Figure 7E, lanes 5 and 7). UbcH7 depletion resulted in a >3.2-fold increase in the steady-state abundance of Ube3a (Figure 7E, lanes 5 and 7), which was comparable to that seen with depletion of Use1 performed in parallel (lane2). To examine any spatial dependency on the two pathways for Ube3a turnover, we performed RNAi depletion of Use1 or UbcH7 or a combination of siRNAs in MEFs and subsequently isolated either cytoplasm or nuclear extracts, as determined by immunoblotting with cytoplasmic (Tubulin) or nuclear (TBP) markers (Figure 7F). Depletion of Use1 (lane 2) led to accumulation of Ube3a specifically in the cytoplasm (compare lanes 2 and 7), consistent with previous studies indicating that Uba6 is present only in the cytoplasm (Lee et al., 2011). The extent of stabilization was similar to that seen with addition of proteasome inhibitor Bortezomib (lane 5). In contrast, depletion of UbcH7 resulted in a strong accumulation of Ube3a in the nucleus and a partial accumulation in the cytoplasm relative to that seen with Bortezomib (compare lanes 3, 5, and 8). Co-depletion of Use1 and UbcH7 led to maximal stabilization in the cytoplasm relative to Bortezomib (compare lanes 7 and 8) but no further increase in the abundance of Ube3a in the nucleus compared to that seen with depletion of UbcH7 alone (compare lanes 8 and 9). These data indicate that both the canonical and non-canonical E1 activating systems are required for control of Ube3a abundance in MEFs, but in a spatially distinct manner.
Discussion
This study provides the first direct analysis of the role of the alternative arm of the ubiquitin activation and transfer cascade in mammalian neural development and function. Several phenotypes were associated with loss of neuronal Uba6 during embryonic development with Nestin-Cre. First, Uba6NKO mice are significantly smaller than control mice, and display significant post-natal lethality. A comprehensive pathological analysis of Uba6NKO mice failed to identify developmental abnormalities outside of the brain that could account for the lethality observed. However, we noticed an absence of food in several mice at necropsy, prompting us to test whether Uba6NKO animals were unable to access food in the standard location. Addition of chow to the bottom of the cage led to a substantial increase in viability, consistent with this notion. Interestingly, mice that are post-natally defective in the Ate1-dependent arm of the N-end rule pathway, which Uba6-Use1 participate in (Lee et al., 2011), are also smaller than control mice and display early lethality (Brower and Varshavsky, 2009). Second, Uba6NKO mice display numerous behavioral phenotypes, including a dramatic reduction in social interactions, an absence of response to fear conditioning paradigms, and evidence of hyperactivity with increased metabolic rate. Defects in Uba6NKO mice in fear conditioning may reflect altered architecture or signaling in the basolateral amygdala, which has been previously linked with fear centers in the brain (Davis and Whalen, 2001). Unlike control mice, Uba6NKO mice fail to habituate in the open field test, traveling significantly farther than control mice with fewer ventures into the center of the field. Hyperkinetic and metabolic rate phenotypes were also seen in Ate1-/- mice (Brower and Varshavsky, 2009). Finally, neurons in both the amygdala and the CA3 region of the hippocampus display a reduction in the number of dendritic spines, indicative of defects in synaptic connectivity in Uba6NKO animals. While these results were obtained through an analysis of female mice, a limited analysis of male mice in both the contextual fear conditioning test and the open field test gave similar results (Figure S3), indicating that the major phenotypes are not sex specific.
These observations provide the first evidence of a role for the alternative arm of the ubiquitin activation and transfer cascade in neurodevelopmental and behavioral disorders in mice, and are consistent with a role for Uba6 in the hippocampus and the amygdala. Currently, we cannot rule out a possible role for glial cells, as Nestin-Cre will also delete the Uba6 gene in these cells as well as neurons. We note however, that no obvious change in glial cell number or morphology was noted based on GFAP staining (data not shown). Further studies are required to understand the morphological defects in neurons in Uba6NKO mice, although we were unable to observe increases in apoptosis as an explanation for reduced neuronal density.
Given the role of Uba6 and its specific E2 conjugating enzyme Use1 in protein turnover, we examined the levels of several proteins previously linked with spine function in extracts from the amygdala of Uba6NKO mice. The levels of both Shank3 and Ube3a were increased in Uba6NKO, independently of changes in mRNA levels, but the levels of several other synaptic proteins was unaltered. This indicates that the changes in Ube3a and Shank3 expression observed do not reflect overall changes in the total levels of spine proteins generally. Uba6 was required for neuronal viability in primary cultures, precluding an analysis of direct regulation of Ube3a and Shank3 by Uba6-Use1. However, using MEFs, we found that deletion of Uba6 or depletion of Use1 led to stabilization of Ube3a, suggesting a potentially general role for Uba1-Use1 in post-translational regulation of Ube3a abundance. Moreover, human USE1 can promote K48-dependent ubiquitin chain formation on UBE3A in vitro in a manner that requires both the catalytic cysteine within the HECT-domain of UBE3A and the catalytic cysteine in USE1. The pattern of ubiquitylation seen with either ubiquitin lacking lysine residues (KO) or K63-only ubiquitin suggests that USE1 targets a single lysine residue in UBE3A, although stochastic ubiquitylation on multiple lysines cannot be excluded. Previous studies have demonstrated that UBCH7, the canonical E2 for substrate ubiquitylation by UBE3A, can also catalyze autoubiquitylation of UBE3A itself (Cooper et al., 2004), a result that was validated here. Thus, we considered the possibility that both Use1 and UbcH7 participate in the regulation of Ube3a levels, and found that depletion of UbcH7 in MEFs results in stabilization of Ube3a to an extent comparable with that seen upon depletion of Use1. Previous studies indicated that the Uba6-Use1 pathway is limited almost exclusively to the cytoplasm, and in this context, the Uba6-Use1 pathway promotes turnover of Rgs5 via the N-end rule pathway in the cytoplasm whereas the canonical arm of the N-end rule pathway involving Uba1-Ube2a/b occurs primarily in the nucleus. Similarly, Uba6-Use1 promotes Ube3a turnover specifically in the cytoplasm while Uba1-UbcH7 promotes Ube3a turnover preferentially in the nucleus and to a lesser extent in the cytoplasm in MEFs. Further work is required to understand precisely how these pathways cooperate to control the abundance of Ube3a and Shank3 in neurons.
Previous studies indicate an association of increased UBE3A copy number with ASD in humans (Glessner et al., 2009) and autism-like behavioral phenotypes in mice, including social deficits, repetitive behavior, and defects in communication (Smith et al., 2011). These UBE3A-dependent phenotypes may involve, at least in part, changes in the abundance of UBE3A targets including Arc (Greer et al., 2010). However, the phenotypes displayed by Uba6NKO and UBE3A-transgenic mice are distinct, as the latter has no obvious phenotypes in the open field or motor performance tests, and Uba6NKO mice lack repetitive behavior characteristic of mice carrying two additional alleles of UBE3A (Smith et al., 2011). While increased levels of Ube3a could impinge on the neurological and behavioral phenotypes seen in Uba6NKO animals, we think it more likely that the observed phenotypes represent the accumulation of multiple Uba6-dependent ubiquitylation targets, possibly including Shank3. Shank3-mutant mice display increased anxiety, defects in social interaction, and a decrease in spine number depending on the precise mutation (Peca et al., 2011a) . Moreover, because Uba6-Use1 also functions in the N-end rule pathway via interaction with the UBR family of E3s (Lee et al., 2011), additional targets downstream of UBRs, including RGS4 and RGS5 may also contribute to the phenotypes seen in Uba6NKO mice. Mutations in RGS4 have been linked to seizures and schizophrenia in humans (Lang et al., 2007), and UBR1 is mutated in Johanson-Blizzard syndrome, which is linked to mental retardation. Finally, Use1 can interact with other RING-E3s (van Wijk et al., 2009) and may therefore function in additional pathways that contribute to the observed phenotypes. A complete understanding of the targets whose accumulation underlies the phenotypes seen in Uba6NKO mice will likely require genetic ablation of candidate substrates in the context of Uba6 deletion followed by behavioral analysis. While the Uba6-Use1 pathway can activate the interferon inducible ubiquitin-like protein Fat10 (Chiu et al., 2007; Aichem et al., 2010; Lee et al., 2011), there is no evidence of a role for Fat10 in neuronal function and the only phenotype observed for Fat10-deficient mice is a defect in an immunological response to infectious agents (Canaan et al., 2006). We did not detect Fat10 in the amygdala although it was specifically detected in MEFs activated by TNFα and γ-interferon for 18 h (Figure S2E). Thus, we think it unlikely that the phenotypes observed here are linked with Fat10 activation via Uba6.
In summary, while the morphological effects seen in Uba6NKO mice define important roles for Uba6 in hippocampal and amygdala development, it is likely that the resulting behavioral effects reflect a complex interplay between multiple targets of the Uba6-Use1 pathway. Nevertheless, these studies indicate that both the canonical and non-canonical arms of the UPS activation and transfer system function independently, and likely cooperatively to control brain development and behavior.
Exprimental Procedures
Animal studies
Conditional Uba6+/Flox mice were produced by Ingenious Technologies (Stony Brook NY) on a hybrid C57/B6 background, the Neo cassette removed by flip-recombinase, and were backcrossed 5 times to B6. Nestin-Cre mice were obtained from Jackson Laboratory. Behavioral studies, quantitative PCR, immunohistochemistry, and Golgi staining are described in Supplemental Methods. All behavioral experiments were blind to genotype.
In vitro ubiquitylation assays
UBE3A was purified from 293T cells stably expressing HA-UBE3A using anti-HA resin, eluted with HA peptide, and dialyzed against 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM DTT. Ubiquitylation reactions (10 μl, 60 min, 37 °C) were performed in 50 mM Tris-HCl, pH 7.4, 50 mM NaCl, 5 mM MgCl2, 2 mM ATP, and 1 mM DTT and contained: 156 μM ubiquitin, 150 nM E1 (UBA6 or UBA1), 500 nM HA-UBE3A, and 5 μM E2 (USE1 or UBCH7) and analyzed by SDS-PAGE and immunoblotting. Where indicated, purified proteasomes (360 nM) (with deubiquitinase activity inhibited by addition of ubiquitin-vinylsulfone) was added.
Cell culture, transfection, and immunoblotting
MEFs were grown in DMEM supplemented with 10% heat inactivated fetal bovine serum. Deletion of the Uba6 gene in Uba6Flox/Flox MEFs was achieved using adenoviral-CRE recombinase (48 h). Cells were lysed in 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5% Nonidet P40, 1 mM EDTA with protease inhibitors (ROCHE) and subjected to immunoblotting (antibody sources provided in Supplemental Methods). siRNAs (30 nM, sequences provided in Supplemental Methods) were transfected with Lipofectin RNAiMax and cells analyzed 72 h post transfection. Brain tissue was homogenized in 50 mM Tris pH 7.4, 150 mM NaCl, 0.5% Triton X-100, 1 mM EDTA supplemented with protease inhibitors (Roche) and phosphatase inhibitors (Sigma), and cleared by centrifugation prior to immunoblotting.
Statistical Analysis
Results are presented as mean ± SEM and were assessed using Student's t tests and one- or two-way ANOVAs. Asterisks denote level of statistical significance (*, P<0.05, **, P<0. 01, ***, P<0.001) and P < 0.05 was considered significant.
Supplementary Material
Highlights
*Uba6 functions with Use1 as an alternative arm of the ubiquitin activation system
*Uba6 is required for neuronal morphogenesis in the hippocampus and amygdala
*Uba6 is required for proper mouse behavior
*Uba6-Use1 promotes ubiquitylation of Ube3a in vitro and Ube3a turnover in MEFs
Acknowledgments
We thank L.-H. Tsai and J. Graeff for advice, P. Howley for reagents, D. Finley for purified proteasomes, the Nikon Imaging Center at Harvard Medical School for microscopy support, and J. Bolen, J. Brownell, L. Dick, and B. Amidon for numerous discussions. This work was supported by grants to J.W.H. from Millennium Pharmaceuticals, Inc. and the NIH (AG011085).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aichem A, Pelzer C, Lukasiak S, Kalveram B, Sheppard PW, Rani N, Schmidtke G, Groettrup M. USE1 is a bispecific conjugating enzyme for ubiquitin and FAT10, which FAT10ylates itself in cis. Nat Commun. 2010;1:1–10. doi: 10.1038/ncomms1012. [DOI] [PubMed] [Google Scholar]
- Ang XL, Seeburg DP, Sheng M, Harper JW. Regulation of Postsynaptic RapGAP SPAR by Polo-like Kinase 2 and the SCF{beta}-TRCP Ubiquitin Ligase in Hippocampal Neurons. J Biol Chem. 2008;283:29424–29432. doi: 10.1074/jbc.M802475200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Schuman EM. Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature. 2006;441:1144–1148. doi: 10.1038/nature04769. [DOI] [PubMed] [Google Scholar]
- Bingol B, Sheng M. Deconstruction for reconstruction: the role of proteolysis in neural plasticity and disease. Neuron. 2011;69:22–32. doi: 10.1016/j.neuron.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Brower CS, Varshavsky A. Ablation of arginylation in the mouse N-end rule pathway: loss of fat, higher metabolic rate, damaged spermatogenesis, and neurological perturbations. PLoS ONE. 2009;4:e7757. doi: 10.1371/journal.pone.0007757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canaan A, Yu X, Booth CJ, Lian J, Lazar I, Gamfi SL, Castille K, Kohya N, Nakayama Y, Liu YC, et al. FAT10/diubiquitin-like protein-deficient mice exhibit minimal phenotypic differences. Mol Cell Biol. 2006;26:5180–5189. doi: 10.1128/MCB.00966-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YH, Sun Q, Chen ZJ. E1-L2 activates both ubiquitin and FAT10. Mol Cell. 2007;27:1014–1023. doi: 10.1016/j.molcel.2007.08.020. [DOI] [PubMed] [Google Scholar]
- Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron. 2006;52:445–459. doi: 10.1016/j.neuron.2006.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EM, Hudson AW, Amos J, Wagstaff J, Howley PM. Biochemical analysis of Angelman syndrome-associated mutations in the E3 ubiquitin ligase E6-associated protein. J Biol Chem. 2004;279:41208–41217. doi: 10.1074/jbc.M401302200. [DOI] [PubMed] [Google Scholar]
- Davis M, Whalen PJ. The amygdala: vigilance and emotion. Mol Psychiatry. 2001;6:13–34. doi: 10.1038/sj.mp.4000812. [DOI] [PubMed] [Google Scholar]
- Dindot SV, Antalffy BA, Bhattacharjee MB, Beaudet AL. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum Mol Genet. 2008;17:111–118. doi: 10.1093/hmg/ddm288. [DOI] [PubMed] [Google Scholar]
- Foo RS, Chan LK, Kitsis RN, Bennett MR. Ubiquitination and degradation of the anti-apoptotic protein ARC by MDM2. J Biol Chem. 2007;282:5529–5535. doi: 10.1074/jbc.M609046200. [DOI] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer PL, Hanayama R, Bloodgood BL, Mardinly AR, Lipton DM, Flavell SW, Kim TK, Griffith EC, Waldon Z, Maehr R, et al. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell. 2010;140:704–716. doi: 10.1016/j.cell.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde AN. The ubiquitin-proteasome pathway and synaptic plasticity. Learn Mem. 2010;17:314–327. doi: 10.1101/lm.1504010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Kinnucan E, Wang G, Beaudenon S, Howley PM, Huibregtse JM, Pavletich NP. Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2-E3 enzyme cascade. Science. 1999;286:1321–1326. doi: 10.1126/science.286.5443.1321. [DOI] [PubMed] [Google Scholar]
- Huibregtse JM, Scheffner M, Beaudenon S, Howley PM. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc Natl Acad Sci U S A. 1995;92:2563–2567. doi: 10.1073/pnas.92.7.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung AY, Sung CC, Brito IL, Sheng M. Degradation of postsynaptic scaffold GKAP and regulation of dendritic spine morphology by the TRIM3 ubiquitin ligase in rat hippocampal neurons. PLoS ONE. 2010;5:e9842. doi: 10.1371/journal.pone.0009842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Li X, Gygi SP, Harper JW. Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature. 2007;447:1135–1138. doi: 10.1038/nature05902. [DOI] [PubMed] [Google Scholar]
- Kuczera T, Stilling RM, Hsia HE, Bahari-Javan S, Irniger S, Nasmyth K, Sananbenesi F, Fischer A. The anaphase promoting complex is required for memory function in mice. Learn Mem. 2011;18:49–57. doi: 10.1101/lm.1998411. [DOI] [PubMed] [Google Scholar]
- Kumar S, Kao WH, Howley PM. Physical interaction between specific E2 and Hect E3 enzymes determines functional cooperativity. J Biol Chem. 1997;272:13548–13554. doi: 10.1074/jbc.272.21.13548. [DOI] [PubMed] [Google Scholar]
- Lang UE, Puls I, Muller DJ, Strutz-Seebohm N, Gallinat J. Molecular mechanisms of schizophrenia. Cellular physiology and biochemistry. International Journal of Experimental Cellular Physiology, Biochemistry, and Pharmacology. 2007;20:687–702. doi: 10.1159/000110430. [DOI] [PubMed] [Google Scholar]
- Lee PC, Sowa ME, Gygi SP, Harper JW. Alternative ubiquitin activation/conjugation cascades interact with N-end rule ubiquitin ligases to control degradation of RGS proteins. Mol Cell. 2011;43:392–405. doi: 10.1016/j.molcel.2011.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litterman N, Ikeuchi Y, Gallardo G, O'Connell BC, Sowa ME, Gygi SP, Harper JW, Bonni A. An OBSL1-Cul7Fbxw8 ubiquitin ligase signaling mechanism regulates Golgi morphology and dendrite patterning. PLoS Biol. 2011;9:e1001060. doi: 10.1371/journal.pbio.1001060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabb AM, Ehlers MD. Ubiquitination in postsynaptic function and plasticity. Annu Rev Cell Dev Biol. 2010;26:179–210. doi: 10.1146/annurev-cellbio-100109-104129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabb AM, Judson MC, Zylka MJ, Philpot BD. Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 2011;34:293–303. doi: 10.1016/j.tins.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak DT, Sheng M. Targeted protein degradation and synapse remodeling by an inducible protein kinase. Science. 2003;302:1368–1373. doi: 10.1126/science.1082475. [DOI] [PubMed] [Google Scholar]
- Peca J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011a;472:437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelzer C, Kassner I, Matentzoglu K, Singh RK, Wollscheid HP, Scheffner M, Schmidtke G, Groettrup M. UBE1L2, a novel E1 enzyme specific for ubiquitin. J Biol Chem. 2007;282:23010–23014. doi: 10.1074/jbc.C700111200. [DOI] [PubMed] [Google Scholar]
- Rial Verde EM, Lee-Osbourne J, Worley PF, Malinow R, Cline HT. Increased expression of the immediate-early gene arc/arg3.1 reduces AMPA receptor-mediated synaptic transmission. Neuron. 2006;52:461–474. doi: 10.1016/j.neuron.2006.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol. 2009;10:319–331. doi: 10.1038/nrm2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52:475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci. 2010;11:490–502. doi: 10.1038/nrn2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SE, Zhou YD, Zhang G, Jin Z, Stoppel DC, Anderson MP. Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci Transl Med. 2011;3:103ra197. doi: 10.1126/scitranslmed.3002627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RF, Spencer WA. Habituation: a model phenomenon for the study of neuronal substrates of behavior. Psychol Rev. 1966;73:16–43. doi: 10.1037/h0022681. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- van Wijk SJ, de Vries SJ, Kemmeren P, Huang A, Boelens R, Bonvin AM, Timmers HT. A comprehensive framework of E2-RING E3 interactions of the human ubiquitin-proteasome system. Mol Syst Biol. 2009;5:295. doi: 10.1038/msb.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Kim AH, Bonni A. The dynamic ubiquitin ligase duo: Cdh1-APC and Cdc20-APC regulate neuronal morphogenesis and connectivity. Curr Opin Neurobiol. 2010;20:92–99. doi: 10.1016/j.conb.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K, Riday TT, Condon KH, Roberts AC, Bernardo DR, Prakash R, Weinberg RJ, Ehlers MD, Philpot BD. Ube3a is required for experience-dependent maturation of the neocortex. Nature neuroscience. 2009;12:777–783. doi: 10.1038/nn.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Rape M. Building ubiquitin chains: E2 enzymes at work. Nat Rev Mol Cell Biol. 2009;10:755–764. doi: 10.1038/nrm2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenker M, Mayerle J, Lerch MM, Tagariello A, Zerres K, Durie PR, Beier M, Hulskamp G, Guzman C, Rehder H, et al. Deficiency of UBR1, a ubiquitin ligase of the N-end rule pathway, causes pancreatic dysfunction, malformations and mental retardation (Johanson-Blizzard syndrome) Nat Genet. 2005;37:1345–1350. doi: 10.1038/ng1681. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.