

Abstract
The steroid hormone, 17β-estradiol (E2) has been reported to enhance executive functions that are known to be mediated by the prefrontal cortex (PFC), although the underlying mechanisms remain unclear. To shed light on the potential mechanisms, we examined the effect of E2 in vivo upon spine density in the rat PFC and the somatosensory cortex (SSC), which has been implicated to be a transient storage site for information that can also contribute to working memory. The results revealed that E2 significantly enhanced the number of dendritic spines in both the SSC and PFC, as well as the expression of spinophilin. In vitro studies revealed further mechanistic insights by demonstrating that E2 enhanced AMPA GluR1 receptor expression and excitatory glutamatergic synapse formation in rat cortical neurons, without an effect upon inhibitory GABAergic synapse formation. Furthermore, E2 rapidly enhanced ERK and Akt activation in cortical neurons, and inhibitors of ERK and Akt activation significantly attenuated E2 induction of excitatory glutamatergic synapses. Administration of E2-BSA likewise significantly enhanced excitatory glutamatergic synapses in cortical neurons, and administration of an ER antagonist, ICI182,780 and a non-NMDA receptor antagonist (NBQX) significantly attenuated the effect of E2 upon enhancement of excitatory glutamatergic synapses, suggesting mediation by extranuclear estrogen receptors and involvement of non-NMDA receptor activation and signaling. As a whole, the studies demonstrate that E2 enhances spine density in both the PFC and SSC, and that E2 enhances excitatory glutamatergic synapse formation in cortical neurons via a rapid extranuclear ER-mediated signaling mechanism that involves up-regulation of GluR1 and mediation by Akt and ERK signaling pathways.
Keywords: Estradiol, nongenomic, synapse, synaptic plasticity, kinase
INTRODUCTION
The steroid hormone 17β-estradiol (E2) exerts regulatory effects in a variety of tissues in the body, including the cardiovascular system, bone, breast and brain [1–5]. Its effects in the brain are of considerable importance, as E2 can exert neuroprotective and cognitive-enhancing effects, as well as modulate homeostasis via neuroendocrine regulatory effects [1, 3, 5–8]. Much work has focused on the cerebral cortex and hippocampus as key E2 target areas in the brain for its neuroprotective and cognitive effects [2, 9–12]. Along these lines, E2 has been shown to profoundly protect the hippocampus CA1 region and cerebral cortex against ischemic damage in focal and global cerebral ischemia animal models and in in vitro hippocampal and cortical primary neuronal cell studies [1, 7, 13–18]. With respect to the cognitive enhancing effects of E2, the majority of studies to date have identified the hippocampus CA1 region as the primary site of E2 action, with E2 shown to up-regulate dendritic spine density in the hippocampus CA1 region and enhance hippocampal-mediated learning and memory [19–24].
Interestingly, the prefrontal cortex (PFC) and somatosensory cortex have also been demonstrated to play a role in learning and memory [25–27]. The PFC has been shown to have a critical role in executive function and is known to be crucial for choosing and implementing encoding strategies that organize the input to, and output from, the hippocampus, and the monitoring necessary for successful retrieval [26–28]. The somatosensory cortex, on the other hand, acts not only as a center for on-line sensory processing, but also as a transient storage site for information that also contributes to working memory [27, 29]. There is now evidence starting to appear which suggests that the cognitive effects of E2 may involve mediation at the level of the cerebral cortex in addition to the well-known actions in the hippocampus. For instance, Keenan et al [30] have demonstrated that postmenopausal women with hormone replacement therapy (HRT) outperformed women without HRT on executive function tests, e.g. tests requiring directed attention, inhibition of inappropriate responses, and cognitive set switching. This led the authors to suggest that the PFC is an important site of action for the cognitive effects of estrogen. Furthermore, Morrison and coworkers [31, 32] have demonstrated that E2 enhances synaptic density in the PFC of monkeys. Thus, in addition to its neuroprotective effects, E2 appears to also exert plasticity and cognitive effects in the cerebral cortex.
To enhance our understanding of the mechanisms underlying E2 actions in the cerebral cortex, we utilized in vivo studies to examine the effect of E2 on spine density in the PFC and SSC of adult ovariectomized rats, and in vitro studies to determine whether E2 regulates glutamatergic and/or GABAergic synapse formation in rat cortical neurons. The studies also assessed the role of rapid E2 signaling via several approaches, which included: 1) use of E2-BSA conjugates in vitro, 2) assessment of E2 activation of Akt and ERK signaling pathways, and 3) examination of the effect of inhibition of Akt and ERK signaling on E2-induced cortical synapse formation. The results of the study revealed that E2 enhanced spine density in both the PFC and SSC of the rat. E2 also enhanced excitatory gluatamatergic but not GABAergic synapse formation in rat cortical neurons in vitro, an effect that was shown to involve extranuclear ER-mediated rapid Akt and ERK signaling and up-regulation of AMPA GluR1 subunit.
EXPERIMENTAL
Drugs
17β-estradiol (E2), E2-6-BSA (17β-estradiol 6-O-carboxymethyloxime-BSA), activated charcoal, dextran and glutamate receptor antagonist (NBQX) were purchased from Sigma Chemical Co., MO, USA. PI3-K inhibitor (LY294002) and MAPK inhibitor (PD98059) were purchased from Alexis Biochemical’s, (South Dakota, USA). Time-release pellets containing 17β-E2 (0.5 mg/pellet, 21-day release) were purchased from Innovative Research of America (Sarasota, FL, USA).
Animals
All animal experiments were approved by the Institutional Committee for Animal Use in Research and Education. Sixty-day-old female Holtzman rats (Harlan, Indianapolis, IN) were individually housed with free access to food and water. Bilaterally ovariectomy (OVX) was performed as described previously by our laboratory [17]. Rats (n=5/group) were immediately implanted with time-release pellets containing placebo, E2 (0.5 mg/pellet, 21-day release). One week following OVX, animals were sacrificed by decapitation and cerebral cortices were dissected and processed for spine density measurements as described below. Serum E2 levels were also measured by a rat E2 ELISA kit (Alpco Diagnostics, Salem NH) and it was shown that the E2 pellets produce serum E2 levels of 301 ± 63.5 pg/ml.
Primary Neuronal Cultures and treatments
Cerebrocortical neurons were cultured from E18 rats, as described previously by our laboratory [33]. For qRT-PCR studies, DIV8-10 neurons were treated for 24 hours with vehicle, E2 or TMX, prior to RNA isolation and quantification of indicated genes. For the plasticity experiments, neurons were grown onto 12mm poly-D-lysine/laminin coated glass cover slips at 60–70K cells/well with 50% media change every third day. At 12–14 days in vitro (DIV), cultures were treated with either vehicle, E2 (10 nM) or E2-6-BSA for 24h at 37°C. Prior to use, all E2-BSA conjugated preparations were treated with charcoal (50mg/ml) and dextran (0.05mg/ml) for 30 min to remove unconjugated steroid moiety. To study the involvement of MAPK/ERK and AKT pathways in E2 actions on synaptic plasticity, cultures were treated with PD98059 (30μM) or LY294002 (20μM) respectively for 15 min at 37°C prior to the addition of E2. Similarly, to study the role of estrogen receptors (ERs) in E2 actions, cultured neurons were treated with a combined ERα/β antagonist, ICI-182780 (1μM) for 15min at 37°C prior to E2 treatment for 24hr. The role of AMPA type glutamate receptor was studied using the AMPA receptor antagonist, NBQX (30μM) for 15 min prior to E2 addition. The effects of various inhibitors/antagonists on the morphology and survival of neurons was determined using MAP2 staining before and after drugs treatments. Treatment of cultures with either MAPK/PI3K inhibitors or NBQX alone did not have any significant effect on cell viability or morphology.
Immunohistochemistry/Immunofluorescence staining
Neurons were treated with E2 in the presence or absence of inhibitors for 24 hours as described above and stained for synaptic proteins. For excitatory synapse measurements using AMPAGluR1, VGLUT1 and synaptophysin (SYN) triple staining, neurons were fixed with methanol for 10min at −20°C and blocked with 5% normal donkey serum for 1hr at room temperature. For inhibitory synapse measurements using GABAR-β2/β3, VGAT and SYN triple staining, neurons were fixed with 4% paraformaldehyde (PFA) containing 4% sucrose for 15min at room temperature, permeated with 0.4% Triton-X100 for 5 min followed by incubation with 5% normal donkey serum for 1hr at room temperature. The cultures were then incubated with primary antibodies overnight at 4°C using a combination of either mouse monoclonal anti-GluR1 (1:500; Santa Cruz Biotechnology, CA), guinea pig polyclonal anti-VGLUT1 (1:2000, Chemicon Inc., CA) and rabbit polyclonal anti-Syn (1:1000; Abcam, MA) or moue monoclonal anti- GABAR-β2/β3 (1:500; Chemicon Inc., CA), guinea pig polyclonal anti-VGAT (1:1500; Calbiochem, CA) and rabbit polyclonal anti-Syn (1:1000; Abcam, MA) all diluted in PBS. Cultures were then washed with PBS followed by incubation with a combination of the following fluorescent secondary antibodies: donkey anti-mouse AlexaFluor488 (1:500), donkey anti-rabbit AlexaFluor647 (1:500; Molecular Probe) and donkey anti-guinea pig TRITC (1:500: Jackson Lab.) for 1hr at room temperature. Cultures were washed with PBS for 3×5min and with dH2O for 1× 10 seconds and mounted using water based mounting medium containing antifading agents (Biomeda).
Golgi Impregnation
A modified version of the rapid Golgi method [34] was used to measure spine density. Animals were anaesthetized with ketamine/xylazine, and transcardially perfused with saline followed by fixation with 300ml solution of 2% PFA and 2.5% glutaraldehyde in 0.1M PBS, pH 7.4. Brains were removed and put in the same fixative for 2h at 4°C. Afterward, brains were incubated in a chromatin solution of 2% potassium dichromate and 5% glutaraldehyde for 36 h followed by metal impregnation in 0.75% silver nitrate solution for 24–36 h. All steps were performed in the dark and repeated twice in succession. Hemispheres were sectioned at 100 μM on a vibratome. Sections were dehydrated in graded ethanol, cleared in xylene and mounted using xylene based mounting medium. Individual neurons were traced under brightfield optics using Axiophot-2 microscope (Carl Zeiss, Germany) at 40×/0.75 oil objective lens and ~5–6 dendritic segments of selected neurons were traced with a 100×/1.33 NA oil-immersion objective with an additional 2.0X magnification lens. Approximately 10 individual neurons were selected from each group of animals, and from each neuron ~4–5 dendrites were analyzed for statistical analysis. The spines on the dendrites counted by three different individuals who were blind to the codes assign to each animal. The data was analyzed by one-way ANOVA, followed by Student-Neuman Kuel post-hoc test, and a p value <0.05 was considered significant.
Spinophilin Silver-Enhanced Nanogold Labeling
Animals were anesthetized with ketamine/xylazine, perfused, and fixed as described above. After fixation, the brains were removed and post-fixed in the same fixative overnight at 4°C. Brains were then cryoprotected in 30% sucrose prepared in 0.1M PB, pH 7.4 for ~96h and coronally sectioned at a thickness of 40μm on a cryostat microtome (Leica, Germany). Sections were stored in chronological order in a cryoprotectant solution (FD Neurotechnology, Baltimore, MD) before processing for immunohistochemistry. For silver gold labeling of spinophilin, sections were thoroughly rinsed first in PBS only followed by PBS containing 0.4% Triton-X100. Sections were then incubated in a solution containing 0.1% coldwater fish gelatin, 0.5% BSA, and 5% normal donkey serum diluted in PBS with 0.1% Triton X-100 at room temperature for 2h followed by incubation with a well characterized rabbit polyclonal anti-spinophilin antibody (1:10,000; Upstate Biotechnology) for 48h at 4°C. Sections were washed with PBS containing 0.1% Triton-X100 and incubated with a gold-labeled secondary antibody (1:200; donkey-anti-rabbit IgG, ultra-small; Aurion, Electron Microscopy Sciences, Hartfield, PA) for 3h at room temperature. Sections were washed with PBS containing 0.1% Triton-X100 and with PBS only, followed by post fixation with 2% glutaraldehyde in PBS for 10min at room temperature. After post-fixation, sections were washed in PBS followed by water and treated with silver enhancement reagent as recommended by the manufacturer. Sections were dehydrated in graded alcohol, cleared in xylene, mounted, and analyzed for spine density.
Stereology
The optical fractionator’s method was used to count the number of spines by the spinophilin silver gold labeling method. This procedure has been successfully used to demonstrate E2 induction of spinophilin-filled spines in hippocampus and prefrontal cortex of monkey brain [32]. All quantitative analyses were performed using a computer-assisted Stereology System equipped with an Olympus microscope, a computer-controlled motorized stage and the StereoInvestigator morphometry and stereology software (MicroBrightfield, Wiliston, VT, USA). This procedure provides an unbiased, efficient sampling technique and thus ultimately estimates the counts of puncta/cell population. Desired regions from prefrontal and somatosensory cortex were selected and the contours were traced at 4× magnification. Optical dissector counting frames were placed in a systematic, random fashion in the delineated regions of the sections, with constant interval in the x and y axes. The x and y distances between sampling fraction was (1.5 μm×1.5 μm)/(100 μm×150 μm). An oil-immersion objective (100×) and condenser were used for spinophilin-labeled-puncta counting. A 2 μm “guard zone” was place at the top surface of the section. Counting was performed with the optical dissector technique through a depth of 10 μm (height of dissector). The total spine counts (gold grains) in each region were calculated and used to determine differences between treatment groups.
Confocal Microscopy and Quantification of Synaptic Clusters
All fluorescent-labeled images were captured on a LSM 510 Meta confocal laser microscope using 63X objective lens. An argon/2 laser was used for the excitation of Alexa Fluor488 (EX max: 592nm) with emission filters set in the wavelength range 505–530nm. HeNe1 laser was used for the excitation of TRITC (EX max: 550nm) with emission filter set in the wavelength range 568–615nm. HeNe2 laser was used for the excitation of Alexa Fluor647 (EX max: 637nm) with emission filter set in the wavelength range 650–800nm. For each individual neuron, typically fourteen Z-stacks (optical slices) were collected at a thickness of 0.30–0.40 μm using optimum pinhole diameter. The Z-stacks were then converted into 3D projection image using LSM 5 Image Examiner (Carl Zeiss, Germany). For the quantification of synaptic protein clusters, Z stacks were transported to Volocity4 image analysis software (Improvision Inc., MA). For quantification of synaptic protein clusters and the density of excitatory/inhibitory synapse, approximately 15–16 neurons of identical morphology were selected from each treatment group for analysis and results were repeated in duplicate. First, neurons of interest were selected and scanned using a 63X objective lens with a further magnification using 2.5X zoom. The center axis of the frame was kept in the middle of the neuronal cell bodies and the whole neuron containing only primary dendrites and axon (under these conditions) was scanned using Z-series scanning generating nearly 12–14 Z-stacks. The Z-stacks were transferred to Volocity4 for counting of red (VGAT or VGLUT1), blue (SYN) or green (GluR1 or GABA) puncta/clusters. The Z-stacks were also converted into 3D images for counting of synapse (white puncta/clusters) manually.
Electron microscopy – spinophilin immunogold labeling
For pre-embedding gold labeling, vibratome sections obtained for light microscopy were cryoprotected in 30% sucrose in 0.1M PB, pH 7.4 for 48h. Subsequently sections were freeze-thawed using nitrogen to enhance the penetration of primary antibodies. Sections were then washed in PBS followed by incubation with primary rabbit anti-spinophilin antibodies (1:50,000, Upstate Biotechnology) for 48h at 4°C. After rinsing in PBS, sections were incubated overnight at 4°C with donkey-anti rabbit IgG coupled to 0.8 nm gold particles (1:100; Aurion). Afterwards, sections were rinsed in PBS and in 0.1M Na acetate (to remove phosphate and chloride ions), followed by silver enhancement with Intense S-EM (Electron Microscopy, PA). Following silver intensification, sections were washed with 0.1M sodium cacodylate buffer x3 for 15 min at room temperature. The tissue was then post-fixed in 4% osmium tetroxide for 1 hr. then rinsed in distilled water and dehydrated in a graded series of ethanol and cleared with 3 changes of propylene oxide. The tissue was infiltrated and polymerized in Embed 812 with Araldite 502 resin overnight in a 60-degree oven. Ultra thin sections were made and stained with uranyl acetate and lead citrate. The sections were examined and photographed using a JEOL 1010 transmission electron microscope.
Western blotting and ELISAs
For Western blot analysis, tissue was homogenized in RIPA buffer (1X Phosphate Buffered Saline, PBS), 1% IGEPAL CA-630 (Sigma Chemicals, St. Louis, MO), 0.5% sodium deoxycholate, 0.1% SDS) and protease inhibitors using a polytron homogenizer. After homogenization, samples were centrifuged at 10,000 rpm for 10 min at 4°C. The supernatant fraction was the total cell lysate. Protein concentration in the lysate was determined using a total protein measurement kit from Sigma Chemicals. Protein samples were denatured in sample buffer containing β-mercaptoethanol in 25mM Tris-glycine buffer and separated on a sodium dodecyl sulfate-polyacrylamide gel after loading equal amount of protein in each lane. Separated proteins were transferred to Immobilon P membrane (Milipore, USA) at 27 V for 15 h in 25 mM Tris-glycine buffer, pH 8.3, 10% methanol using a Transblot apparatus (Bio-Rad Laboratories, Inc., Hercules, CA). Following the transfer, the membranes were rinsed twice with T-TBS (20 mM Tris, 137 mM NaCl, 0.1% Tween-20) for 5 min each rinse and then incubated with 5% non-fat dry milk for 1 h at room temperature to block non specific/unbound surface. The membrane was incubated overnight with appropriate primary antibodies (at dilutions in the range 1:2000–40000). The membrane was then washed with T-TBS to remove unbound antibody, followed by incubation with secondary HRP-conjugated donkey anti-rabbit or anti-mouse IgG (Transduction Laboratories, San Diego, CA) for 1 h at room temperature. The signal was detected using an ECL detection kit (Amersham, UK) and the membranes were exposed to Kodak Biomax MR film. For MAPKinase assay, cells were lysed in RIPA buffer supplemented with PMSF, aprotinin, and NaVO4 and centrifuged at 12000g for 10min at 4°C. Supernatant was collected and protein concentration was determined using a modified Lowry assay. pMEK1/2S218/S222 and total MEK1/2 protein were determined using TiterZyme EIA kits (Assay Designs, Ann Arbor, MI). The pMEK1/2 EIA displays less than 0.1% cross-reactivity with pJNK, ERK2 and pERK2. Purified pMEK1/2 and MEK1 protein were used as positive controls. pERKT185/Y187 and total ERK1/2 levels was determined by EIA (Biosource, Camarillo, CA), which are up to 20 times more sensitive than Western blotting, with sensitivities of <0.8 U/mL and <16 pg/mL, respectively. Purified ERK protein and pERK standards were used as positive controls.
Biotinylation assay of surface proteins
Surface expression of postsynaptic synaptic proteins following E2 treatment was analyzed by the biotinylation method using a commercially available biotinylation kit (Pierce Biotechnology, Inc.). Twelve days old cultured cortical neurons were treated with E2 (10nM) for various durations at 37°C. Cells were then washed with PBS and incubated at 4°C with sulfo-NHS-SS-biotin reagent (Pierce Biotechnology, Inc.) for 30 min. Reaction was quenched using a quenching solution and the cells were washed twice with PBS followed by lyses with RIPA buffer (0.15 mM NaCl/0.05 mM Tris·HCl, pH 7.2/1% Triton X-100/1% sodium deoxycholate/0.1% SDS). The cell lysate was centrifuged at 12000g for 10min at 4°C and the supernatant was collected and analyzed for proteins concentration for Lowry (Pierce). Biotinylated proteins were precipitated with immobilized streptavidin beads as described by manufacturer’s protocol, and analyzed for AMPA GluR1, GABAAR2/3 and β-actin expression using Western blotting. Two controls were run simultaneously to verify the specificity of biotinylation reaction as suggested by the manufacturer’s protocols, and no specific or nonspecific proteins were detected in any of the controls analyzed.
RESULTS
Estrogen Enhances Spine Density in the PFC and SSC
Since the SSC acts not only as a center for on-line sensory processing, but also as a transient storage site for information that also contributes to working memory, we examined whether E2 can modulate spine density in the SSC of ovariectomized adult rats. Golgi staining was performed on SSC sections collected from OVX rats after one week of E2 treatment. Figure 1A&B shows representative Golgi staining results from layer II–III of SSC of vehicle- and E2-treated ovariectomized rats. Quantification of spine density revealed that E2 treatment significantly increased spine density as compared to vehicle-treated animals (Fig. 1C). To further confirm the changes in spine density as revealed by the Golgi staining results, we utilized the spinophilin immunogold labeling method, an alternative method to measure spine density. This method uses immunogold labeling for the spine-specific marker, spinophilin, and has been used previously by others to show that E2 enhances spine density in the PFC in the monkey [32]. In our studies, we used the method to analyze spine density changes in both the SSC and PFC of ovariectomized rats treated with placebo or E2. Representative photomicrographs are shown for spinophilin-immunogold labeling in the SSC (Fig. 2A) and PFC (Fig. 2B). Fig. 2C shows electron microscopy results demonstrating that spinophilin gold particles are localized at postsynaptic densities and spines. Quantification of the number of spines in the PFC and SSC are presented in Fig. 2D. As shown in Fig 2D, E2 significantly increased spine density in both the PFC and SSC, as compared to vehicle control, further confirming and extending the Golgi staining results. Fig. 2E shows Western blot results demonstrating that E2 increases spinophilin protein levels in both the SSC and PFC.
Figure 1. Spine density measurement by Golgi impregnation method.

(A) Shows a light micrographic picture of a pyramidal neuron from layer II–III of the somatosensory cortex (inset grey box) stained by Golgi-impregnation methods. (B) Shows light microscopic pictures of individual dendrites bearing spines from Veh and E2 treated animals. Approximately 10 individual neurons were selected from each group of animals and from each neuron ~4–5 dendrites were analyzed for statistical analysis. (C) Shows statistical analysis of spine numbers per 50μm length of the dendrite from Veh (light dotted bars) and E2 treated (dark dotted bars) animals (N=3 from each group). Statistical significance was determined by One Way ANOVA using Newman Keuls post test and ** = P<0.001.
Figure 2. Spine density measurement by spinophilin-immunogold labeling.

(A and B) Show light microscopic pictures of silver-enhanced immunogold labeling of spinophilin from somatosensory cortex (SSC) and prefrontal cortex (PFC) of Veh and E2 treated animals. (C) Shows high resolution electron micrograph of spinophilin positive gold particles from SSC area. Spinophilin was specifically localized in the postsynaptic spines/dendrites (PSDs) as shown by various arrows but not in the presynaptic terminals (PSs) as shown by asterisks. Spine density in the SSC and PFC was determined by measuring spinophilin labeled gold particles from Veh and E2 treated animals using Optical fractionator’s method. (D) Shows statistical bar diagram of spine density from the SSC and PFC from Veh (light grey bars) and E2 treated (dark grey bars) animals. Significance was determined by One Way ANOVA using Newman-Keuls post test. As can be seen from the bar diagram (D), E2-treated animals (dark grey bars) show significantly higher spine density in both SSC (P<0.001) and PFC (P<0.002) compared to Veh-treated animals (light grey bars). (E) Western blot results showing E2 increases expression of spinophilin in the SSC and PFC. Scale bars represent 5μm in (A and B) and 200 nm in (C).
Estrogen Enhances Excitatory Glutamatergic Synapse Formation in Cortical Neurons
We next sought to examine whether E2 enhances excitatory and/or inhibitory synapse formation in cultured embryonic rat cortical neurons in vitro. Toward this end, we first performed triple-immunoflorescent staining for excitatory synaptic proteins – e.g. for the postsynaptic proteins, synaptophysin (SYN) and AMPA GluR1 receptor (GluR1), and the presynaptic vesicular glutamate transporter protein-1 (VGLUT1) (Fig. 3 A&B), and for inhibitory synaptic proteins – postsynaptic gamma-aminobutyric acid receptor subunit (GABAAR2/3), synaptophysin (SYN) and presynaptic vesicular GABA transporter protein (VGAT) (Fig. 3C). As shown in Fig. 3 A&B, the merged figures show SYN, GluR1 and VGLUT1-positive clusters on dendrites of the cortical neuron, indicating excitatory synapses. Likewise, Fig. 3C shows SYN, GABAAR2/3, and VGAT-positive clusters on dendrites, indicating inhibitory synapses. In addition, biotinylation of surface proteins of the cortical neurons confirmed GluR1 and GABAAR2/3 receptor localization on the membrane of cortical neurons (Fig. 3D). We next used the triple-staining for excitatory and inhibitory synapses and biotinylated surface protein Western blotting procedures to determine whether E2 regulates excitatory and/or inhibitory synapses in cultured cortical neurons at 24h post-treatment, and whether E2 enhances GluR1 or GABAAR2/3 protein expression at the surface (membrane) of cortical neurons. As shown in Fig. 4 A&B, E2 treatment significantly increased SYN, GluR1 and VGLUT1 clusters, as well as the number of excitatory synapses in cortical neurons. In contrast, E2 did not significantly increase GABAAR2/3 and VGAT clusters, nor did E2 increase the number of inhibitory synapses in cortical neurons (Fig. 4C&D). Fig. 4E shows confirmation by biotinylation studies and Western blot analysis that E2 increased the level of GluR1, but not GABAAR2/3 on the surface of cortical neurons in vitro.
Figure 3. Organization of excitatory and inhibitory synaptic proteins in cultured rat cortical neurons.
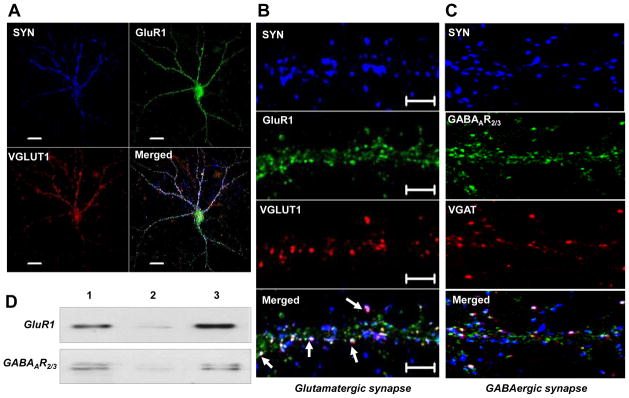
(A) 3D Confocal images of 14DIV cortical neurons showing expression of excitatory synaptic proteins; synaptophysin (SYN; blue), AMPA GluR1 (green), presynaptic vesicular glutamate transporter protein-1 (VGLUT1; red) and their colocalization (Merged) on the synapse/dendrites. (B) An enlarged view of a dendrite showing organization of SYN (blue), GluR1 (green), VGLUT1 (red) positive clusters and their colocalization (Merged) at the synapse (white arrows). (C) Organization of inhibitory synapse where; SYN (blue), postsynaptic γ-animobutyric acid receptor subunit (GABAAR2/3; green) and vesicular GABA transporter protein (VGAT; red) positive clusters are expressed along the dendrite and spines and colocalize at the synapse (white arrows). (D) Shows surface expression of GluR1 and GABAAR2/3 receptors detected by biotinylation method. Cultured neurons were biotinylated and homogenized as describe in method section. Total cell homogenate either directly leaded on to SDS gel (Lane 1) or after passing through a column of resin without conjugated anti-biotin antibodies (Lane 2) or after conjugation with anti-biotin antibodies (Lane 3). After washing protein were eluted from the column and detected by Western blotting. Several controls were also run (not shown) to verify the specificity of the biotinylation reaction as mentioned in the method section of the text.
Figure 4. E2 enhances excitatory synapse but not inhibitory synapse formation in cultured cortical neurons.

(A) Shows statistical analysis of synaptic proteins; SYN, VGLUT1 and AMPA GluR1 receptor and the density of AMPA GluR1 positive synapses compared to Veh treated group (light grey bars). Statistical analysis performed by One Way ANOVA with Newman Keuls post test, revealed that 24 hr E2 treatment (dark grey bars) significantly increases (***P<0.0001 vs. Veh) clustering of all the synaptic proteins and the number of excitatory glutamatergic synapse. (B) Shows confocal images of 14DIV cortical neurons showing excitatory synapse (white arrows) were significantly increased in E2 (24hr) treated neurons (right panel) compared to Veh treated neurons (left panel). (C) Statistical analysis, performed as in (B), revealed that 24 hr E2 treatment (dark grey bars) significantly increases (P<0.0001 vs. Veh) SYN positive clusters but does not increase (P>0.05 vs. Veh) VGAT or GABAAR2/3 positive clusters and the density of inhibitory GABAergic synapse compared to Veh treated neurons (light grey bars). (D) Shows confocal images of 14DIV cortical neurons showing inhibitory synapse (white arrows) were not increased by 24h E2 treatment (right panel) compared to Veh treated neurons (left panel). (E) Western blot results show that surface expression of AMPA GluR1 receptor was highly increased (upper panel) by 24hr E2 treatment compared to vehicle, whereas; surface expression of GABAAR2/3 receptor subunit (lower panel) was not changed by 24h E2 treatment. Scale bar represent 10 μm in A and C.
Extranuclear Estrogen Receptors and Rapid Kinase Signaling Mediate E2 Plasticity Effects in Cortical Neurons
It is well known that E2 can also have rapid effects at the cell membrane to activate various kinase signaling pathways, which could participate in its plasticity effects. We thus examined whether E2 exerts rapid signaling to enhance ERK and Akt cell signaling pathways and whether E2 can rapidly enhance GluR1 surface protein levels in cultured cortical neurons in vitro. As shown in Fig. 5A, Western blot analysis revealed that E2 rapidly increased phosphorylation of ERK and Akt in cultured cortical neurons, with peak levels observed at 2–7 min for pERK, and 10–30 min for pAkt. The cell impermeable, E2-BSA analogue also increased pERK at 5 min post treatment (Fig 5A). ELISA results for pERK also show a rapid enhancement by E2 treatment of pERK levels in cortical neurons and that pretreatment with the MEK inhibitor, PD98059 completely prevented the E2 enhancement of ERK activation in cortical neurons (Fig. 5B). The effects of E2 on ERK activation involved estrogen receptor activation, as treatment with the ER antagonist, ICI182,780 strongly attenuated both ERK activation (Fig. 5C) and MEK activation (Fig. 5D). Finally, biotinylation studies further revealed that E2 rapidly enhances surface GluR1 protein levels in cultured cortical neurons from 1–3h after E2 treatment (Fig. 5E). Fig. 6 shows the effect of various cell signaling pathway inhibitors (MEK inhibitor, PD98059; PI3K inhibitor, LY294002) or receptor antagonists (ER antagonist, ICI192,780; AMPA receptor antagonist, NBQX) upon the ability of E2 or the cell impermeable E2 conjugate, E2-BSA to enhance excitatory synapse formation in cultured cortical neurons when examined at 24h after E2/E2-BSA treatment. Representative photomicrographs are presented in Fig 6A, while the average number of SYN (B), VGLUT1 (C) and GLuR1 (D) clusters/neurons and excitatory synapse density (E&F) is presented in Fig. 6 B–F. The results show that E2 enhancement of SYN, VGLUT1 and GluR1 clusters (panels B–D) and excitatory synapse density (E2-induced, panel E; E2-BSA-induced, panel F) is significantly inhibited by pretreatment with inhibitors of the MEK-ERK, PI3K-Akt signaling pathway, or by estrogen receptor or AMPA receptor antagonists (Fig 6 B–F).
Figure 5. E2 rapidly enhances activation of ERKs and Akt, as well as enhances surface expression of AMPA GluR1 in cultured cortical neurons.

(A) Western blot shows that E2 treatment rapidly enhanced phosphorylation of ERKs1/2 (upper lane) and Akt (lower lane) in the cultured neurons. E2-BSA (10nM) also induces rapid phosphorylation of ERKs in cultured neurons (upper lane last band). Various numbers in the upper lane indicate the duration of E2 treatments. (B) A rapid effect of E2 on the phosphorylation of ERKs (light grey bars) was also detected by ELISA which was identical to the pattern observed by Western blot (above). E2-induced ERKs phosphorylation was completely blocked by prior treatment of neuronal cultures with the MEK inhibitor PD98059 (dark grey bars). (C) Rapid effects of E2 on ERKs activation was estrogen receptor-mediated as pretreatment of cultured neuron by ICI-182780, an estrogen receptor antagonist, completely blocked E2-induced rapid activation of ERKs. (D) Pretreatment of cultured neurons with ICI-182780 also blocked E2-induced activation of MEK. (E) E2 also rapidly enhances surface expression of AMPA GluR1 in cultured neurons as detected by the biotinylation method. The numbers indicates time (1h-3h) of E2 treatment followed by biotinylation.
Figure 6. E2-induced excitatory synapse formation involves mediation by estrogen receptors and AMPA receptors, as well as ERK and Akt signaling pathways.

(A) shows representative confocal images of 14DIV cortical neurons after various treatment conditions: Veh (treated with vehicle for 24h), E2 (treated with 10nM E2 for 24h), PD+E2 (treated with 30μM PD98059 for 20min followed by 10nM E2 for 24h), LY+E2 (treated with 15μM LY294002 for 20min followed 10nM E2 for 24h), ICI182,780+E2 (treated with 1μM ICI-182780 for 20min followed 10nM E2 for 24h), NBQX +E2 (treated with 30μM NBQX for 20min followed by 10nM E2 for 24h). (B–F) show statistical analysis of the average number of clusters/10μm for: SYN (B), VGLUT1 (C), GluR1 (D), and excitatory synapses (E). The effect of E2-BSA upon excitatory synapses is also shown in panel F. **P < 0.001 vs. vehicle control, and *P <0.05–0.001 vs. E2.
DISCUSSION
Using both in vivo and in vitro approaches, the current study demonstrates that E2 enhances synaptic plasticity in the adult ovariectomized female PFC and SSC and synapse formation in cultured embryonic cortical neurons, and sheds light on the mechanisms that potentially underlie the effects. Both classical Golgi staining and spinophilin nanogold particle immunostaining with stereological analysis were used to determine changes in spine density in the PFC and SSC in vivo. Both approaches demonstrated a significant enhancement of spine density in the PFC and SSC by E2 treatment. It should be noted that the E2 pellet used in our in vivo studies produces high supraphysiological serum E2 levels. However, our in vitro studies which used a low physiologically-relevant dose of E2 confirmed an enhancing effect of E2 on synaptic plasticity in rat cortical neurons. Our finding of E2 enhancement of spine density in the rat PFC is consistent with previous reports in the monkey, where E2 also enhanced PFC spine density [31, 32]. With respect to other studies on E2 and SSC plasticity regulation, Horvath et al have previously reported that E2 can increase cholinergic fiber density in the SSC following excitotoxic lesion of the rat nucleus basalis magnocellularis [35]. Furthermore, a critical role for E2 in regulating SSC neuron development and survival has also been suggested based on the fact that ERβ knockout mice display atrophy of the SSC region [36]. With respect to functional significance of the cortical plasticity effects of E2, it has been well documented that E2 can enhance working memory and executive function [30, 37–40], key cognitive functions known to require PFC mediation. E2 has also been implicated to modulate sensory processing in the SSC, including such sensory modalities as auditory, pain and visual cues [41–43].
In the current study, in vitro studies revealed that E2 enhances excitatory glutamatergic synapses, but not inhibitory GABAergic synapses in cultured cortical neurons. Furthermore, immunocytochemical and biotinylation studies revealed that E2 up-regulated AMPA GluR1 receptor subunit at the membrane of cortical neurons in vitro, an effect that occurred rapidly - within 1–3 hours. The mechanisms underlying E2 up-regulation of GluR1 expression at the membrane is unclear. However, previous work showed that E2 can similarly increase membrane GluR1 levels in hippocampal slices [44], and that the GluR1 enhancing effect [44] involved an increase in actin polymerization via a Rho-ROCK-LIM kinase-cofilin pathway [45]. It is possible that a similar mechanism may underlie the increased membrane GluR1 levels and plasticity effects we observed in the cerebral cortex, although this remains to be determined.
The results of our study further revealed that the E2 up-regulation of GluR1 and excitatory synapses in the cerebral cortex involved estrogen receptor (ER)-mediation, as it was blocked by the ER antagonist, ICI182,780. Furthermore, our study provided evidence that an extranuclear ER may help mediate the E2 effects upon increased excitatory synapse density, as the cell impermeable E2 conjugate, E2-BSA was also capable of increasing excitatory synapse density in the cultured cortical neurons. It should be mentioned that some studies suggest that E2-BSA may not exclusively activate extranuclear ER and that it may also activate nuclear ER and genomic signaling due to non-conjugated free E2 present in the conjugate reagent. However, we filtered our E2-BSA with charcoal to remove free unconjugated E2 prior to use. Furthermore, the effects of E2 and E2-BSA on excitatory synapse density were blocked by inhibitors of MAPK and PI3K-Akt kinase signaling pathways, as well as by an AMPA receptor antagonist, which suggests that rapid kinase signaling pathways and AMPA receptors play an important role in mediating E2 synaptic plasticity effects in the cortex. While the E2 plasticity effects appeared to be ER-mediated, it remains unclear which subtype of ER (ERα, ERβ or GPER1) mediates the E2 plasticity effects in the cerebral cortex. Previous studies in the hippocampus have suggested that ERβ may play a key role in mediating E2 effects upon synaptic plasticity, rapid release of glutamate and hippocampal-dependent cognitive function [46, 47]. However, other studies using ER selective agonists have implicated both ERα and ERβ in mediating E2 plasticity effects in the hippocampus [44, 48]. Since ERβ is expressed at a much higher level than ERα in the cerebral cortex, it is possible it may mediate E2 plasticity effects in the cortex. Further studies are needed to address this question. To our knowledge, there are no studies addressing the potential role of GPER1/GPR30 in mediating E2-induced synaptic plasticity effects in the brain, and thus studies to address its potential role are also needed.
Finally, our study found that E2 increased excitatory glutamatergic synapses but not GABAergic inhibitory synapses in rat cortical neurons in vitro. One caveat to consider is that these studies were performed in vitro in cultured embryonic cortical neurons. Thus, one cannot rule out that E2 may exert regulation of GABAergic inhibitory synapses in vivo and/or in adult animals. Indeed, we recently reported that on proestrus, when E2 levels are high in the blood, excitatory glutamatergic synapses on GnRH neurons are increased in the adult rat hypothalamus, while GABAergic inhibitory synapses on GnRH neurons are decreased [49]. Clearly, additional in vivo studies will be needed to address this issue more fully in the cerebral cortex. Nevertheless, it should be noted that our in vitro findings of increased glutamatergic but not GABAergic synapses in cortical neurons following E2 treatment is consistent with previous work demonstrating that E2 can enhance excitatory synaptic neurotransmission in the brain [24, 44, 45]. It is also consistent with Affymetrix microarray results from our laboratory, which revealed that in vivo E2 treatment enhances many genes in the rat cerebral cortex involved in excitatory glutamatergic neurotransmission, including GluR1, GLT-1, GLAST, PSD-93, SAP-97, SAP-102, synaptotagmin IV, and syntaxin (Dhandapani K, Khan MM, Wade M, Brann D, unpublished data). The fact that E2 enhances many genes implicated in synaptic plasticity suggests that both nongenomic and genomic signaling effects of E2 are likely to play a key role in mediating its synaptic plasticity effects in the cerebral cortex.
In conclusion, the current study advances our understanding of E2 effects upon synaptic plasticity in the rat PFC and SSC, and provides evidence for a rapid extranuclear ER-mediated signaling mechanism for E2 enhancement of cortical excitatory synapses. As a whole, the studies add to a growing literature suggesting that E2 has important plasticity-enhancing effects in the cerebral cortex, and may help explain previous reports of E2 enhancement of cortical functions, such as executive function and sensory processing.
Highlights.
Estradiol enhanced spine density in the prefrontal and somatosensory cortex
Estradiol increased excitatory glutamatergic synapse formation in cortical neurons
Extranuclear estrogen receptor signaling mediates estradiol effects on plasticity
Acknowledgments
This research was supported by a Research Grant (NS050730) from the National Institutes of Neurological Disorders and Stroke, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jover-Mengual T, Zukin RS, Etgen AM. MAPK signaling is critical to estradiol protection of CA1 neurons in global ischemia. Endocrinology. 2007;148:1131–43. doi: 10.1210/en.2006-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brann DW, Dhandapani K, Wakade C, Mahesh VB, Khan MM. Neurotrophic and neuroprotective actions of estrogen: basic mechanisms and clinical implications. Steroids. 2007;72:381–405. doi: 10.1016/j.steroids.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gibson CL, Gray LJ, Murphy SP, Bath PM. Estrogens and experimental ischemic stroke: a systematic review. J Cereb Blood Flow Metab. 2006;26:1103–13. doi: 10.1038/sj.jcbfm.9600270. [DOI] [PubMed] [Google Scholar]
- 4.Zhang YQ, Shi J, Rajakumar G, Day AL, Simpkins JW. Effects of gender and estradiol treatment on focal brain ischemia. Brain Res. 1998;784:321–4. doi: 10.1016/s0006-8993(97)00502-7. [DOI] [PubMed] [Google Scholar]
- 5.Kelly MJ, Qiu J, Ronnekleiv OK. Estrogen signaling in the hypothalamus. Vitam Horm. 2005;71:123–45. doi: 10.1016/S0083-6729(05)71005-0. [DOI] [PubMed] [Google Scholar]
- 6.Ishii H, Tsurugizawa T, Ogiue-Ikeda M, Asashima M, Mukai H, Murakami G, et al. Local production of sex hormones and their modulation of hippocampal synaptic plasticity. Neuroscientist. 2007;13:323–34. doi: 10.1177/10738584070130040601. [DOI] [PubMed] [Google Scholar]
- 7.Jover T, Tanaka H, Calderone A, Oguro K, Bennett MV, Etgen AM, et al. Estrogen protects against global ischemia-induced neuronal death and prevents activation of apoptotic signaling cascades in the hippocampal CA1. J Neurosci. 2002;22:2115–24. doi: 10.1523/JNEUROSCI.22-06-02115.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, et al. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci U S A. 2001;98:1952–7. doi: 10.1073/pnas.041483198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maki PM. Estrogen effects on the hippocampus and frontal lobes. Int J Fertil Womens Med. 2005;50:67–71. [PubMed] [Google Scholar]
- 10.Simpkins JW, Singh M. More than a decade of estrogen neuroprotection. Alzheimers Dement. 2008;4:S131–6. doi: 10.1016/j.jalz.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 11.Dumitriu D, Rapp PR, McEwen BS, Morrison JH. Estrogen and the aging brain: an elixir for the weary cortical network. Ann N Y Acad Sci. 2010;1204:104–12. doi: 10.1111/j.1749-6632.2010.05529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maki PM. Hormone therapy and cognitive function: is there a critical period for benefit? Neuroscience. 2006;138:1027–30. doi: 10.1016/j.neuroscience.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 13.Woolley CS. Acute effects of estrogen on neuronal physiology. Annu Rev Pharmacol Toxicol. 2007;47:657–80. doi: 10.1146/annurev.pharmtox.47.120505.105219. [DOI] [PubMed] [Google Scholar]
- 14.Merchenthaler I, Dellovade TL, Shughrue PJ. Neuroprotection by estrogen in animal models of global and focal ischemia. Ann N Y Acad Sci. 2003;1007:89–100. doi: 10.1196/annals.1286.009. [DOI] [PubMed] [Google Scholar]
- 15.Lebesgue D, Chevaleyre V, Zukin RS, Etgen AM. Estradiol rescues neurons from global ischemia-induced cell death: multiple cellular pathways of neuroprotection. Steroids. 2009;74:555–61. doi: 10.1016/j.steroids.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simpkins JW, Rajakumar G, Zhang YQ, Simpkins CE, Greenwald D, Yu CJ, et al. Estrogens may reduce mortality and ischemic damage caused by middle cerebral artery occlusion in the female rat. J Neurosurg. 1997;87:724–30. doi: 10.3171/jns.1997.87.5.0724. [DOI] [PubMed] [Google Scholar]
- 17.Zhang QG, Raz L, Wang R, Han D, De Sevilla L, Yang F, et al. Estrogen attenuates ischemic oxidative damage via an estrogen receptor alpha-mediated inhibition of NADPH oxidase activation. J Neurosci. 2009;29:13823–36. doi: 10.1523/JNEUROSCI.3574-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang QG, Wang R, Khan M, Mahesh V, Brann DW. Role of Dickkopf-1, an antagonist of the Wnt/beta-catenin signaling pathway, in estrogen-induced neuroprotection and attenuation of tau phosphorylation. J Neurosci. 2008;28:8430–41. doi: 10.1523/JNEUROSCI.2752-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woolley CS, Weiland NG, McEwen BS, Schwartzkroin PA. Estradiol increases the sensitivity of hippocampal CA1 pyramidal cells to NMDA receptor-mediated synaptic input: correlation with dendritic spine density. J Neurosci. 1997;17:1848–59. doi: 10.1523/JNEUROSCI.17-05-01848.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McEwen B, Akama K, Alves S, Brake WG, Bulloch K, Lee S, et al. Tracking the estrogen receptor in neurons: implications for estrogen-induced synapse formation. Proc Natl Acad Sci U S A. 2001;98:7093–100. doi: 10.1073/pnas.121146898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McEwen BS. Invited review: Estrogens effects on the brain: multiple sites and molecular mechanisms. J Appl Physiol. 2001;91:2785–801. doi: 10.1152/jappl.2001.91.6.2785. [DOI] [PubMed] [Google Scholar]
- 22.Li C, Brake WG, Romeo RD, Dunlop JC, Gordon M, Buzescu R, et al. Estrogen alters hippocampal dendritic spine shape and enhances synaptic protein immunoreactivity and spatial memory in female mice. Proc Natl Acad Sci U S A. 2004;101:2185–90. doi: 10.1073/pnas.0307313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spencer JL, Waters EM, Romeo RD, Wood GE, Milner TA, McEwen BS. Uncovering the mechanisms of estrogen effects on hippocampal function. Front Neuroendocrinol. 2008;29:219–37. doi: 10.1016/j.yfrne.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smejkalova T, Woolley CS. Estradiol acutely potentiates hippocampal excitatory synaptic transmission through a presynaptic mechanism. J Neurosci. 2010;30:16137–48. doi: 10.1523/JNEUROSCI.4161-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dash PK, Moore AN, Kobori N, Runyan JD. Molecular activity underlying working memory. Learn Mem. 2007;14:554–63. doi: 10.1101/lm.558707. [DOI] [PubMed] [Google Scholar]
- 26.Jung MW, Baeg EH, Kim MJ, Kim YB, Kim JJ. Plasticity and memory in the prefrontal cortex. Rev Neurosci. 2008;19:29–46. doi: 10.1515/revneuro.2008.19.1.29. [DOI] [PubMed] [Google Scholar]
- 27.Linden DE. The working memory networks of the human brain. Neuroscientist. 2007;13:257–67. doi: 10.1177/1073858406298480. [DOI] [PubMed] [Google Scholar]
- 28.D’Esposito M, Detre JA, Alsop DC, Shin RK, Atlas S, Grossman M. The neural basis of the central executive system of working memory. Nature. 1995;378:279–81. doi: 10.1038/378279a0. [DOI] [PubMed] [Google Scholar]
- 29.Harris JA, Miniussi C, Harris IM, Diamond ME. Transient storage of a tactile memory trace in primary somatosensory cortex. J Neurosci. 2002;22:8720–5. doi: 10.1523/JNEUROSCI.22-19-08720.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keenan PA, Ezzat WH, Ginsburg K, Moore GJ. Prefrontal cortex as the site of estrogen’s effect on cognition. Psychoneuroendocrinology. 2001;26:577–90. doi: 10.1016/s0306-4530(01)00013-0. [DOI] [PubMed] [Google Scholar]
- 31.Hao J, Rapp PR, Leffler AE, Leffler SR, Janssen WG, Lou W, et al. Estrogen alters spine number and morphology in prefrontal cortex of aged female rhesus monkeys. J Neurosci. 2006;26:2571–8. doi: 10.1523/JNEUROSCI.3440-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang Y, Janssen WG, Hao J, Roberts JA, McKay H, Lasley B, et al. Estrogen replacement increases spinophilin-immunoreactive spine number in the prefrontal cortex of female rhesus monkeys. Cereb Cortex. 2004;14:215–23. doi: 10.1093/cercor/bhg121. [DOI] [PubMed] [Google Scholar]
- 33.Dhandapani K, Brann D. Neuroprotective effects of estrogen and tamoxifen in vitro: a facilitative role for glia? Endocrine. 2003;21:59–66. doi: 10.1385/endo:21:1:59. [DOI] [PubMed] [Google Scholar]
- 34.Mervis RF, Pope D, Lewis R, Dvorak RM, Williams LR. Exogenous nerve growth factor reverses age-related structural changes in neocortical neurons in the aging rat. A quantitative Golgi study. Ann N Y Acad Sci. 1991;640:95–101. doi: 10.1111/j.1749-6632.1991.tb00198.x. [DOI] [PubMed] [Google Scholar]
- 35.Horvath KM, Hartig W, Van der Veen R, Keijser JN, Mulder J, Ziegert M, et al. 17beta-estradiol enhances cortical cholinergic innervation and preserves synaptic density following excitotoxic lesions to the rat nucleus basalis magnocellularis. Neuroscience. 2002;110:489–504. doi: 10.1016/s0306-4522(01)00560-7. [DOI] [PubMed] [Google Scholar]
- 36.Wang L, Andersson S, Warner M, Gustafsson JA. Morphological abnormalities in the brains of estrogen receptor beta knockout mice. Proc Natl Acad Sci U S A. 2001;98:2792–6. doi: 10.1073/pnas.041617498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Voytko ML, Murray R, Higgs CJ. Executive function and attention are preserved in older surgically menopausal monkeys receiving estrogen or estrogen plus progesterone. J Neurosci. 2009;29:10362–70. doi: 10.1523/JNEUROSCI.1591-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marinho RM, Soares JM, Jr, Santiago RC, Maganhin CC, Machado F, de Miranda Cota AM, et al. Effects of estradiol on the cognitive function of postmenopausal women. Maturitas. 2008;60:230–4. doi: 10.1016/j.maturitas.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 39.Wang AC, Hara Y, Janssen WG, Rapp PR, Morrison JH. Synaptic estrogen receptor-alpha levels in prefrontal cortex in female rhesus monkeys and their correlation with cognitive performance. J Neurosci. 2010;30:12770–6. doi: 10.1523/JNEUROSCI.3192-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang LC, Zhang QG, Zhou CF, Yang F, Zhang YD, Wang RM, et al. Extranuclear estrogen receptors mediate the neuroprotective effects of estrogen in the rat hippocampus. PLoS One. 2010;5:e9851. doi: 10.1371/journal.pone.0009851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohkura S, Fabre-Nys C, Broad KD, Kendrick KM. Sex hormones enhance the impact of male sensory cues on both primary and association cortical components of visual and olfactory processing pathways as well as in limbic and hypothalamic regions in female sheep. Neuroscience. 1997;80:285–97. doi: 10.1016/s0306-4522(97)00103-6. [DOI] [PubMed] [Google Scholar]
- 42.Tremere LA, Jeong JK, Pinaud R. Estradiol shapes auditory processing in the adult brain by regulating inhibitory transmission and plasticity-associated gene expression. J Neurosci. 2009;29:5949–63. doi: 10.1523/JNEUROSCI.0774-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kis Z, Budai D, Imre G, Farkas T, Horvath S, Toldi J. The modulatory effect of estrogen on the neuronal activity in the barrel cortex of the rat. An electrophysiological study. Neuroreport. 2001;12:2509–12. doi: 10.1097/00001756-200108080-00044. [DOI] [PubMed] [Google Scholar]
- 44.Zadran S, Qin Q, Bi X, Zadran H, Kim Y, Foy MR, et al. 17-Beta-estradiol increases neuronal excitability through MAP kinase-induced calpain activation. Proc Natl Acad Sci U S A. 2009;106:21936–41. doi: 10.1073/pnas.0912558106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kramar EA, Chen LY, Brandon NJ, Rex CS, Liu F, Gall CM, et al. Cytoskeletal changes underlie estrogen’s acute effects on synaptic transmission and plasticity. J Neurosci. 2009;29:12982–93. doi: 10.1523/JNEUROSCI.3059-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu F, Day M, Muniz LC, Bitran D, Arias R, Revilla-Sanchez R, et al. Activation of estrogen receptor-beta regulates hippocampal synaptic plasticity and improves memory. Nature neuroscience. 2008;11:334–43. doi: 10.1038/nn2057. [DOI] [PubMed] [Google Scholar]
- 47.Smejkalova T, Woolley CS. Estradiol acutely potentiates hippocampal excitatory synaptic transmission through a presynaptic mechanism. J Neurosci. 2010;30:16137–48. doi: 10.1523/JNEUROSCI.4161-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Waters EM, Mitterling K, Spencer JL, Mazid S, McEwen BS, Milner TA. Estrogen receptor alpha and beta specific agonists regulate expression of synaptic proteins in rat hippocampus. Brain Res. 2009;1290:1–11. doi: 10.1016/j.brainres.2009.06.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khan M, De Sevilla L, Mahesh VB, Brann DW. Enhanced glutamatergic and decreased GABAergic synaptic appositions to GnRH neurons on proestrus in the rat: modulatory effect of aging. PLoS One. 2010;5:e10172. doi: 10.1371/journal.pone.0010172. [DOI] [PMC free article] [PubMed] [Google Scholar]