

Abstract
Purpose.
The goal of this study was to determine whether Compound 49b stimulates insulin-like growth factor binding protein-3 (IGFBP-3) activation in retinal endothelial cells (REC) through DNA-dependent protein kinase (DNA-PK).
Methods.
REC were grown in a normal glucose (5 mM) or high glucose medium (25 mM). Some cells were transfected with protein kinase A (PKA) siRNA, following treatment with 50 nM Compound 49b, a novel β-adrenergic receptor agonist. Cell proteins were extracted and analyzed for DNA-PK expression by Western blotting. Additional cells were treated with or without NU7441 (a specific DNA-PK inhibitor) prior to Compound 49b treatment. Cell lysates were processed for IGFBP-3 ELISA analyses and Western blotting to measure casein kinase 2 (CK2). Immunoprecipitation for total and phospho–IGFBP-3, cell proliferation and cell death measurements were done after transfection with the S156A IGFBP-3 mutation (key phosphorylation site involved in DNA-PK) plasmid DNA.
Results.
Compound 49b required DNA-PK to activate IGFBP-3 in REC. IGFBP-3 activation was significantly reduced following treatment with either the DNA-PK inhibitor or following transfection with the IGFBP-3 S156A mutant plasmid (P < 0.05). Significant increases in cell death and decreases in cell proliferation were also observed in cells transfected with the IGFBP-3 S156A mutant plasmid (P < 0.05). Casein kinase levels were not altered after treatment with NU7741 or Compound 49b.
Conclusions.
Our findings suggest Compound 49b induces DNA-PK levels through PKA activity. DNA-PK is required for Compound 49b–induced IGFBP-3 expression, leading to inhibition of REC cell death.
Keywords: apoptosis, IGFBP-3, DNA-PK, retinal endothelial cell
Beta-adrenergic receptor agonists increase protein kinase A, leading to phoshorylation of DNA-dependent protein kinase (DNA-PK). DNA-PK phoshorylates insulin-like growth factor binding protein-3 on Ser 156 to regulate apoptosis in retinal endothelial cells.
Introduction
Diabetic retinopathy (DR) is the leading cause of adult blindness. Despite tight glycemic control, the number of DR patients keeps growing, with limited therapeutic options.1 Insulin-like growth factor-1 (IGF-1) is a fundamental mitogen involved in cell survival, proliferation, and differentiation in the retina.2 IGF-1 exerts its action primarily through the type 1 IGF receptor (IGF1R) and its binding proteins.3 Poorly controlled diabetic patients exhibit many abnormalities of the circulating growth hormone (GH)-IGF-insulin–like growth factor binding proteins (IGFBP) axis, including GH hypersecretion and reduced circulating levels of both IGF-1 and IGFBP-3.4
Based on the reported role of IGFBP-3 in supporting cell growth, we previously investigated a role for IGFBP-3 in DR.5–7 In those studies, we demonstrated that (1) β-adrenergic receptor agonists stimulate endogenous IGFBP-3 activity in retinal endothelial cells (REC), (2) β-adrenergic receptor stimulation of IGFBP-3 requires activation of protein kinase A (PKA), and (3) the β-adrenergic-PKA-IGFBP-3 pathway protects against apoptosis in DR, both in whole retinal lysates from rodents or in REC cultured under high glucose conditions.8
While it was clinically important to find that β-adrenergic receptor agonists can protect against retinal damage in diabetes through IGFBP-3 actions, the cellular pathways involved were not clear. IGFBP-3 serves as a carrier protein for IGF-1, but has recently been shown to have additional actions independent of IGF-1.9,10 These IGF-independent actions involve regulation of autocrine/paracrine pathways to stimulate cell growth and inhibit apoptosis.11 Among its many IGF-independent actions, IGFBP-3 binds to proteins on the cell surface, such as glycosaminoglycans,12,13 heparin,14 fibrinogen,15 and specific cell surface receptor low density lipoprotein receptor-related protein 1 (LRP-1) or TGF-β.16 IGFBP-3 activity is modulated by proteolysis, glycosylation, ubiquitination, methylation,17–20 and phosphorylation.21,22 Phosphorylation appears to be an important posttranslational modification of IGFBP-3, as IGFBP-3 can be phosphorylated at casein kinase 2 (CK2) sites: Ser111 and Ser113, or DNA-PK sites: Ser156, Ser165, and Thr170 to regulate IGFBP-3 actions on cell growth.21,23 The cellular regulation of IGFBP-3 in the retina remains unknown.
Our goal in these studies was to determine the cellular mechanism by which β-adrenergic receptor regulates IGFBP-3, specifically whether phosphorylation by DNA-PK is a key step. Our overall goal is to further establish the pathways that play a critical role in the growth-promoting actions of IGFBP-3 and their role in DR.
Materials and Methods
Reagents
IGFBP-3 antibodies, CK2, and phospho-serine antibodies were purchased from Upstate (Lake Placid, NY). DNA-PK antibodies were purchased from ABNOVA (Littleton, CO). Protein A/G PLUS-Agarose beads and actin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Human IGFBP-3 immunoassay ELISA kit was purchased from R&D Systems (Minneapolis, MN). Human PRKACA siRNA and Non-Targeting siRNA # 1 were purchased from Dharmacon RNAi Technologies (Chicago, IL). Primers, RNAimax, and Lipofectamine 2000 were purchased from Invitrogen (Carlsbad, CA). Secondary antimouse and antirabbit antibodies conjugated with horseradish peroxidase were purchased from Promega (Madison, WI). Enhanced chemiluminescence (ECL) for immunoblot development and signal detection was purchased from Amersham Biosciences (Piscataway, NJ). NU7441 was purchased from Tocris Bioscience (Minneapolis, MN). Cell death ELISA kit was bought from Roche (Mannheim, Germany). Cell proliferation assay kit was bought from Millipore (Billierca, MA). QuikChange II XL Site-Directed Mutagenesis Kit was purchased from Stratagene (La Jolla, CA). IGFBP-3 NB plasmid was a gift from Maria Grant, MD (University of Florida).
Cell Culture
Primary human retinal endothelial cells (REC) were acquired from Cell Systems Corporation (CSC, Kirkland, WA). Cells were grown in M131 medium containing microvascular growth supplements (MVGS; Invitrogen), 10 μg/μL gentamycin and 0.25 μg/μL amphotericin B. For high glucose conditions, cells were transferred to high glucose (25 mM) (Cell Systems Corporation) medium, supplemented with MVGS and antibiotics for 3 days. Only primary cells within passages 6 were used. Cells were starved by incubating in high or normal glucose medium without growth factors for 24 hours and used to perform the experiments unless otherwise indicated.
Transfection of siRNA and Plasmid DNA
RECs were transfected with siRNA (PKA siRNA or scrambled siRNA [control]) at a final concentration of 20 nM using RNAiMAX transfection reagent (Invitrogen) according to the manufacturer's instructions. The cells were used for experiments 24 hours after transfection. The cells were also transfected with IGFBP-3 NB plasmid DNA at 1 μg/mL using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. The cells were used for experimentation 24 hours after transfection.
ELISA Analysis
An ELISA for IGFBP-3 levels was performed using an IGFBP-3 ELISA assay kit (R&D Systems) according to the manufacturer's instructions to evaluate the IGFBP-3 expression. Briefly, confluent RECs were starved for 24 hours, treated with NU 7441 (10 μM) for 30 minutes,24 followed by treatment with 50 nM Compound 49b for 30 minutes under either normal or high glucose conditions. Cell death ELISA analyses of whole cell pellets were used to quantify apoptosis. For all ELISA analyses, equal protein amounts were loaded into each well, allowing for comparisons using optical density (OD).
Cell Proliferation Analysis
To study how Compound 49b treatment affected proliferation, 50,000 cells with or without IGFBP-3 S156A mutation transfection were plated into each well of a 96-well dish and incubated for 24 hours. Following treatment with or without Compound 49b, the cellular proliferation was determined using tetrazolium salt WST-1 and a microplate reader (UQuant Reader; BioTek, Winooski, VT) according to the assay manufacturer's instructions (Cell Proliferation Assay Kit, WST dye, ELISA based; Millipore) at 450 nm. The absorbance at 450 nm (recorded in OD) is directly correlated with cellular proliferation.
Western Blot Analysis
After appropriate treatments and rinsing with cold PBS, RECs were scraped into lysis buffer containing the protease and phosphatase inhibitors. Equal amounts of protein from the cell extracts were separated on the precast tris-glycine gel (Invitrogen), blotted onto a nitrocellulose membrane. After blocking in TBST (10 mM Tris-HCl buffer, pH 8.0, 150 mM NaCl, 0.1% Tween 20) containing 5% (wt/vol) BSA, the membrane was treated with anti-CK2, DNA-PK, IGFBP-3, phospho–IGFBP-3, and actin antibodies (1:500). After primary antibody application, membranes were incubated with horseradish peroxidase labeled secondary antibodies. The antigen-antibody complexes were detected using chemilluminescence reagent kit (Thermo Scientific, Pittsburgh, PA). Mean densitometry of immunoreactive bands was assessed using Kodak 4000R software (Carestream Molecular Imaging, Rochester, NY), and results were expressed in densitometric units and compared with control groups for each individual experiment.
Immunoprecipitation
After rinsing with PBS, cells were lysed by freeze-thawing in lysis buffer containing the protease and phosphatase inhibitors for 20 minutes on ice. The cell pellets were transferred into 1.5 μL Eppendorf tubes (Fisher, Pittsburgh, PA) and cleared by centrifugation at 12,000 rpm for 20 minutes at 4°C. The cells containing an equal amount of protein from control and each treatment were incubated with appropriate antibodies overnight at 4°C, at which time Protein A/G PLUS-Agarose beads were added and incubation continued for 2 hours with gentle rocking. The beads were washed three times with lysis buffer and once with PBS and the immunocomplexes were released by heating in Laemmli sample buffer and analyzed by Western blotting for the indicated molecules using their specific antibodies.
Mutagenesis
The IGFBP-3 NB plasmid DNA was a gift from Maria Grant, MD. The phosphorylation Ser156 site was mutated to alanine to prevent their phosphorylation using the forward primer: 5′-GGGCATGCTAAAGACGCCCAGCGCTACAAAGTTGACTACG-3′ and the reverse primer 5′-CGTAGTCAACTTTGTAGCGCTGGGCGTCTTTAGCATGCCC-3′ according to the QuikChange II XL Site-Directed Mutagenesis Kit manufacturer. The mutants were verified by DNA sequencing (University of Tennessee Health Science Center Molecular Science Core).
Statistics
All the experiments were repeated three times, and the data are presented as mean ± SEM. Data were analyzed by a nonparametric Kruskal-Wallis one-way ANOVA, followed by a Dunn's test with P values less than 0.05 considered statistically significant. In the case of Western blotting, one representative blot is shown. The control was normalized to one and the treatment was compared with control by fold change.
Results
Compound 49b Requires PKA to Activate DNA-PK
RECs were cultured in normal glucose (5 mM) or high glucose (25 mM) for 3 days. On the second day, cells were transfected with PKA siRNA for 24 hours, followed by treatment with Compound 49b (50 nM) for 30 minutes. Cell extracts were analyzed for protein levels of DNA-PK by Western blotting. Data indicate that Compound 49b resulted in DNA-PK activation in high glucose, with DNA-PK activation dramatically decreased if PKA was knocked down (Fig. 1).
Figure 1. .

Compound 49b–induced DNA-PK expression is through PKA activation in high ambient glucose. Bar graph of DNA-PK levels measured by Western blot (A) in REC with PKA siRNA transfection for 24 hours, following treatment with Compound 49b for 30 minutes in 5 mM glucose and 25 mM glucose. (B) is the bar graph of PKA level after PKA siRNA transfection. (A) *P < 0.05 versus HG Sc siRNA. #P < 0.05 versus HG Sc siRNA + 49b. N = 4. (B) *P < 0.05 versus NG Sc siRNA. #P < 0.05 versus HG Sc siRNA.
Compound 49b–Induced IGFBP-3 Expression Requires DNA-PK
To determine the role of DNA-PK in Compound 49b's induction of IGFBP-3 expression, RECs were treated with 10 μM NU7441 (DNA-PK inhibitor) 30 minutes prior to stimulation with Compound 49b. Results show that Compound 49b induces IGFBP-3 levels through DNA-PK actions, since NU7741 inhibited the ability of Compound 49b to increase IGFBP-3 (Fig. 2).
Figure 2. .

Compound 49b–induced IGFBP-3 expression is through DNA-PK in RECs in high ambient glucose. Bar graph of IGFBP-3 levels measured by the ELISA kit (A) in RECs treated with NU7441 for 30 minutes and following treatment with Compound 49b for 30 minutes. *P < 0.05 versus NG control. #P < 0.05 versus HG control. $P < 0.05 versus HG NU7441. N = 4.
CK2 Was not Involved in Compound 49b– or DNA-PK–Induced IGFBP-3 Activation
CK2 has been reported to be a key regulator of the IGFBP-3 expression.25 To determine whether CK2 was involved in Compound 49b regulation of IGFBP-3, RECs were stimulated with Compound 49b or NU7441+Compound 49b in normal or high glucose. Our results showed NU7441 could effectively inhibited DNA-PK expression and CK2 levels were similar in both normal and high glucose conditions (Fig. 3). Neither Compound 49b nor NU7441 altered CK2 levels, suggesting that CK2 is not involved in Compound 49b's regulation of IGFBP-3.
Figure 3. .
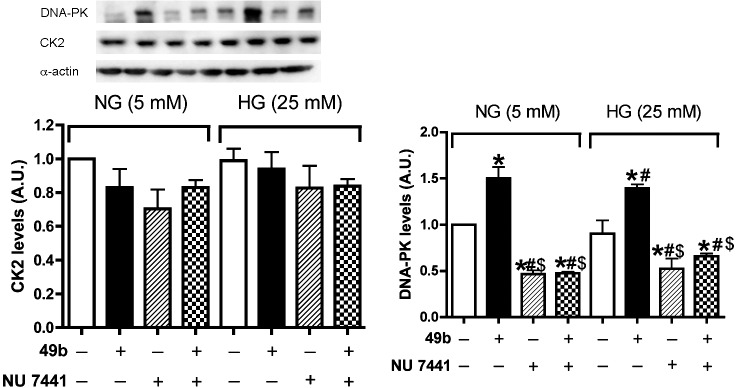
CK2 expression did not change with either Compound 49b or NU7441 treatment in normal or high glucose medium. Western blot analysis of CK2 levels and DNA-PK levels in RECs treated with Compound 49b or NU7441 in normal glucose (NG-5 mM) or high glucose (HG-25 mM) medium. *P < 0.05 versus NG control. #P < 0.05 versus HG control. $P < 0.05 versus HG NU7441. N = 3.
DNA-PK Is Involved in Compound 49b–Induced IGFBP-3 Expression to Inhibit Apoptosis in High Ambient Glucose in RECs
Since data suggest Compound 49b activates DNA-PK to regulate IGFBP-3, we wanted to determine which phosphorylation site is key to DNA-PK expression of IGFBP-3. Based on findings in the literature,21 we chose to mutate Ser156 to Ala using site-directed mutagenesis. We then transfected the cells with the mutated plasmid DNA, followed by treatment with Compound 49b. Cell apoptosis was measured by both DNA fragmentation and cleaved caspase 3 levels after transfection with the mutant versus no transfection. Successful transfection was verified by an IGFBP-3 Western blot. Figure 4A shows high expression of IGFBP-3 after the IGFBP-3 S156A mutation transfection, demonstrating successful transfection. Transfection of the mutation caused a decrease in IGFBP-3 activation (Fig. 4A). Compound 49b could not inhibit cell apoptosis following transfection with the mutant (Figs. 4B, 4C), demonstrating that this phosphorylation site is key for DNA-PK regulation of IGFBP-3.
Figure 4. .

Compound 49b could not inhibit cell death or stimulate IGFBP-3 activation with IGFBP-3 S156A plasmid DNA transfection. (A) Bar graph of pIGFBP-3/IGFBP-3 of RECs in normal or high glucose transfected with IGFBP-3 S156A plasmid DNA following treatment with Compound 49b. Proteins from RECs transfected with IGFBP-3 S156A plasmid DNA was immunoprecipitated with anti–IGFBP-3 or antiphospho-serine antibodies. The immunoprecipitates were analyzed by Western blotting using anti–IGFBP-3 antibodies. *P < 0.05 versus NG control plasmid, #P < 0.05 versus HG control plasmid. N = 3. (B, C) Bar graph of DNA fragmentation (B) and cleaved caspase 3 expression (C) of RECs in normal or high glucose transfected with IGFBP-3 S156A plasmid DNA following treatment with Compound 49b. *P < 0.05 versus NG control plasmid. #P < 0.05 versus HG control plasmid. $P < 0.05 versus HG control mutant IGFBP-3 plasmid. N = 3.
DNA-PK Is Involved in Compound 49b–Induced REC Proliferation in High Ambient Glucose
To determine whether DNA-PK/IGFBP-3 is involved in Compound 49b–induced cell proliferation, we detected cell proliferation assay after RECs transfected with mutated IGFBP-3 S156A plasmid DNA, followed by treatment with Compound 49b. With mutant transfection, Compound 49b could not induce cell proliferation (Fig. 5), suggesting that DNA-PK stimulation by Compound 49b is required in cell proliferation.
Figure 5. .

Compound 49b could not stimulate cell proliferation following IGFBP-3 S156A plasmid DNA transfection. Bar graph of ratio to NG control of REC proliferation in normal or high glucose transfected with IGFBP-3 S156A plasmid DNA following treatment with Compound 49b. *P < 0.05 versus NG control plasmid. #P < 0.05 versus HG control plasmid. $P < 0.05 versus HG control mutant IGFBP-3 plasmid. N = 8.
Discussion
DR is a major complication of both type 1 and type 2 diabetes mellitus.1 In our previous studies using various diabetic models, we discovered that β-adrenergic receptor signaling contributes to the maintenance of retinal homeostasis despite hyperglycemia. Specifically, we found that the β-adrenergic receptor agonist, Compound 49b, inhibits the neuronal and vascular changes seen in DR and that its mechanism of action involves PKA-dependent activation of retinal IGFBP-3 levels.8 These results suggest that IGFBP-3 acts as a novel mediator of the etiopathophysiology underlying DR, particularly the vascular changes. There are reports that IGFBP-3 promotes either cell proliferation or apoptosis in different cell systems. Likewise, IGFBP-3 expression has been shown to be upregulated by both pro-apoptotic agents, as well as by survival factors such as IGF-1, in RECs,26–28 and epithelial cells.29 The findings suggest the hypothesis that IGFBP-3 is a multifunctional protein whose role in the cells is dependent on cellular context. The experiments described herein were designed to further examine the mechanisms regulating IGFBP-3 actions in the retina.
In this study, we provide evidence that DNA-PK plays a role in β-adrenergic receptor-PKA induced expression of IGFBP-3 in retinal endothelial cells cultured in high glucose. This conclusion is based on our results that show: (1) Compound 49b stimulation of DNA-PK expression is PKA-dependent, (2) Compound 49b stimulates IGFBP-3 activation through DNA-PK, and (3) Phosphorylation of Ser156 on IGFBP-3 is a critical step in the ability of IGFBP-3 to prevent apoptosis and induce cell proliferation in RECs under high glucose conditions.
Recent evidence suggests that IGFBP-3 not only serves as binding site for IGF, but it also has actions that are independent of IGF. Several novel binding partners for IGFBP-3 have been proposed to explain its IGF-independent actions. Figure 6 shows that S111 and S113 are the potential sites of CK2 phosphorylation, which may influence IGFBP-3 role in regulating cell apoptosis; residues S156, S165, and T170 are the potential sites of DNA-PK phosphorylation, which may be involved in DNA damage, replication, and gene transcription.30 To clarify the detailed mechanisms regarding to IGFBP-3 in cell apoptosis, we used RECs in culture and investigated changes in CK2 when cells were treated with Compound 49b or DNA-PK inhibitor. Our data show that CK2 levels did not change following Compound 49b or DNA-PK inhibitor treatment in RECs under high glucose conditions. Thus, we concluded that CK2 phosphorylation site of IGFBP-3 is not involved in β-adrenergic receptor agonist inhibition of cell apoptosis.
Figure 6. .

Amino acid sequences of the S156A mutation in IGFBP-3. Upper line: Potential sites for CK2 phosphorylation (blue color) and DNA-PK phosphorylation (green color) in IGFBP-3. Lower line: the S156 mutation in IGFBP-3 what we developed (yellow color).
In our experiments, Compound 49b could not activate IGFBP-3 to inhibit cell apoptosis or induce cell proliferation in RECs when cells were treated with the DNA-PK inhibitor or following transfection with the mutant IGFBP-3 Ser156, in addition to Compound 49b. This strongly suggests the significance of phosphorylation of IGFBP-3 by DNA-PK in the protection of Compound 49b in DR. However, phosphorylation by other kinases may also occur in specific condition31–33 and phosphorylation of IGFBP-3 by DNA-PK could modulate directly the effects of IGFBP-3.31 Additional studies on the actions of regulation of IGFBP-3 are warranted, particularly cytosolic versus nuclear actions.
Conclusions
In summary, this study shows that phosphorylation Ser156 site of IGFBP-3 by DNA-PK is of vital importance in the function of IGFBP-3 in inhibiting apoptosis and inducing cell proliferation in RECs in diabetic conditions. Further studies to better understand mechanisms that regulate IGFBP systems in the retina may be useful in developing drug targets that could halt the progression of DR.
Acknowledgments
Supported by the National Eye Institute Vision Grant R01EY022045 (JJS); Juvenile Diabetes Research Foundation Grant (2-2011-597; JJS); Oxnard Foundation (JJS); Research to Prevent Blindness Award; and National Eye Institute Vision Core Grant PHS 3P30 EY013080.
Disclosure: Q. Zhang, None; J.J. Steinle, P
References
- 1. Ioacara S, Lichiardopol R, Ionescu-Tirgoviste C, et al. Improvements in life expectancy in type 1 diabetes patients in the last six decades. Diabetes Res Clin Pract. 2009; 86: 146–151 [DOI] [PubMed] [Google Scholar]
- 2. Bartke A, Chandrashekar V, Dominici F, et al. Insulin-like growth factor 1 (IGF-1) and aging: controversies and new insights. Biogerontology. 2003; 4: 1–8 [DOI] [PubMed] [Google Scholar]
- 3. Thrailkill KM. Insulin-like growth factor-I in diabetes mellitus: its physiology, metabolic effects, and potential clinical utility. Diabetes Technol Ther. 2000; 2: 69–80 [DOI] [PubMed] [Google Scholar]
- 4. Touskova V, Trachta P, Kavalkova P, et al. Serum concentrations and tissue expression of components of insulin-like growth factor-axis in females with type 2 diabetes mellitus and obesity: the influence of very-low-calorie diet. Mol Cell Endocrinol. 2012; 361: 172–178 [DOI] [PubMed] [Google Scholar]
- 5. Chang KH, Chan-Ling T, McFarland EL, et al. IGF binding protein-3 regulates hematopoietic stem cell and endothelial precursor cell function during vascular development. Proc Natl Acad Sci U S A. 2007; 104: 10595–10600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Giannini S, Cresci B, Pala L, et al. IGFBPs modulate IGF-I– and high glucose-controlled growth of human retinal endothelial cells. J Endocrinol. 2001; 171: 273–284 [DOI] [PubMed] [Google Scholar]
- 7. Liu LQ, Sposato M, Liu HY, et al. Functional cloning of IGFBP-3 from human microvascular endothelial cells reveals its novel role in promoting proliferation of primitive CD34+CD38- hematopoietic cells in vitro. Oncol Res. 2003; 13: 359–371 [DOI] [PubMed] [Google Scholar]
- 8. Zhang Q, Guy K, Pagadala J, et al. Compound 49b prevents diabetes-induced apoptosis through increased IGFBP-3 levels. Invest Ophthalmol Vis Sci. 2012; 53: 3004–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jarajapu YP, Cai J, Yan Y, et al. Protection of blood retinal barrier and systemic vasculature by insulin-like growth factor binding protein-3. PloS One. 2012; 7: e39398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim JH, Choi DS, Lee OH, et al. Antiangiogenic antitumor activities of IGFBP-3 are mediated by IGF-independent suppression of Erk1/2 activation and Egr-1–mediated transcriptional events. Blood. 2011; 118: 2622–2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yamada PM, Lee KW. Perspectives in mammalian IGFBP-3 biology: local vs. systemic action. Am J Physiol Cell Physiol. 2009; 296: C954–976 [DOI] [PubMed] [Google Scholar]
- 12. Baricevic I, Masnikosa R, Lagundzin D, Golubovic V, Nedic O. Alterations of insulin-like growth factor binding protein 3 (IGFBP-3) glycosylation in patients with breast tumours. Clin Biochem. 2010; 43: 725–731 [DOI] [PubMed] [Google Scholar]
- 13. Firth SM, Baxter RC. Characterisation of recombinant glycosylation variants of insulin-like growth factor binding protein-3. J Endocrinol. 1999; 160: 379–387 [DOI] [PubMed] [Google Scholar]
- 14. Beattie J, Phillips K, Shand JH, et al. Molecular recognition characteristics in the insulin-like growth factor (IGF)-insulin-like growth factor binding protein −3/5 (IGFBP-3/5) heparin axis. J Mol Endocrinol. 2005; 34: 163–175 [DOI] [PubMed] [Google Scholar]
- 15. Campbell PG, Durham SK, Hayes JD, Suwanichkul A, Powell DR. Insulin-like growth factor-binding protein-3 binds fibrinogen and fibrin. J Biol Chem. 1999; 274: 30215–30221 [DOI] [PubMed] [Google Scholar]
- 16. Huang SS, Leal SM, Chen CL, Liu IH, Huang JS. Identification of insulin receptor substrate proteins as key molecules for the TbetaR-V/LRP-1-mediated growth inhibitory signaling cascade in epithelial and myeloid cells. FASEB J. 2004; 18: 1719–1721 [DOI] [PubMed] [Google Scholar]
- 17. Chang YS, Kong G, Sun S, et al. Clinical significance of insulin-like growth factor-binding protein-3 expression in stage I non-small cell lung cancer. Clin Cancer Res. 2002; 8: 3796–3802 [PubMed] [Google Scholar]
- 18. Hanafusa T, Yumoto Y, Nouso K, et al. Reduced expression of insulin-like growth factor binding protein-3 and its promoter hypermethylation in human hepatocellular carcinoma. Cancer Lett. 2002; 176: 149–158 [DOI] [PubMed] [Google Scholar]
- 19. Bang P, Brismar K, Rosenfeld RG. Increased proteolysis of insulin-like growth factor-binding protein-3 (IGFBP-3) in noninsulin-dependent diabetes mellitus serum, with elevation of a 29-kilodalton (kDa) glycosylated IGFBP-3 fragment contained in the approximately 130- to 150-kDa ternary complex. J Clin Endocrinol Metab. 1994; 78: 1119–1127 [DOI] [PubMed] [Google Scholar]
- 20. Santer FR, Bacher N, Moser B, et al. Nuclear insulin-like growth factor binding protein-3 induces apoptosis and is targeted to ubiquitin/proteasome-dependent proteolysis. Cancer Res. 2006; 66: 3024–3033 [DOI] [PubMed] [Google Scholar]
- 21. Cobb LJ, Liu B, Lee KW, Cohen P. Phosphorylation by DNA-dependent protein kinase is critical for apoptosis induction by insulin-like growth factor binding protein-3. Cancer Res. 2006; 66: 10878–10884 [DOI] [PubMed] [Google Scholar]
- 22. Hollowood AD, Stewart CE, Perks CM, et al. Evidence implicating a mid-region sequence of IGFBP-3 in its specific IGF-independent actions. J Cell Biochem. 2002; 86: 583–589 [DOI] [PubMed] [Google Scholar]
- 23. Cobb LJ, Mehta H, Cohen P. Enhancing the apoptotic potential of insulin-like growth factor-binding protein-3 in prostate cancer by modulation of CK2 phosphorylation. Mol Endocrinol. 2009; 23: 1624–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hardcastle IR, Cockcroft X, Curtin NJ, et al. Discovery of potent chromen-4-one inhibitors of the DNA-dependent protein kinase (DNA-PK) using a small-molecule library approach. J Med Chem. 2005; 48: 7829–7846 [DOI] [PubMed] [Google Scholar]
- 25. Kosuge S, Sawano Y, Ohtsuki K. A novel CK2-mediated activation of type II cAMP-dependent protein kinase through specific phosphorylation of its regulatory subunit (RIIalpha) in vitro. Biochem Biophys Res Commun. 2003; 310: 163–168 [DOI] [PubMed] [Google Scholar]
- 26. Spoerri PE, Caballero S, Wilson SH, Shaw LC, Grant MB. Expression of IGFBP-3 by human retinal endothelial cell cultures: IGFBP-3 involvement in growth inhibition and apoptosis. Invest Ophthalmol Vis Sci. 2003; 44: 365–369 [DOI] [PubMed] [Google Scholar]
- 27. Kielczewski JL, Jarajapu YP, McFarland EL, et al. Insulin-like growth factor binding protein-3 mediates vascular repair by enhancing nitric oxide generation. Circ Res. 2009; 105: 897–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang Q, Soderland C, Steinle JJ. Regulation of retinal endothelial cell apoptosis through activation of the IGFBP-3 receptor. Apoptosis. 2013; 18: 361–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Leibowitz BJ, Cohick WS. Endogenous IGFBP-3 is required for both growth factor-stimulated cell proliferation and cytokine-induced apoptosis in mammary epithelial cells. J Cell Physiol. 2009; 220: 182–188 [DOI] [PubMed] [Google Scholar]
- 30. Baxter RC, Skriver L. Altered ligand specificity of proteolysed insulin-like growth factor binding protein-3. Biochem Biophys Res Commun. 1993; 196: 1267–1273 [DOI] [PubMed] [Google Scholar]
- 31. Schedlich LJ, Nilsen T, John AP, Jans DA, Baxter RC. Phosphorylation of insulin-like growth factor binding protein-3 by deoxyribonucleic acid-dependent protein kinase reduces ligand binding and enhances nuclear accumulation. Endocrinology. 2003; 144: 1984–1993 [DOI] [PubMed] [Google Scholar]
- 32. Coverley JA, Martin JL, Baxter RC. The effect of phosphorylation by casein kinase 2 on the activity of insulin-like growth factor-binding protein-3. Endocrinology. 2000; 141: 564–570 [DOI] [PubMed] [Google Scholar]
- 33. de Silva HC, Firth SM, Twigg SM, Baxter RC. Interaction between IGF binding protein-3 and TGFβ in the regulation of adipocyte differentiation. Endocrinology. 2012; 153: 4799–4807 [DOI] [PubMed] [Google Scholar]