

Abstract
The study of gene function in developmental biology has been significantly furthered by advances in antisense technology made in the early 2000s. This was achieved, in particular, by the introduction of morpholino (MO) oligonucleotides. The introduction of antisense MO oligonucleotides into cells enables researchers to readily reduce the levels of their protein of interest without investing huge financial or temporal resources, in both in vivo and in vitro model systems. Historically, the African clawed frog Xenopus has been used to study vertebrate embryological development, due to its ability to produce vast numbers of offspring that develop rapidly, in synchrony, and can be cultured in buffers with ease. The developmental progress of Xenopus embryos has been extensively characterized and this model organism is very easy to maintain. It is these attributes that enable MO-based knockdown strategies to be so effective in Xenopus. In this chapter, we will detail the methods of microinjecting MO oligonucleotides into early embryos of X. laevis and X. tropicalis. We will discuss how MOs can be used to prevent either pre-mRNA splicing or translation of the specific gene of interest resulting in abrogation of that gene’s function and advise on what control experiments should be undertaken to verify their efficacy.
Keywords: Xenopus, Laevis, Tropicalis, Morpholino, Microinjection, Knockdown, Inhibition, Translation, Splicing, Splice, Antisense
1. Introduction
Understanding of the molecular and genetic control of cardiogenesis has advanced rapidly in recent years through the use of several complementary model systems, including oviparous vertebrate model organisms such as the zebrafish (Danio rerio) and the frog (principally Xenopus laevis). Xenopus species have numerous advantages in addition to the external nature of their embryonic development and ease of use. They are largely transparent at tadpole stages, allowing anatomical defects in the heart to be easily seen; their hearts undergo the process of atrial septation similarly to higher vertebrates; they can survive to advanced developmental stages in the absence of a functioning circulatory system, allowing for more detailed studies of early cardiovascular defects; and they have a well-defined fate map at the 32-cell stage (6 h post-fertilization) that allows the blastomeres that will give rise to the heart to be identified and manipulated. These features have enabled many investigators to examine events in cardiac development in the frog, but it has only been with the advent of effective antisense techniques that significant advances have been made linking these events to the genes that control them. The most extensively used of these techniques is the use of morpholino (MO) oligonucleotides to inhibit the function of specific genes by preventing translation or splicing of their corresponding messenger RNA (mRNA). This has led to the publication of many studies of heart development in Xenopus that have advanced our understanding of this process in vertebrates (1–9).
The use of MOs began in the mid-1990s, when morpholine-based oligonucleotides were introduced into cultured cells to achieve inhibition of gene function (Partridge et al., 1996). Since then, MOs have been utilized extensively in vertebrate embryos, particularly those of the frog Xenopus and the zebrafish (Danio rerio) to achieve successful inhibition of specific genes (10–12). This type of experiment is invaluable in characterizing the function of a gene and also for confirming the results of genetic mapping studies by phenocopying the biological effects of mapped loss-of-function mutations isolated in genetic screens.
MO antisense oligonucleotides are synthetic nucleic acid analogs that consist of a six-membered morpholine ring instead of the normal five-membered sugar ring (13). These neutrally charged oligonucleotides are thereby stable, soluble, and bind to RNA with high-affinity. In addition, they are resistant to nuclease-degradation and have limited interaction with proteins (14–16). Consisting of 25 bases, MOs are designed to reduce gene function in two ways. First, the MO can be designed to target sequences in the 5′ untranslated region (UTR), close to the translation initiation codon of the gene. By binding to this location, the MO can sterically block the attachment of the ribosomal machinery and inhibit protein translation (11). Alternatively, they can be made to target the splice junctions within the pre-mRNA strand. When a MO complimentary to the splice donor site is used, this typically prevents the associated intron from being spliced out of the mRNA. This results in the incorporation of intron-encoded amino acids and, in many cases, early termination of translation due to the presence of stop codons present within the intron itself or created by a shift in the reading frame of subsequent sequences. MOs complimentary to the splice acceptor site tend to cause exon skipping, which occurs when the splice donor of the preceding exon cannot splice with the targeted acceptor site and instead splices with the acceptor of a more 3′ exon. This results in the targeted exon and its flanking introns being spliced out. Other aberrant splicing events have also been detected and the results of mRNA mis-splicing can vary in different instances, so these events should always be characterized for the particular MO used (17).
The decision to use a translation-blocking versus a splice-blocking MO strategy is often based on the availability of effective antibodies against the targeted gene product, as this determines the type of assay that can be used to demonstrate MO efficacy in vivo. When an antibody is available, a reduction in endogenous protein levels can be assayed by western blotting of electrophoresed embryo lysates (see Subheading 2.3). This is perhaps the best assay of MO efficacy and is ideal for translation-blocking strategies. When antibodies are not available, specific activity of a translation-blocking MO against its targeted sequence can be demonstrated in vivo through co-injection of targeted mRNA encoding a fusion with an epitope tag to which an antibody is available, or in vitro through the use of cell-free translation systems (see Subheading 2.4). Alternatively, a suitable splice-blocking MO may be used. In this case, reverse transcription polymerase chain reaction (RT-PCR) is used to monitor mis-splicing of the endogenous targeted mRNA (see Subheading 2.5).
Proper design of MOs is a critical factor in their effectiveness. Translation-blocking MOs should be designed against 25-bp target sequences within the 50-bp region centered on the translation initiation site. Similarly, splice-blocking MOs should be designed against 25-bp target sequences within a 50-bp region centered on either the splice donor or splice acceptor site. It is recommended that self-complementarity be avoided in order to prevent intrastrand pairing and/or dimer formation. In most cases, MO sequences will be suggested by the vendor based on these considerations and the submitted sequence. To demonstrate that a phenotype results from depletion of a particular targeted mRNA, it is advisable to use at least two independent MOs designed to target unique regions of the same mRNA, as these are unlikely to share off-target effects. Control MOs are also useful for validating the observations made in MO experiments. MOs containing five mismatched nucleotides distributed across their sequence are commonly used in negative control experiments, as are MOs targeting sequences from other species with no significant similarity to orthologous sequences in Xenopus, such as MOs against the human β-globin mRNA. Well-characterized MOs such as those against β-catenin may serve as positive controls for the MO microinjection procedure, should these be necessary.
Strategies other than MOs have been employed to inhibit gene function. These include RNA interference (RNAi), whereby the RNA is targeted for degradation by the binding of small-inhibitory RNA (siRNA) molecules and recruitment of the RNA-induced silencing complex (RISC), phosphorothioate-linked DNA (S-DNA) that employs cellular RNase H to cleave the target RNA strand, and peptide nucleic acid nucleotides (PNAs) that, similarly to MOs, sterically block RNA translation (14, 18). However, due to their few off-target effects, low cost, and binding success, MOs have become the favored tool for studying gene knockdown in vertebrate models (14, 19). In this chapter, we describe methods for conducting MO-mediated knockdown experiments in both Xenopus laevis and Xenopus tropicalis, together with associated methods for determining their efficacy.
2. Materials
2.1. Obtaining X. laevis and X. tropicalis Embryos
1-mL syringe.
Syringe needles (25-gauge, 5/8-in.).
Human chorionic gonadotropin (hCG) (Sigma, St. Louis, Missouri). 1,000 U/mL stock in dH2O. Store at 4°C.
Benzocaine (ethyl ρ-amino benzoate). 10% (w/v) stock solution in ethanol. Dilute to 0.05% in distilled water (dH2O) for use.
Dejellying solution: 2% (w/v) cysteine hydrochloride in water, pH adjusted to 8.0 with sodium hydroxide. Make fresh each day.
Kitchen shears.
Testis buffer: Leibovitz L-15 medium supplemented with 0.3 g/L l-glutamine (Sigma), 10% (v/v) bovine calf serum (defined, iron-supplemented, sterile-filtered; e.g., Hyclone), 50μg/mL gentamycin sulfate. Store at 4°C.
10× Marc’s Modified Ringers (MMR) solution (20): 0.1 M NaCl, 2 mM KCl, 1 mM MgSO4, 2 mM CaCl2, 5 mM HEPES pH 7.4. Store at room temperature. Dilute stock to 1× in dH2O for use. Store at room temperature.
10× Modified Barth’s Saline (MBS) pH 7.8: 880 mM NaCl, 10 mM KCl, 10 mM MgSO4, 50 mM HEPES pH 7.8, 25 mM NaHCO3. 1× MBS solution is made by mixing 100 mL of 10× stock solution with 700 μL 1 M CaCl2 and adjusting the volume to 1 L with dH2O. Dilute to 0.1× working solution with dH2O for storing developing embryos. Store at room temperature.
2.2. Microinjection of Morpholinos
Microinjection buffer: 1× MBS, 4% (w/v) Ficoll 400 (Sigma). Store at 16°C for up to a week.
Sterile nuclease-free H2O.
Mineral oil.
Glass capillaries (0.8–1.0 × 102-mm Kwik-Fil Borosilicate glass capillaries, 1B100F-4, World Precision Instruments).
Micropipette puller (Sutter Instrument Co., model P-87).
Micromanipulator (Narishige M-152 and Kanetec USA Corp. magnetic base, or Singer Instruments Mk1).
Microinjector (Narishige IM 300).
Stereo dissecting microscope (Leica MZ6).
Petri dishes (35-mm, 5-cm, and 10-cm).
Microinjection dish: 5-cm dish with nylon mesh glued to inner surface (approximate mesh diameter 0.8 mm, Small Parts, Inc.).
Plastic transfer pipettes.
Parafilm.
Graticule with 100-μm divisions.
Fine-tipped forceps.
2.3. Embryo Lysis and Western Blotting
Embryo lysis buffer: 50 mM Tris pH 7.6, 150 mM NaCl, 6 mM EDTA, 10% Triton X-100, protease inhibitor cocktail tablets (Roche) in distilled water. Use within 1 day if adding protease inhibitors. Store at 4°C.
Sonicator (Bioruptor, Diagenode.)
Loading buffer: 4× NuPAGE LDS Sample buffer (Invitrogen). Dilute to 1× with lysis sample and add 1 μL β-mercaptoethanol per sample.
Precast SDS-PAGE gel, e.g., NuPAGE 4–12% Bis-Tris Gel, 1.0 mm × 12 well (Invitrogen).
Xcell SureLock Mini-Cell electrophoresis system (Invitrogen).
Running buffer: 20× MES buffer (Invitrogen).
Protein molecular weight standards.
Nitrocellulose membrane, PVDF membrane.
Transfer apparatus, Mini Trans-blot Cell (BioRad).
Transfer buffer: 15 g Glycine, 3 g Tris-base in 1 L dH2O. Add 200 mL 100% methanol once dissolved. Make fresh on the day and chill before use.
20× TBST: 200 mM Tris–HCl pH 8, 3 M NaCl, 2% v/v Tween-20 in distilled water. Dilute in 1 L distilled water for 1× TBST working solution.
Blocking buffer: 5% skimmed milk powder in 1× TBST.
Whatman filter paper (Whatman Ltd.).
Appropriate primary and secondary antibodies.
Chemiluminescent substrate, e.g., ECL chemiluminescent visualization (Thermo Scientific).
Autoradiography film.
2.4. In Vitro Translation Blocking Assay
m7G-capped mRNA of gene of interest.
Nuclease-free sterile water.
Dry-block heater.
RNasin RNase inhibitor (Promega).
Nuclease-treated rabbit reticulocyte lysate translation system (Promega).
2.5. Total RNA Purification and RT-PCR
TRIzol reagent (Invitrogen) or RNeasy mini column(s) (Qiagen).
DNase I, e.g., RQ1 DNase (Promega).
SuperScript reverse transcription system (Invitrogen).
RNasin RNase inhibitor (Promega).
Magnesium chloride solution, 25 mM in nuclease-free water, filter-sterilized.
Deoxynucleotide triphosphate (dNTP) mix: dATP, dCTP, dTTP, and dGTP, each at 10 μM in nuclease-free water.
10× Taq polymerase buffer: 500 mM potassium chloride, 100 mM Tris hydrochloride, pH 9.0, 1% (v/v) Triton X-100, 25 mM magnesium chloride.
Taq polymerase.
Thin-walled PCR tubes.
DNA molecular weight standards.
Thermal cycler, e.g., GeneAmp PCR System 9700 (Applied Biosystems).
3. Methods
3.1. Obtaining X. laevis and X. tropicalis Embryos
The day before you intend to inject embryos, adult females must be induced to lay eggs by administration of hCG, a procedure termed “priming.” This procedure is similar for both X. laevis and X. tropicalis. Once collected, eggs are fertilized in vitro to provide synchronously cleaving early embryos. Haste is important when fertilizing Xenopus embryos, therefore all materials required should be made ready beforehand.
For X. laevis, inject 500 U of hCG into a dorsal lymph sac of each adult female (see Note 1) and keep them in aquatic system water overnight (approximately 16 h) at 16°C in the dark.
For X. tropicalis, inject 20 U of hCG into a dorsal lymph sac of each adult female and male required, then keep in aquatic system water overnight (approximately 20 h) at 23–28°C (see Notes 2 and 3). Inject a second dose of 100 U of hCG the next morning.
The next morning, replace the water in the tanks with 1× MMR solution (3 inches depth for X. laevis, 2 inches depth for X. tropicalis) and leave the frogs at room temperature for ovulation. Remove debris or feces from tank to limit pollution of eggs. The eggs can remain in the tank until there are a sufficient number to fertilize.
Kill a male by immersion in 0.05% benzocaine for 30 min, followed by decapitation and pithing. Dissect out the testes into 10 mL of testis buffer. Testes may be stored at 16°C for up to a week in 10 mL testis buffer.
Collect eggs from the tank into a 10-cm Petri dish (approximately 300–500 X. laevis eggs, approximately 600–800 X. tropicalis eggs) and remove as much MMR as possible, allowing the eggs to form a monolayer (see Note 4).
Take 1/3rd X. laevis testis, or both X. tropicalis testes and macerate using forceps.
Carefully pass the macerated testis tissue over the eggs, ensuring that all eggs come into contact with it. Once done, allow the eggs to sit undisturbed for 3 min.
Flood the fertilized eggs with 0.1× MBS (approximately 30 mL) and allow to sit undisturbed for 10–15 min at room temperature. Contraction of the dark pigment at the animal pole and/or rotation of the egg within its vitelline membrane so that the animal pole is uppermost are both signs of fertilization.
Once fertilization has occurred, the outer jelly coat should be removed by treatment with dejellying solution. Add approximately 20–30 mL of solution to the embryos and gently agitate to mix. Do not swirl as this may lead to the formation of a duplicated anteroposterior axis in the embryos (see Note 5). Once the jelly coats have been removed, pour off the solution and wash the embryos thoroughly (approximately five times) in 0.1× MBS. X. laevis embryos may be incubated at 16°C, X. tropicalis at 23°C, prior to microinjection (see Notes 6 and 7).
3.2. Microinjection of Morpholinos
We utilize microinjection techniques to introduce MOs into the fertilized egg at the one-cell stage; however, it is also possible to use other transfection methods, e.g., Endo-porter (21) and electroporation (22).
Prepare several glass capillary needles using a micropipette puller at settings to give a fine gauge (Fig. 1b). We use a Sutter Instrument Co. model P-87 flaming/brown micropipette puller at the following settings: heat 781, pull 50, velocity 80, time 150, and pressure 200. The settings required may vary between instruments and between individual tungsten filaments and may need adjustment. Store needles in a safe box to limit breakage.
Set up instruments for microinjection beside a suitable stereo dissecting microscope. Place the needle in the holder of a three-axis micromanipulator mounted onto a magnetic base (Fig. 1a).
Lower the needle onto the floor of the dish gently so that its tip slightly bends. Using fine-tipped forceps, break the tip of the glass pipette at this point. Figure 1b shows a needle suitable for microinjecting Xenopus embryos.
After breaking the tip, equilibrate (or “balance”) the pressure in the needle using the injector apparatus before drawing up any solution (see Note 8).
Fill the needle with approximately 1 μL of sterile water dispensed onto Parafilm on the microscope stage.
To calibrate the needle by droplet volume: Fill a 35-mm petri dish with a thin layer of mineral oil. Place the dish on top of a graticule. Lower the tip of the needle into the oil and inject a droplet of water. Measuring the diameter (d) of the droplet against the graticule allows its volume to be calculated as 4/3π(1/2d)3, assuming that the droplet is spherical. A 10-nL volume is suitable for microinjection into Xenopus, corresponding to a droplet with a diameter of 268 μm. Adjust the volume by either altering the injection time (typically 200–500 ms) or pressure (typically 9–12 psi). Calibration may be verified by comparing the actual and expected numbers of injections required to expel a known volume of solution from the needle.
Once calibrated, fill the needle with an appropriate volume of MO solution from a drop dispensed onto Parafilm.
Transfer embryos into the microinjection dish containing microinjection buffer. The embryos will orient themselves with their animal pole uppermost.
Inject into the animal hemisphere at an angle of approximately 30° from the equator of the embryo (see Note 9). Withdraw the needle gently from the embryo after each injection to minimize damage.
Collect injected eggs and place into a petri dish filled with microinjection solution.
Incubate the embryos at 16°C while they undergo early cleavage cycles, then transfer into 10 cm petri dishes containing 0.1× MBS. Culture embryos at 16–18°C until gastrulation is complete, and at 16–23°C thereafter.
Fig. 1.

Microinjection apparatus and glass capillary needle tip gauge. (a) Photograph of the assembled microinjection apparatus with a stereo dissecting microscope. (b) Image of a pulled glass capillary needle. The optimal breakage position is indicated (arrowhead). The needle is stained for visualization. Scale bar is 2 mm.
3.3. Embryo Lysis and Western Blotting
The efficacy with which a MO inhibits translation of a specific mRNA in embryos can be demonstrated directly using several approaches, all of which utilize the immunoblotting (or western blotting) technique. When a suitable antibody raised against the targeted gene product is available, the levels of the endogenous protein can be assayed in embryo lysates. If an antibody is not available, mRNA encoding an epitope-tagged form of the protein may be coinjected with the MO, followed by western blotting of embryo lysates with an antibody against the epitope tag. In this case, the injected mRNA must contain the target site for MO binding. The following details the methods for preparing whole-embryo lysates of X. laevis and X. tropicalis, and for western blotting analysis of protein levels.
Collect 10–15 embryos per stage, per condition to be analyzed, and remove as much buffer as possible using a micropipette. At this point, embryos may be snap-frozen in a dry ice-ethanol bath and stored at −80°C for later processing.
Add 100 μL lysis buffer per sample. Pipette to homogenize the embryos and sonicate at 4°C for 15 min with a 30-s on/30-s off cycle to shear genomic DNA (see Note 10).
Centrifuge samples at 18,000 × g for 5 min at 4°C to remove cellular debris. Transfer the supernatant to a clean 1.5-mL microfuge tube and either keep on ice (if it will be used immediately) or store at −80°C.
Prepare SDS-PAGE gels in the apparatus. Remove the protective strip and well comb. Pour 800 mL 1× MES buffer into the apparatus. Gently wash any excess storage buffer/residual acryl-amide out of the wells with 1× MES buffer using a pipette.
10–15 μg of embryo-equivalent protein lysate should be adequate to visualize protein depletion by western analysis. Mix the sample with 4× NuPAGE sample buffer in a new tube. Add 1 μL β-mercaptoethanol to each tube and mix. Boil sample in a water bath at 95°C for 5 min. Centrifuge briefly to collect the sample.
Load the samples into the wells, alongside appropriate protein molecular weight standards.
Run the gel at 150 V for approximately 1 h until the standard has separated appropriately.
While the gel is running, cut Whatman filter paper and PVDF membrane to an appropriate size, slightly larger than the gel. Prepare transfer buffer and prechill. Before the gel has finished running, pour some of the transfer buffer into a tray large enough to assemble the transfer sandwich cassette.
Once the gel is run, crack open the gel casting and discard. Gently place the gel in the tray with chilled transfer buffer. Excise and discard the wells and the thickened gel at the base.
Prewet the PVDF membrane for 10 s in 100% methanol, then store in transfer buffer. Use tweezers to handle the membrane.
Place the transfer cassette in the tray. The proteins will transfer from the gel onto the nitrocellulose membrane electrophoretically, hence the proteins will move from the cathode (usually marked black) to the anode (red) in the transfer apparatus. You must ensure to correctly prepare the transfer sandwich with the nitrocellulose membrane between the gel and the anode. First, lay down a sponge prewet with transfer buffer, then one piece of prewet filter paper. Roll out any bubbles with a glass rod from the center outwards. Place the gel gently on the filter paper, ensuring no bubbles are trapped. With tweezers, place the membrane onto the gel and add another prewet filter paper on top. Roll out any bubbles, and then add the last prewet sponge. Close the cassette securely and place into the transfer module, ensuring the gel-membrane sandwich is in the correct orientation with the membrane towards the anode.
Pour in the remaining transfer buffer and place at 4°C (see Note 11). Run at a current of 400 mA for 1 h.
Before the transfer is complete, prepare a washing container with 1× TBST. Prepare a sufficient amount of blocking buffer to cover the membrane.
Once the transfer is complete, open up the cassette and discard the filter paper. If using a prestained protein standard, the transfer of these bands to the membrane will indicate a successful transfer (see Note 12).
Quickly place the membrane into 1× TBST. Ensure it does not dry out. Rinse gently with 1× TBST, then replace the TBST buffer with sufficient blocking buffer to cover the membrane (approximately 10 mL for a 100 × 80 mm membrane). Incubate for 1 h at room temperature on a rocking platform.
Discard the blocking buffer, dilute the primary antibody in an appropriate volume of fresh blocking buffer, and add it to the membrane, ensuring it is sufficiently covered (see Note 13). Incubate for 1 h at room temperature or overnight at 4°C on a rocking platform.
Wash the membrane in 30 mL 1× TBST at room temperature on a rocking platform for 20 min. Repeat four times (see Note 14).
Prepare a 1:10,000 dilution of secondary horseradish peroxi-dase-conjugated antibody in an appropriate volume of blocking buffer. Discard the final 1× TBST wash. Add the blocking buffer with secondary antibody to the membrane and incubate for 20–60 min at room temperature on a rocking platform.
Discard antibody solution and wash the membrane as in step 17.
To visualize the protein, place the membrane on a dry plastic surface. With haste, pipette 500 μL of each ECL substrate component directly onto the membrane. Gently hand-rock the membrane to mix the solutions and ensure complete coverage. Wait 30 s before gently blotting the excess substrate from the membrane. Place the membrane between acetate sheet protectors or Saran-wrap and place into a light-tight developing cassette.
In a darkroom with safelight, carefully place a piece of autoradiography film on top of the membrane and close the cassette. An initial exposure time of 1 min will indicate whether subsequent exposures, of shorter or longer duration, are required. An example of a western blot of X. laevis embryos coinjected with mRNA encoding a V5-tagged protein and specific translation-blocking MOs is given in Fig. 2.
Fig. 2.

An example of translation-blocking MOs targeting the 5′UTR region of X. laevis genes Tbx5 and Tbx20. (a) Schematics showing the MO target locations on Tbx5 and Tbx20 mRNA. (b–e) Western blot analysis of Tbx5 and Tbx20 translation inhibition by specific MOs using V5 antibody. (b) Tbx5 MOs inhibit the in vitro translation of Tbx5 RNA fused with V5 epitope in a dose-dependent manner. Both Tbx20 (T20) MO and Con MO are unable to inhibit Tbx5-V5 translation. (c) Tbx20 MOs inhibit the in vitro translation of Tbx20 RNA tagged with V5 epitope in a dose-dependent manner. Both Tbx5 (T5) MO and Con MO cannot inhibit Tbx20-V5 translation. (d) Embryos were injected with 2 ng Tbx5-V5 RNA and Tbx5 MOs. Translation of Tbx5 was inhibited in vivo by Tbx5 MO in X. laevis animal caps in a dose-dependent manner as assessed by anti-V5 western blot. Tbx20 and Con MO are unable to reduce Tbx5-V5 protein expression. The membrane was reprobed with anti-Shp2 antibody as a loading control. (e) Translation of 2 ng Tbx20-V5 RNA is inhibited by Tbx20 MOs in X. laevis animal caps in a dose-dependent manner. Tbx5 and Con MO are unable to reduce Tbx20-V5 protein expression, assayed by western blot with anti-V5 antibody. Reproduced from (24) with permission from The Company of Biologists.
3.4. In Vitro Translation-Blocking Assay
The ability of a MO to bind to its target mRNA and inhibit translation can be tested in vitro through the use of cell-free translation systems, such as reticulocyte lysate or wheatgerm extract, providing an alternative to the in vivo assays described in Subheading 3.3. The method described here uses purified m7G-capped mRNA in a reticulocyte lysate translation system, but can be easily adapted to use coupled transcription-translation systems (e.g., TnT, Promega). A second MO designed to target an unrelated mRNA, or a 5-bp mismatched MO serves as negative controls to demonstrate the sequence specificity of the inhibitory effect.
Approximately 1.5 μg of m7G-capped mRNA encoding the targeted protein (including the site for MO binding) should be denatured before assembling the translation reaction. Mix the capped mRNA with nuclease-free water to a final volume of 11 μL. Heat at 95°C in a dry-block heater for 2 min, then chill immediately in an ice-water bath. Keep on ice until adding it to the reaction.
Add 2 μL of MO (0.5–5 μg) to 35 μL of nuclease-treated rabbit reticulocyte lysate on ice and mix by gentle pipetting (see Note 15).
Add 1 μL amino acid mix (see Note 16) and 1 μL RNasin to the reaction. Mix by gentle pipetting.
Add the 11 μL of denatured capped mRNA prepared in step 1 and incubate at 30°C for 90 min (see Notes 16 and 17). Samples may be stored at −80°C.
Analyze the 5–15 μL samples of translation products by SDS-PAGE and western blotting (see Note 16) (Fig. 2), as described in Subheading 3.3, steps 4–21.
3.5. Total RNA Purification and RT-PCR
If using splice-blocking MOs, the reverse transcription polymerase chain reaction (RT-PCR) can be used to characterize mis-splicing of the transcript in vivo. The design of primers for RT-PCR will depend upon the intron-exon structure of the gene and the predicted mis-splicing products, but in general they should amplify a minimum of 150 bp of the correctly spliced transcript along with either larger fragments resulting from intron inclusion (when the splice donor site is targeted), or differently sized fragments resulting from exon skipping (when the splice acceptor is targeted). This may necessitate the use of more than one pair of primers simultaneously in a multiplex RT-PCR reaction and may also require primers to be designed against introns as well as exons. These primers should be tested against cDNA or genomic DNA templates to ensure that they effectively amplify their predicted targets. It should be noted that standard RT-PCR can only provide a semiquantitative assay of the relative abundance of different splice forms of a transcript. Quantitative RT-PCR (qPCR) techniques may provide a more accurate measure, if required. The products of RT-PCR reactions may be cloned, sequenced, and aligned with a reference sequence from the targeted gene to confirm their origin.
Collect ten embryos at an appropriate stage (based on the temporal expression profile of the targeted gene) per condition for RT-PCR analysis.
Isolate total RNA using either TRIzol reagent or RNeasy mini columns, following the manufacturer’s protocol for RNA purification from animal cells.
Following RNA isolation, treat samples with DNase to remove all genomic DNA. Add 2 U of DNase (e.g., RQ1 DNase, Promega) to each sample, mix, centrifuge briefly, and incubate at 37°C for 1 h. To heat-inactivate the enzyme, incubate at 65°C for 10 min. Place samples on ice. Subsequent RNA-clean up may be required using RNeasy spin columns following manufacturer’s protocol.
Dilute RNA samples to 250 ng/μL final concentration with sterile, nuclease-free water.
To synthesize first-strand cDNA with SuperScript reverse transcriptase: Mix 1 μL of 250 ng/μL random primers, 2 μL 250 ng/μL total RNA, 1 μL 10 mM dNTP mix, 8 μL sterile, nuclease-free water. Incubate at 65°C for 5 min. Chill in ice water and centrifuge briefly (<20 s) before adding 4 μL first-strand buffer, 2 μL 0.1 M DTT and 1 μL RNase inhibitor. Incubate at 42°C for 2 min before adding 1 μL of reverse transcriptase (see Note 18). Incubate for 50 min at 42°C, then for 15 min at 70°C. Add 1 μL of RNase H and incubate at 37°C for 20 min. Store the reaction at −20°C until ready to carry out PCR.
To perform PCR: Select thin-walled PCR tubes of an appropriate size for the thermal cycler to be used and assemble the reactions on ice. Mix 0.5 μL of first-strand cDNA reaction, 5 μL of 10× Taq polymerase buffer, 5 μL 25 mM μL 10 mM MgCl2, 1 dNTP mix, 25 picomoles of each sequence-specific primer, sufficient nuclease-free water to bring the final volume to 49 μL, and 1 μL Taq polymerase. Cycle as appropriate for synthesis.
Analyze the reaction products by electrophoresis on a 1% (w/v) agarose gel, alongside suitable molecular weight standards. An example of how RT-PCR can be used to determine the efficacy of splice-blocking MOs is given in Fig. 3.
Fig. 3.
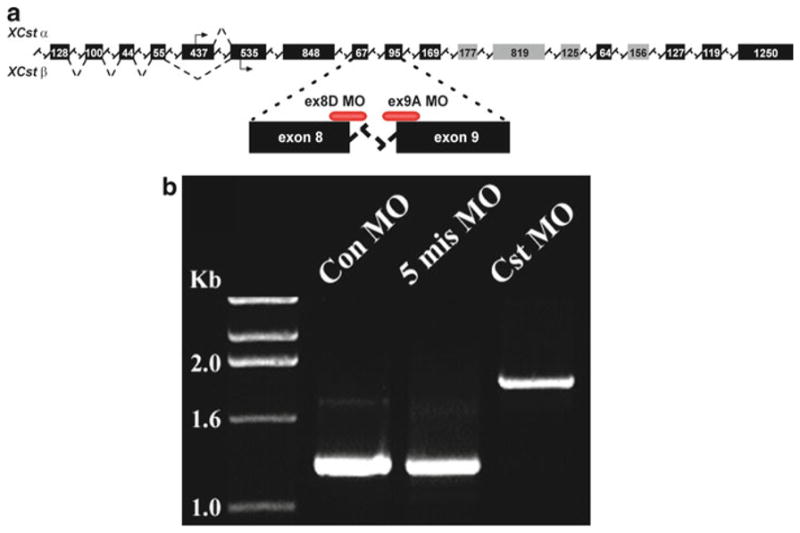
An example of a splice-blocking MO targeting the X. laevis gene Castor (Cst). (a) The position of the Cst splice junction morpholinos relative to the pre-mRNA transcript. These MOs target the splice donor site on exon 8 (ex8D MO) and the splice acceptor site on exon 9 (ex9A MO). (b) RT-PCR analysis of stage 42 X. laevis tadpoles injected at the one-cell stage with both the ex8D and ex9A MOs, demonstrating the inhibition of proper splicing of Cst pre-mRNA and the inclusion of a 679-bp intron, as indicated by the larger amplified product compared to control (Con) MO and 5-mismatch (5-mis) MO. Abrogation of splicing in this context introduced a stop codon after the second amino acid following exon 8. Reprinted from Christine and Conlon (23), Copyright (2008), with permission from Elsevier.
Footnotes
Chorionic gonadotropin is considered biohazardous so caution must be used, gloves must be worn at all times when handling the hormone, and needles must be disposed of in the correct manner.
Mild anesthesia can help to subdue very active X. tropicalis for injection, reducing the risk of injury. Immerse frogs for 25 min in a 0.025% (w/v) solution of ethyl 3-aminobenzoate methanesulfonate salt in water. Monitor anesthesia carefully and remove frogs from the anesthetic solution once they become sufficiently inactive for safe handling and injection.
Exposing X. tropicalis to temperatures below 23°C can be lethal to them.
Check egg quality under a microscope to assess for dead or dying eggs (larger and white), color density on animal pole (should be solid and not speckly, with a noticeable defined border between the two poles), and shape abnormalities (eggs should be roughly spherical).
Watch closely to see the jelly coats dissolve from the embryos. This should take approximately 3–5 min at room temperature. Embryos will appear to cluster more tightly without the jelly coats. Do not leave embryos in dejellying solution for too long as this may damage them and can also affect the firmness of the embryo, making microinjection more difficult.
Eggs that did not successfully fertilize will normally stick to the plate and have a slightly flattened appearance. Fertilized eggs will generally be free-floating and spherical.
The time window between dejellying and the first embryonic cleavage is typically 30–40 min for X. laevis, 25–30 min for X. tropicalis (depending upon temperature). Subsequent cleavages occur at approximately 30 and 20 min intervals for X. laevis and X. tropicalis, respectively. This should be taken into account when planning microinjections.
Each time the needle is replaced or rebroken, it must be rebalanced to ensure that there is no net positive or negative pressure on the injection solution in the needle. Positive pressure causes the injection solution to leak from the tip of the needle, while negative pressure causes the needle to take up buffer from the injection dish resulting in a decreased concentration of MO in the injection solution.
Generally it is not advised to inject >80 ng of MO per embryo. We test our MOs by initially injecting a range of concentrations, e.g., 20, 40, and 80 ng per X. laevis embryo in a 10-nL volume, and assessing the efficacy of the MO by the methods described. MOs are aliquoted and stored at −20°C. Before injection allow the aliquot to reach room temperature to resolubilize the oligonucleotide. Other sources state storing MOs at room temperature is adequate. Should the MO come out of solution, it is suggested to heat the aliquot to 65°C for 5 min and allow to cool to room temperature. Centrifuge the aliquot for 30 s at 18,000 × g prior to injection (http://www.gene-tools.com/node/25).
Unsheared genomic DNA in the samples will lead to significant difficulties when loading the polyacrylamide gel.
Alternatively, the apparatus may be set in a tray of ice with an ice pack placed in the module for transferring at room temperature.
When unstained standards are used, other staining procedures can be performed on the membrane to determine transfer success, such as Coomassie and Ponceau staining.
An antibody against a standard protein can serve as an internal loading control. This incubation can be performed simultaneously with the target antibody if the sizes of the corresponding proteins are known to be sufficiently different, otherwise it may be necessary to either run two separate gels to transfer and blot separately, or to strip the blot and reprobe with the second primary antibody after ECL visualization. To strip the blot, there are two methods; low and high stringency incubations. First, wash the membrane in water for 5 min. For low stringency stripping, incubate the membrane in 0.2 M NaOH for 20 min at room temperature; for high stringency prepare a 62.5 mM Tris pH6.8 buffer with 2% w/v SDS. Before use, add β-mercaptoethanol to a final concentration of 100 mM. Incubate the membrane in this buffer for 30 min at 55–65°C. After incubation, wash the membrane in water for 5 min, followed by three washes in 1× TBST, before proceeding with the western procedure (blocking and primary antibody incubation).
The primary antibody-blocking solution can be reused at least once. Store at 4°C for subsequent uses.
The MO should be added to the translation reaction before adding the capped mRNA to ensure maximal inhibition of translation.
If a suitable antibody is not available for western blotting, the translation products can be radiolabeled by incorporation of 35S methionine (in presence of amino acid mix lacking methionine) and analyzed by SDS-PAGE and autoradiography.
If radiolabeling will be used, reduce the volume of the capped mRNA in step 1 to compensate for 35S methionine added in step 3, maintaining a final reaction volume of 50 μL.
Include a “minus RT” reaction in which the reverse transcriptase is omitted. This provides a control to demonstrate that PCR products are derived from reverse-transcribed RNA, not from sources of contamination such as genomic DNA carried over from the RNA purification procedure.
References
- 1.Movassagh M, Philpott A. Cardiac differentiation in Xenopus requires the cyclin-dependent kinase inhibitor, p27Xic1. Cardiovasc Res. 2008;79:436–447. doi: 10.1093/cvr/cvn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nagao K, Taniyama Y, Kietzmann T, Doi T, Komuro I, Morishita R. HIF-1alpha signaling upstream of NKX2.5 is required for cardiac development in Xenopus. J Biol Chem. 2008;283:11841–11849. doi: 10.1074/jbc.M702563200. [DOI] [PubMed] [Google Scholar]
- 3.Kumano G, Ezal C, Smith WC. ADMP2 is essential for primitive blood and heart development in Xenopus. Dev Biol. 2006;299:411–423. doi: 10.1016/j.ydbio.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 4.Inui M, Fukui A, Ito Y, Asashima M. Xapelin and Xmsr are required for cardiovascular development in Xenopus laevis. Dev Biol. 2006;298:188–200. doi: 10.1016/j.ydbio.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 5.Zhang C, Basta T, Klymkowsky MW. SOX7 and SOX18 are essential for car-diogenesis in Xenopus. Dev Dyn. 2005;234:878–891. doi: 10.1002/dvdy.20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garriock RJ, D’Agostino SL, Pilcher KC, Krieg PA. Wnt11-R, a protein closely related to mammalian Wnt11, is required for heart morphogenesis in Xenopus. Dev Biol. 2005;279:179–192. doi: 10.1016/j.ydbio.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 7.Small EM, Warkman AS, Wang DZ, Sutherland LB, Olson EN, Krieg PA. Myocardin is sufficient and necessary for cardiac gene expression in Xenopus. Development. 2005;132:987–997. doi: 10.1242/dev.01684. [DOI] [PubMed] [Google Scholar]
- 8.Hilton EN, Manson FD, Urquhart JE, Johnston JJ, Slavotinek AM, Hedera P, Stattin EL, Nordgren A, Biesecker LG, Black GC. Left-sided embryonic expression of the BCL-6 corepressor, BCOR, is required for vertebrate laterality determination. Hum Mol Genet. 2007;16:1773–1782. doi: 10.1093/hmg/ddm125. [DOI] [PubMed] [Google Scholar]
- 9.Bartlett HL, Weeks DL. Lessons from the lily pad: Using Xenopus to understand heart disease. Drug Discov Today Dis Models. 2008;5:141–146. doi: 10.1016/j.ddmod.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Draper BW, Morcos PA, Kimmel CB. Inhibition of zebrafish fgf8 pre-mRNA splicing with morpholino oligos: a quantifiable method for gene knockdown. Genesis. 2001;30:154–156. doi: 10.1002/gene.1053. [DOI] [PubMed] [Google Scholar]
- 11.Heasman J, Kofron M, Wylie C. Beta-catenin signaling activity dissected in the early Xenopus embryo: a novel antisense approach. Dev Biol. 2000;222:124–134. doi: 10.1006/dbio.2000.9720. [DOI] [PubMed] [Google Scholar]
- 12.Nutt SL, Bronchain OJ, Hartley KO, Amaya E. Comparison of morpholino based translational inhibition during the development of Xenopus laevis and Xenopus tropicalis. Genesis. 2001;30:110–113. doi: 10.1002/gene.1042. [DOI] [PubMed] [Google Scholar]
- 13.Moulton JD. Using morpholinos to control gene expression. Curr Protoc Nucleic Acid Chem Chapter. 2007;4(Unit 4):30. doi: 10.1002/0471142700.nc0430s27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Summerton JE. Morpholino, siRNA, and S-DNA compared: impact of structure and mechanism of action on off-target effects and sequence specificity. Curr Top Med Chem. 2007;7:651–660. doi: 10.2174/156802607780487740. [DOI] [PubMed] [Google Scholar]
- 15.Bill BR, Petzold AM, Clark KJ, Schimmenti LA, Ekker SC. A primer for morpholino use in zebrafish. Zebrafish. 2009;6:69–77. doi: 10.1089/zeb.2008.0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eisen JS, Smith JC. Controlling morpholino experiments: don’t stop making antisense. Development. 2008;135:1735–1743. doi: 10.1242/dev.001115. [DOI] [PubMed] [Google Scholar]
- 17.Morcos PA. Achieving targeted and quantifiable alteration of mRNA splicing with Morpholino oligos. Biochem Biophys Res Commun. 2007;358:521–527. doi: 10.1016/j.bbrc.2007.04.172. [DOI] [PubMed] [Google Scholar]
- 18.Dagle JM, Weeks DL. Oligonucleotide-based strategies to reduce gene expression. Differentiation. 2001;69:75–82. doi: 10.1046/j.1432-0436.2001.690201.x. [DOI] [PubMed] [Google Scholar]
- 19.Knudsen H, Nielsen PE. Antisense properties of duplex- and triplex-forming PNAs. Nucleic Acids Res. 1996;24:494–500. doi: 10.1093/nar/24.3.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ubbels GA, Hara K, Koster CH, Kirschner MW. Evidence for a functional role of the cytoskeleton in determination of the dorsoventral axis in Xenopus laevis eggs. J Embryol Exp Morphol. 1983;77:15–37. [PubMed] [Google Scholar]
- 21.Summerton JE. Endo-Porter: a novel reagent for safe, effective delivery of substances into cells. Ann N Y Acad Sci. 2005;1058:62–75. doi: 10.1196/annals.1359.012. [DOI] [PubMed] [Google Scholar]
- 22.Falk J, Drinjakovic J, Leung KM, Dwivedy A, Regan AG, Piper M, Holt CE. Electroporation of cDNA/Morpholinos to targeted areas of embryonic CNS in Xenopus. BMC Dev Biol. 2007;7:107. doi: 10.1186/1471-213X-7-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christine KS, Conlon FL. Vertebrate CASTOR is required for differentiation of cardiac precursor cells at the ventral midline. Dev Cell. 2008;14:616–623. doi: 10.1016/j.devcel.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown DD, Martz SN, Binder O, Goetz SC, Price BM, Smith JC, Conlon FL. Tbx5 and Tbx20 act synergistically to control vertebrate heart morphogenesis. Development. 2005;132:553–563. doi: 10.1242/dev.01596. [DOI] [PMC free article] [PubMed] [Google Scholar]