

Abstract
The carboxylic acid functional group can be an important constituent of a pharmacophore, however, the presence of this moiety can also be responsible for significant drawbacks, including metabolic instability, toxicity, as well as limited passive diffusion across biological membranes. To avoid some of these shortcomings while retaining the desired attributes of the carboxylic acid moiety, medicinal chemists often investigate the use of carboxylic acid (bio)isosteres. The same type of strategy can also be effective for a variety other purposes, for example, to increase the selectivity of a biologically active compound or to create new intellectual property. Several carboxylic acid isosteres have been reported, however, the outcome of any isosteric replacement cannot be readily predicted as this strategy is generally found to be dependent upon the particular context (i.e., the characteristic properties of the drug and the drug–target). As a result, screening of a panel of isosteres is typically required. In this context, the discovery and development of novel carboxylic acid surrogates that could complement the existing palette of isosteres remains an important area of research. The goal of this Minireview is to provide an overview of the most commonly employed carboxylic acid (bio)isosteres and to present representative examples demonstrating the use and utility of each isostere in drug design.
Keywords: bioisosteres, carboxylic acids, drug design, isosteric replacement
Introduction
The carboxylic acid functional group plays a cardinal role in the biochemistry of living systems as well as in drug design. A wide variety of endogenous substances, such as amino acids, triglycerides and prostanoids, possess the carboxylic acid moiety. Furthermore, this functional group is often part of the pharmacophore of diverse classes of therapeutic agents.[1] Indeed, a large number (>450) of carboxylic acid-containing drugs have been marketed worldwide, including widely used nonsteroidal anti-inflammatory drugs (NSAIDs), antibiotics, anticoagulants, and cholesterol-lowering statins, among others. The acidity, combined with the ability to establish relatively strong electrostatic interactions and hydrogen bonds, is the reason this functional group is often a key determinant in drug-target interactions. However, despite the success of carboxylic acid drugs, the presence of a carboxylic acid residue in a drug or a drug candidate can represent a liability. For instance, a diminished ability to passively diffuse across biological membranes can raise a significant challenge, particularly in the context of central nervous system (CNS) drug discovery, where the blood–brain barrier (BBB) can be relatively impermeable to negatively charged carboxylates.[2] Furthermore, idiosyncratic drug toxicities arising from the metabolism of the carboxylic acid moiety (e.g., glucuronidation)[3] have been linked to withdrawals of marketed drugs.[4] Thus, to avoid these and other possible shortcomings, the replacement of the carboxylic acid functional group with a suitable surrogate, or (bio)isostere, can represent an effective strategy.
The concept of bioisosterism is based on the notion that single atoms, groups or whole molecules that exhibit similar volume, shape, and/or physicochemical properties can produce broadly similar biological effects.[5] However, as occasionally observed, isosteric replacements can result in derivatives that exhibit different or even opposite pharmacology relative to the parent carboxylic acids. The outcome of any isosteric replacement cannot be accurately predicted, however, the physicochemical properties of both the isostere and the corresponding binding partner(s) in the biological system are often a key factor. In addition to the acidity of the carboxylic acid surrogate, several other parameters, such as size, shape, charge distribution and lipophilicity, play important roles. The relative importance of each of these properties depends on the particular context considered. Thus, for instance, whereas lipophilicity of the isostere might be one of the most important properties when designing analogues for improved passive diffusion across biological membranes, geometry and charge distribution of the acidic moiety can be critical aspects if the carboxylic acid isostere is required to establish two-point interactions with an arginine side chain in the binding site. Several carboxylic acid surrogates have been reported that display utility in drug design (Figure 1). Herein, we summarize the key properties and provide representative examples describing the use of each of the carboxylic acid (bio)isosteres in drug design.
Figure 1.

Representative examples of a) acyclic and b) cyclic carboxylic acid bioisosteres and their corresponding predicted logD values as determined using Pipeline Pilot version 8.0 (Accelrys, Inc., San Diego, USA).
Hydroxamic Acids
Hydroxamic acids exhibit moderately acidic characteristics, with pKa values in the range of ~8–9,[6] and strong metal-chelating properties.[7] This functional group is found in a variety of natural products[8] and in a number of biologically active compounds.[9] Although hydroxamic acids are most commonly employed in drug design for their metal-chelating properties,[7] this functional group has also been employed successfully as a carboxylic acid bioisostere. Deprotonation of the hydroxamic acid moiety can lead either to the N or to O anion (Scheme 1),[10] depending on the nature of the substituents and solvent.[6,11] In polar solvents, and at physiological conditions, deprotonation of the hydroxamic acid yields both the N and O anions. In vivo, hydroxamic acids can undergo relatively rapid hydrolysis[12] to the corresponding carboxylic acids, although the stability of these compounds towards hydrolysis can be significantly improved by introducing relatively large substituents at the nitrogen atom of the hydroxylamine fragment.[13] Such modifications would also prevent the formation of the N anion. In addition, like carboxylic acids, hydroxamic acids can be metabolized via sulfation and glucuronidation, which ultimately could lead to the formation of toxic chemically reactive metabolites.[14] Recent examples of hydroxamic acids and esters employed as carboxylic acid bioisosteres include benzhydroxamic acids and esters as MAP/ERK kinase (MEK) inhibitors[15] that exhibit favorable activity profiles in the biochemical and cellular assays, as well as acceptable absorption, distribution, metabolism, and excretion (ADME)/pharmacokinetic (PK) properties (e.g., 3 and 4; Table 1).
Scheme 1.

Deprotonation equilibrium of hydroxamic acid, and the tautomerisation between the N and O anion.
Table 1.
Comparison of hydroxamate and carboxylate MAP/ERK kinase inhibitors in a cell-free kinase cascade assay.[15]
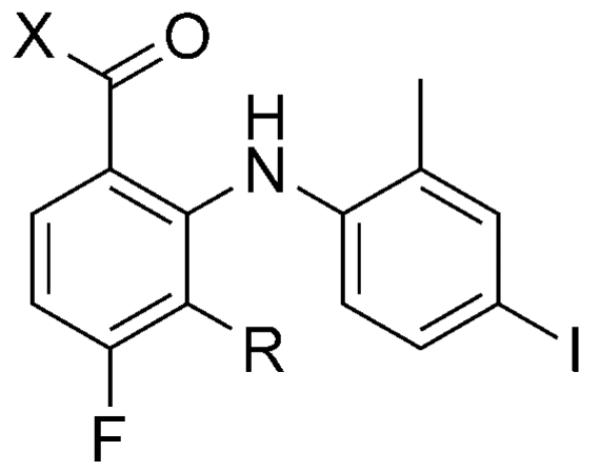
| |||
|---|---|---|---|
| Compd | X | R | IC50 [nm] |
| 1 | OH | H | 15 |
| 2 | OH | F | 5.8 |
| 3 | NHOH | H | 7.9 |
| 4 | NHOCH2cPr | F | 7.0 |
Phosphonic and Phosphinic Acids
The main characteristics of both phosphonic and phosphinic acids are the relatively high acidity, pKa1 value of ~1–3, and their nonplanar geometries.[17] Compared with carboxylic acids, these acidic moieties are more polar, thus these isosteres usually lead to a lowering of the partition coefficient (logP).[17] The use of these acidic moieties as replacements of the carboxylic acid functional group has been exploited successfully in the design of analogues of neurotransmitters γ-aminobutyric acid (GABA, 5) and glutamic acid (6) (Figure 2).[16,18] In many cases, while the affinity of the isosteric analogue is generally equivalent to that of the parent carboxylic acid compound, the instrinstic activity, however, is changed. One such example includes phaclofen (7),[19] the phosphonate derivative of the GABA agonist baclofen (8). In this case, while baclofen is a GABAB agonist, phaclofen (7) acts as an antagonist. In the case of mono- or di-alkylphosphinic acid derivatives of GABA, however, isosteric replacement of the carboxylic acid moiety resulted in highly potent agonists (e.g., 9, and 10; Table 2) that, unlike the natural ligand (GABA), are selective for GABAB receptors.[16] Similar outcome was observed for some glutamic acid derivatives, such as l-AP4 (11; Figure 2), which selectively activate the group III metabotropic glutamate receptors (mGluR).[18]
Figure 2.

The structures of GABA, glutamic acid and selected analogues.
Table 2.
Inhibition of binding of [3H]baclofen to GABAB receptors (cat cerebellum) and [3H]muscimol to GABAA receptors (rat cortex).[16]
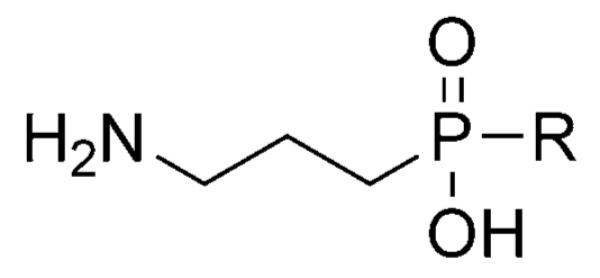
| |||
|---|---|---|---|
| Compd | R | IC50 [μm] | |
| GABAB | GABAA | ||
| GABA (5) | – | 0.025 | 0.128 |
| 9 | H | 0.0024 | 1.7 |
| 10 | Me | 0.0066 | inactive |
Sulfonic and Sulfinic Acids
Like phosphonic acids, sulfonic and sulfinic acids are nonplanar as well as both more polar and acidic (pKa<1) than carboxylic acids. Amino acids analogues bearing either sulfonic or sulfinic acids have been found to be relatively potent activators of the glutamate receptors.[20] Moreover, sulfonate derivatives of baclofen (8), such as saclofen (12; Figure 2), have been reported that, like phaclofen (7), exhibit GABA receptor antagonist properties.[21] Other notable examples of sulfonic acids being employed as carboxylic acid isosters include derivatives of the cholecystokinin-B (CCK-B) receptor antagonist 13. Whereas planar, heterocyclic carboxylic acid bioisosteres (e.g., 3-hydroxy-isoxazole 14) exhibited similar activity to 13, sulfonic acid analogue 15 was the only derivative bearing a nonplanar carboxylic acid isostere to produce low nanomolar IC50 values (cf., 15, 16, and 17; Table 3).[22]
Table 3.
Comparison of sulfonic and carboxylic acid CCK-B receptor antagonists.[22]
| ||||
|---|---|---|---|---|
| Compd | X | IC50 [nm] | pKa | |
| CCKB | CCKA | |||
| 13 | CH2COOH | 1.7 | 4500 | 5.6 |
| 15 | CH2SO3Na | 1.3 | 1010 | <1 |
| 14 |

|
2.6 | 1700 | 6.5 |
| 16 | CH2P(O)(OH)2 | 23 | 2700 | 3.4, 7.8 |
| 17 | CH2P(O)(OH)Me | 23 | 4400 | 3.7 |
Sulfonamides, Acylsulfonamides, and Sulfonylureas
Sulfonamides were among the first examples of carboxylic acid isosteres to show utility in drug design. Classical examples of this type of carboxylic acid surrogate include the sulfonamide class of antibacterial agents developed in the 1930s and 40s, such as sulfadiazine (18) and sulfanilamide (19), the active form of prontosil (20).[23] Compounds of this class work by inhibiting tetrahydropteroate synthetase. The similarities between the sulfanilamide anion in 19 and the carboxylate of the natural substrate, para-aminobenzoic acid (PABA; 21) were found to be critical to the bacteriostatic properties of the sulfanilamides.[24] Although nonplanar and generally only weakly
 acidic (pKa= ~10)[17] compared with PABA (pKa= ~6.5), the sulfonamide moiety can in fact establish a similar geometry of hydrogen bonds compared with carboxylic acids as the distance between the two oxygen atoms is similar to that found between the two oxygen atoms of a carboxylate. Furthermore, incorporation of appropriate substituents can lower the pKa value of the sulfonamide moiety to a value essentially equal to that of PABA, for example, sulfadiazine (18) that exhibits a pKa value of approximately 6.5.
acidic (pKa= ~10)[17] compared with PABA (pKa= ~6.5), the sulfonamide moiety can in fact establish a similar geometry of hydrogen bonds compared with carboxylic acids as the distance between the two oxygen atoms is similar to that found between the two oxygen atoms of a carboxylate. Furthermore, incorporation of appropriate substituents can lower the pKa value of the sulfonamide moiety to a value essentially equal to that of PABA, for example, sulfadiazine (18) that exhibits a pKa value of approximately 6.5.
In addition to sulfonamides, acylsulfonamides (also known as sulfonimides) and sulfonylureas have been used extensively as carboxylic acid surrogates. The pKa values of acysulfonamides and sulfonylureas fall within the range of carboxylic acids (4–5).[25] Notable examples include several cysteinyl leukotriene (LTE4) receptor antagonists, such as compounds 22 and 23 (Table 4), which exhibit improved activity compared with the parent carboxylic acid (24) and other derivatives bearing commonly used carboxylic acid bioisosteres, such as tetrazole (25).[26] Other interesting examples were reported by scientists at GlaxoSmithKline where replacement of the carboxylic acid moiety of β3-adrenergic receptor agonist 26 (Table 5) led to derivatives (27–29) with higher potency and greater functional selectivity for the β3-adrenergic receptor.[27]
Table 4.
Antagonists of the cysteinyl leukotriene (LTE4) receptor.[26]
| ||||
|---|---|---|---|---|
| Compd | X | Concn [μm] |
Inhibition[a] [%] |
pKd[b] |
| 24 | COOH | 3.3 | 100 | 6.6 |
| 25 |
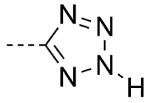
|
1 | 50 | 6.6 |
| 22 | CONHSO2Ph | 0.1 | 100 | 8.6 |
| 23 | CONHSO2nBu | 1.0 | 100 | – |
Inhibition of LTE4.
pKd determined in guinea pig tracheal spirals using LTE4 as an agonist.
Table 5.
Stimulation of cAMP accumulation in CHO cells expressing human β3, β2, and β1 adrenergic receptors by acylsulfonamides and sulfonylureas.[27]
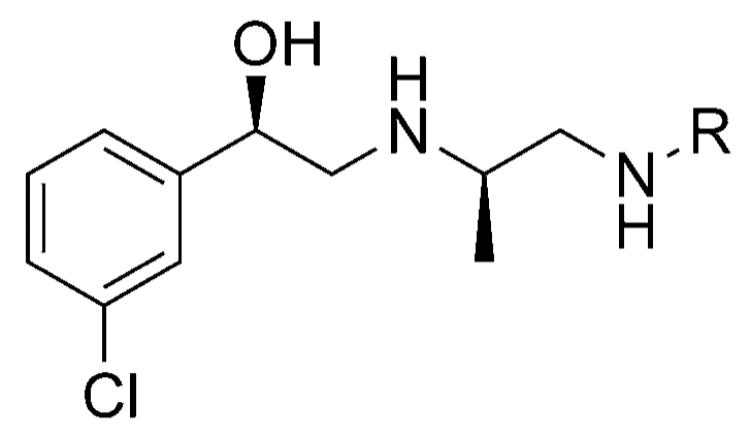
| ||||
|---|---|---|---|---|
| Compd | R | pEC50 | ||
| β 3 | β 2 | β 1 | ||
| 26 |
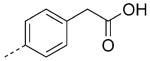
|
7.8 | 7.3 | 7.1 |
| 27 |
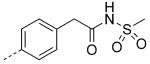
|
8.3 | 7.7 | 7.0 |
| 28 |
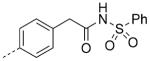
|
9.1 | 7.5 | 6.1 |
| 29 |
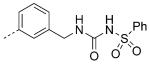
|
7.8 | 6.6 | 6.4 |
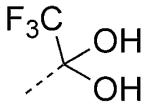
2,2,2-Trifluoroethan-1-ol and Trifluoromethylketones
Highly fluorinated alcohols and ketones comprise viable carboxylic acid bioisosteres. For example, 2,2,2-trifluoroethan-1-ol is a weakly acidic (pKa= ~12), nonplanar carboxylic acid bioisostere (Scheme 2). The most interesting feature of this carboxylic acid surrogate is the relatively high lipophilicity, which makes this moiety potentially attractive for CNS drug discovery programs, where the presence of a carboxylic acid moiety in a drug candidate can result in a limited rate of passive diffusion across the BBB.[2] Similar overall properties can be ascribed to the trifluoromethylketone moiety. Under aqueous physiological conditions, this functional group is in equilibrium with the hydrate form, with the latter being bioisosteric with the carboxylic acid moiety (Scheme 2).
Scheme 2.

Structure of carboxylic acid bioisosteres based on fluorinated alcohol and ketones. In the latter case, it is the hydrate form, which is present under physiological conditions, that is bioisosteric with the carboxylic acid moiety.
An interesting case study comparing these two carboxylic acid surrogates was reported by scientists at Merck in the design of brain-penetrant prostaglandin E receptor 1 (EP1) antagonists. Whereas carboxylic acid 30 (Table 6) was potent in the functional assay, this and other related carboxylic acid-containing analogues exhibited limited brain penetration as well as short half-lives in plasma.[28] Replacement of the carboxylic acid moiety with either the 2,2,2-trifluoroethan-1-ol or the trifluormethylketone moiety led to derivatives 31 and 32, respectively (Table 6) that retain excellent binding affinity for the EP1 receptor, but exhibit favorable ADME/PK properties including oral bioavailability and brain penetration.[28]
Table 6.
Examples of EP1 receptor antagonists bearing a carboxylic acid or 2,2,2-trifluoroethan-1-ol or trifluoro-methylketone hydrate isostere.[28]
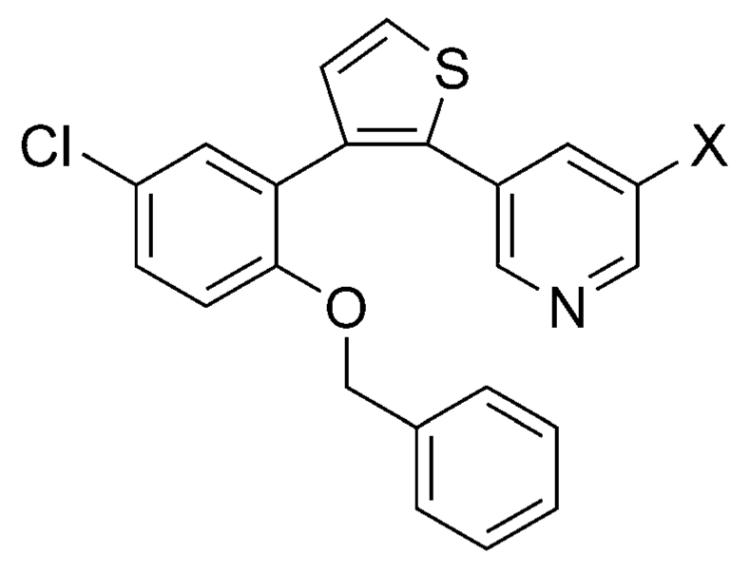
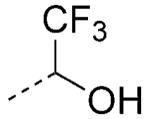
EP1 inhibitory constant.
Brain/plasma (B/P) ratio.
Half-life (t1/2) in rat.
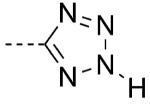
Tetrazoles
Tetrazoles are amongst the most commonly employed carboxylic acid isosteres; for recent Review articles, see Herr et al.[29] and Myznikov et al.[30] The 5-substituted tetrazoles exist in equilibrium between the 1H and the 2H tautomers (Scheme 3). The most important features of the tetrazolic acids are the planarity and acidity, which closely resemble those of carboxylic acids (pKa = ~4.5–4.9).[17,31] However, relative to carboxylic acids, tetrazoles are slightly larger.[32,33] Moreover, analysis of crystal structures and computational studies indicate that the hydrogen-bond environment surrounding the tetrazolates extends further than in the case of carboxylates.[33] In addition, tetrazolate anions are more lipophilic than the corresponding carboxylates,[34] and compared with the latter, they exhibit slightly different electrostatic potential[35] and charge distribution due to the delocalization of the negative charge over the five-membered ring system. Interestingly, experimental evidence suggests that tetrazoles, like carboxylic acids, are capable of forming two-point interactions with amidines.[36] However, the stability of the tetrazolate–amidine complex is found to be lower than the corresponding carboxylate–amidine salt.[36] Metabolically, tetrazoles can also undergo N-glucuronidation,[3] however, these adducts are not as chemically reactive as the O-glucuronides derived from carboxylic acids and hydroxamic acids and, therefore, have not been linked to toxic effects in humans.
Scheme 3.

Tautomers of 5-substituted tetrazoles.
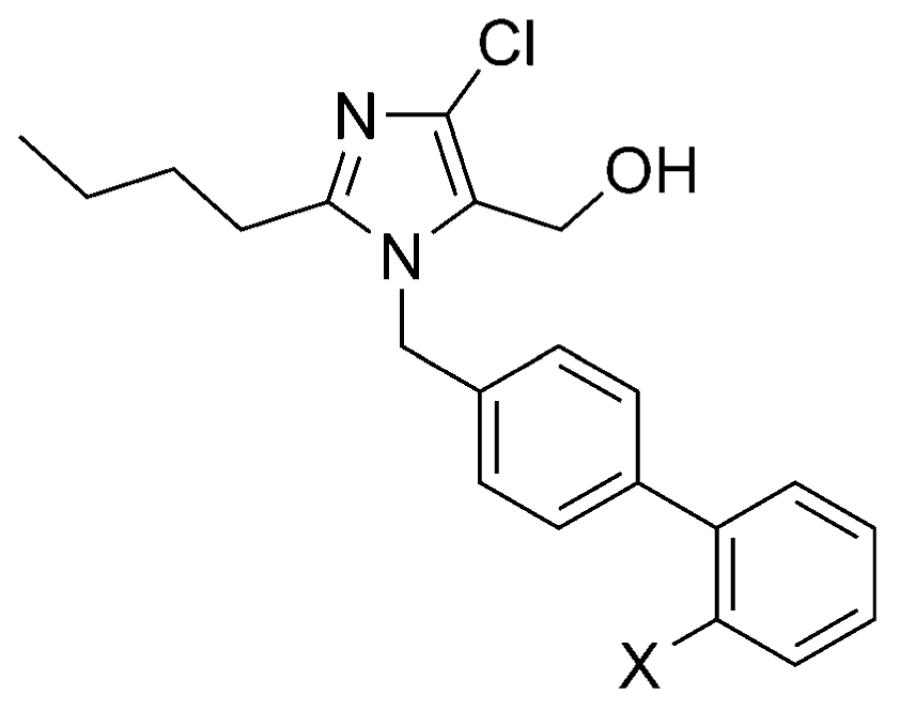
In drug design, the most important examples showcasing the utility of tetrazoles as carboxylic acid isosteres include several nonpeptidic angiotensin II type 1 (AT1) receptor antagonists.[37] The AT1 receptor is a member of the G protein-coupled receptor (GPCR) superfamily that plays an important role in vasoconstriction.[38] In a series of N-(biphenylylmethyl)imidazole AT1 receptor antagonists, where both carboxylic acid 33 and several bioisosteres (34–37) exhibited potent biological activity in vitro, only the tetrazole derivative, losartan (Cozaar, 38), was found to be effective after oral administration (Table 7).[39] Interestingly, site-directed mutagenesis and modeling studies indicated that the tetrazole moiety of losartan (38) and related antagonists occupy the same space within the AT1 receptor as the carboxylic acid terminus of the natural ligand, angiotensin, although the tetrazole ring appears to be involved in more complex sets of interactions with the surrounding amino acid residues, namely Lys119 and His256.[40] Notably, five out of six of the AT1 receptor antagonists used clinically for the treatment of high blood pressure contain a tetrazole moiety.[39,41]
Table 7.
AT1 receptor antagonist and in vivo activities of carboxylic acid 33 and a series of related isosteres.[39]
| |||||
|---|---|---|---|---|---|
| Compd | X | pKa estimated |
IC50[a] [μm] | Dose[b] [mgkg−1] | |
| iv | po | ||||
| 33 | COOH | 5 | 0.23 | 3 | 11[c] |
| 34 | CONHOH | 10.5 | 4.1 | 3 | >30[d] |
| 35 | CONHOCH3 | 10.9 | 2.9 | 10 | inactive[e] |
| 36 | CONHSO2Ph | 8.4 | 0.14 | >3[d] | 30 |
| 37 | NHSO2CF3 | 4.5 | 0.083 | 10 | 100 |
| 38 |
|
5–6 | 0.019 | 0.80[c] | 0.59[c] |
Inhibition of specific binding of [3H]angiotensin II (2 nm) to rat adrenal cortical microsomes.
Intravenous (iv) and oral (po) dose at which statistically significant drops in blood pressure were observed (>15 mmHg) in renal hypertensive rats.
Value given is the effective dose that lowers the blood pressure by 30 mmHg (ED30).
At this dose, no statistically significant drop in blood pressure was observed.
Compound failed to cause a significant drop in blood pressure even at 100 mgkg−1.
5-Oxo-1,2,4-oxadiazole and 5-Oxo-1,2,4-thiadiazole
The 5-oxo-1,2,4-oxadiazole and related thiadiazole systems (Figure 1) are planar acidic heterocycles (pKa= ~6–7),[42] which, like tetrazoles, have been successfully employed in the synthesis of AT1 receptor antagonists (Table 8).[43] Interestingly, these heterocycles are reported to be comparatively more lipophilic than the tetrazole; this property led to increased oral bioavailability of potent AT1 receptor antagonists (cf., 39, 40, and 41; Table 8).[43]
Table 8.
Inhibitory effects on the specific binding of [125I]AT1 and AT1-induced pressor responses in rats.[43]
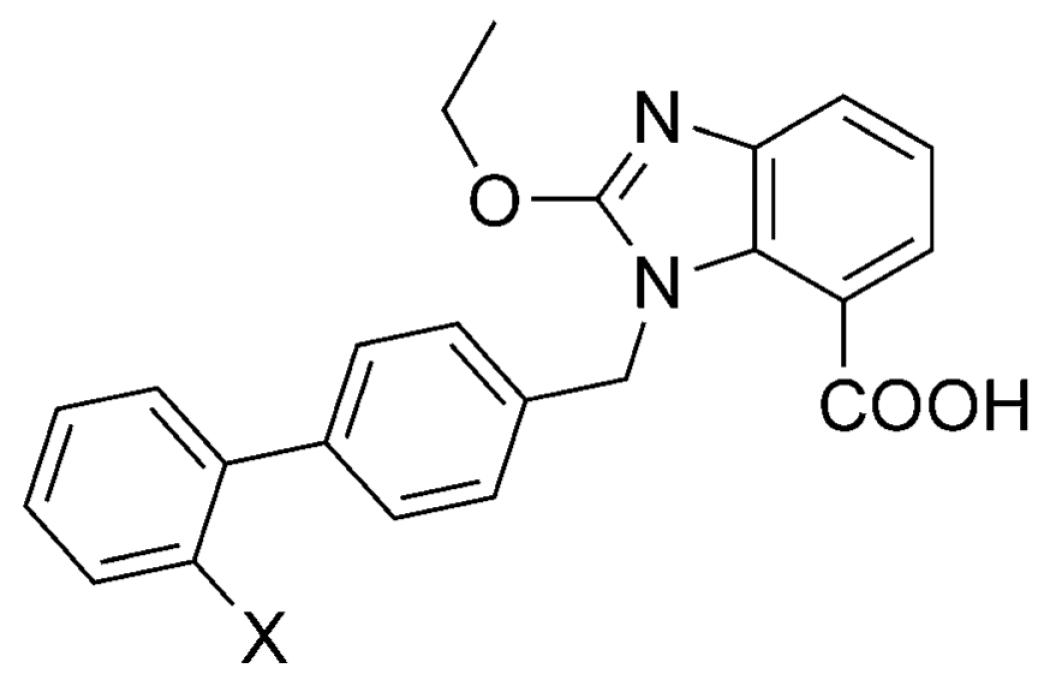
| ||||||
|---|---|---|---|---|---|---|
| Compd | X | Properties of X | BA[a] [%] |
IC50[b] [μm] |
Inhibition[c] [%] 3h/7 h |
|
| pKa | logP | |||||
| 39 |
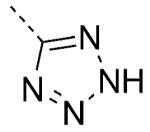
|
5.3 | 0.32 | 5 | 0.11 | 100/92 |
| 40 |
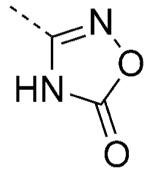
|
6.1 | 0.9 | 20 | 0.42 | 100/100 |
| 41 |
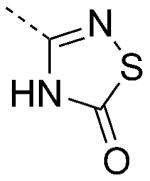
|
6.6 | 1.58 | 51 | 0.25 | 100/100 |
Oral bioavailability (BA).
Receptor affinity as determined by the inhibition of specific binding of [125I]AT1 (0.2 nm) to bovine adrenal cortex. The IC50 value is the concentration of compound which inhibits [125I]AT1 binding by 50%.
Percent inhibition of AT1 (0.1 μgkg−1 iv)-induced pressor response at 3 and 7 h after administration of the test compounds (1 mgkg−1 po) in conscious male Sprague–Dawley rats.
Thiazolidinedione, Oxazolidinedione, and Oxadiazolidine-dione
The thiazolidinedione heterocycle (Figure 1) comprises a planar, moderately acidic (pKa= ~6–7), and relatively lipophilic carboxylic acid isostere.[44] This heterocycle is found in several potent agonists of the peroxisome proliferator activated receptor γ (PPARγ) that are used for the treatment of noninsulin-dependent type 2 diabetes mellitus (e.g., pioglitazone (42); Figure 3).[45] Since several endogenous compounds, as well as drug candidates that are known to activate PPARg, are carboxylic acids,[46] it seems likely that the thiazolidinedione moiety of the “glitazone” drugs are likely acting as carboxylic acid surrogate. Thiazolidinediones and related heterocycles have also been employed as carboxylic acid bioisosteres in the design of GPR40 agonists (e.g., 43–46; Figure 3).[47] GPR40 is a GPCR that enhances insulin secretion when activated by naturally occurring, medium- to long-chain fatty acids (FFA),[48] or by synthetic carboxylic acid compounds, such as several aryl propionic acids derivatives.[49]
Figure 3.

Biologically active thiazolidinedione-containing compounds.
Metabolically, the thiazolidinedione ring is hypothesized to undergo oxidative cytochrome P450 (CYP450)-mediated ring opening (Scheme 4),[3] which could in turn lead to the formation of electrophilic metabolites thereby causing idiosyncratic adverse reactions.[50] Indeed, the first marketed thiazolidinedione drug, troglitazone, was withdrawn from the market because of numerous reports of liver failure that might have been caused, at least in part, by bioactivation of the thiazolidinedione ring. Similar problems, however, have not been observed with other glitazones.
Scheme 4.

Cytochrome P450-mediated oxidation and ring opening of the thiazolidinedione heterocycle.
3-Hydroxyisoxazole and 3-Hydroxyisothiazole
The 3-hydroxy derivative of isoxazole and the corresponding isothiazole (Figure 1) are planar carboxylic acid isosteres that exhibit pKa values of ~4–5. The 3-hydroxyisoxazole heterocycle is found in naturally occurring amino acids, such as ibotenic acid (47; Figure 4), which is an agonist of the N-methyl-d-aspartic acid (NMDA) receptor.[51] This type of carboxylic acid bioisostere has been employed extensively in the development of several potent derivatives of GABA and glutamate[51] neurotransmitters.
Figure 4.

Examples of 3-hydroxyisoxazoles.
As summarized in Figure 4, a number of agonists of GABA receptors, such as muscimol (48),[52] and 4,5,6,7-tetrahydroisoxa-zolo[5,4-c]pyridin-3-ol (THIP, 49),[53] as well as antagonists (e.g., 50) have been developed.[54] In addition, among 3-hydroxyisoxazole-containing glutamate analogues are agonists and antagonists of the ionotropic glutamate AMPA receptor, such as compounds 51–53 (Figure 4),[51] while examples of 3-hydroxyisothiazole-containing glutamate derivatives include NMDA receptor agonist 54 (Figure 4), an isothiazole derivative of the naturally occurring ibotenic acid.[55]
Substituted Phenols
Although unsubstituted phenols are weakly acidic (pKa= ~9.9), the appropriate choice of electron-withdrawing substituents can lower the pKa values to within or even below the range of aliphatic carboxylic acids.[56] Representative examples include 2,6-difluorophenols (pKa = ~7.1),[57] which have been employed as lipophilic bioisosteres of the carboxylic acid in the design of competitive inhibitors of GABA aminotransferase (e.g., 55 and 56; Figure 5) that exhibit comparable binding affinity for the enzyme as the natural substrate (GABA).[58] The same bioisostere has been employed to obtain potent noncarboxylate aldose reductase inhibitors (cf., 57 and 58; Table 9).[59]
Figure 5.

2,6-Difluorophenol-containing GABA aminotransferase inhibitors.
Table 9.
Aldose reductase inhibitory activity of a noncarboxylate derivative (58) compared with the carboxylic acid (57).[58]
| Compd | Structure | IC50 [μm] |
|---|---|---|
| 57 |
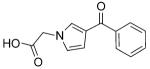
|
1.9 |
| 58 |
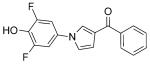
|
0.4 |
Squaric Acid
Squaric acid (59), a planar, diprotic four-membered ring oxocarbon, exhibits remarkable acidity (i.e., pKa1=0.5, pKa2=3.5).[56] Although the acidity of squaric acid, like that of other cyclic polyones, such as tetronic acid or the cyclopentane-1,3-diones (see below), can be explained at least in part by the principle of vinylogy;[60,61] in the case of the squaric acid and related oxocarbons, the 2π-pseudoaromaticity of the ring system is believed to contribute to the stabilization of the doubly deprotonated form (Scheme 5).[62] Derivatives of squaric acids, particularly squaramides (e.g., 60), have been exploited as bioisosteres of the carboxylic acid and phosphate[63] moieties. An interesting application of this bioisostere was reported by Kinney and co-workers, who developed NMDA receptor antagonists, such as 61[64] and 62[65] (Table 10), in which the squaramide moiety was used to mimic the a-amino acid moiety of phosphonate 63 and glutamic acid.[64]
Scheme 5.

Squaric acid (59), squaramide (60), and the general structures of related compounds.
Table 10.
NMDA affinity (IC50) and in vivo efficacy as determined by NMDA-induced lethality (ED50) of selected phosphonoalkyl-3,4-diamino-3-cyclobutene-1,2-dione derivatives.[64, 65]
| Compd | Structure | IC50 [μm] | ED50 [mgkg−1] |
|---|---|---|---|
| 63 |
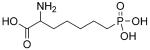
|
0.39 | 38 |
| 61 |
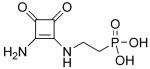
|
0.47 | 29 |
| 62 |

|
0.03 | 2.1 |
3- and 4-Hydroxyquinolin-2-ones
Notable examples of bioisosterism with the carboxylic acid moiety have been reported for series of 3- and 4-hydroxyquinolin-2-ones.[66-68] Although considerably less acidic than carboxylic acids (pKa=8.7), scientists at Pfizer discovered that 3-hydroxyquinolin-2-one-containing d-amino acid oxidase inhibitors can in fact establish hydrogen-bonding interactions within the enzyme active site, in a very similar fashion as carboxylic acid-containing inhibitors (cf., 64 and 65; Figure 6).[68] Examples of 4-hydroxyquinolin-2-ones as carboxylic acid isosteres have been reported by scientists at Merck with a series of potent NMDA antagonists with improved brain penetration.[66,67] Although effective in vitro, 5-iodo-7-chlorokynurenic acid (66) and related analogue 67 exhibit limited brain penetration,
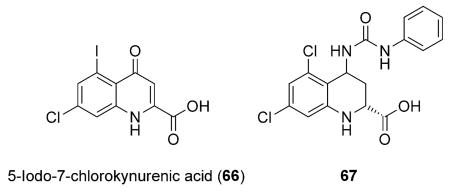 and thus could not be considered as potential candidates for the treatment of CNS diseases. By, replacement of the carboxylic acid with vinylogous-type acids of noncarboxylated 4-hydroxyquinolin-2-ones (66–68), a series of NMDA antagonists with potent binding affinity as well as improved brain penetration resulted (Table 11).[66]
and thus could not be considered as potential candidates for the treatment of CNS diseases. By, replacement of the carboxylic acid with vinylogous-type acids of noncarboxylated 4-hydroxyquinolin-2-ones (66–68), a series of NMDA antagonists with potent binding affinity as well as improved brain penetration resulted (Table 11).[66]
Figure 6.
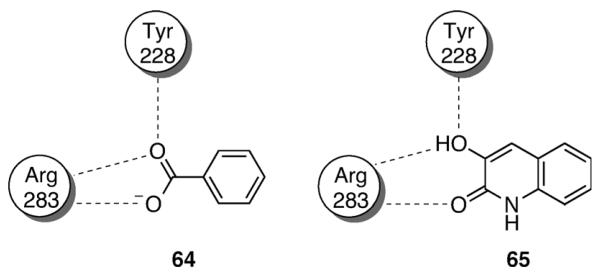
Schematic representation of hydrogen-bonding interactions of carboxylic acid 64 and hydroxyquinolin-2-one 65 within the binding site of d-amino acid oxidase.
Table 11.
NMDA receptor activities of 3′-substituted-4-hydroxy-2-quinolones.[65]
Inhibition of the binding of [3H]-4 to the strychnine-insensitive glycine site on rat brain membranes.
Protection from audiogenic seizure in DBA/2 mice (21–23 days old, weighing 5–9 g) after oral administration of test compounds.
Tetronic and Tetramic Acids
Tetronic (71) and tetramic (72) acids are naturally occurring acidic heterocycles.[70] Despite the similar structure, the two heterocycles exhibit markedly different acidity. Whereas 71 is considerably acidic with a pKa value of ~3.8, the corresponding pyrrolidine-2,4-dione (i.e., tetramic acid) exhibits a pKa value of ~6.4.[71] This difference has been explained by the fact that tetronic acid exists predominantly in the enol tautomeric form (71B; Scheme 6) and, as such, behaves more as an oxoacid, whereas the tautomeric equilibrium of tetramic acid is believed to be shifted more towards the di-oxo tautomer (72A; Scheme 6), leading this heterocycle to exhibit pKa values of a carbon acid.[56]
Scheme 6.

The keto-enol tautomerisation of tetronic (71) and tetramic (72) acids.
Although tetronic and tetramic acids are found in numerous biologically active compounds, their use as carboxylic acid isosteres is not particularly common. Examples include a series of tetramic acid-containing NMDA antagonists (e.g., 73; Table 12) that were designed based on the same pharmacophore model used in the design of 3′-substituted-4-hydroxy-2-quinolones described above (Table 11). Other studies also reported tetronic and tetramic acid-containing derivatives of known NSAIDs as γ-secretase modulators with in vitro activities comparable to the parent carboxylic acid compound.[72]
Table 12.
NMDA antagonist activity of tetramic acid derivative 73.[69]
Inhibition of the binding of [3H]-l-689,560 to the strychnine insensitive glycine site on rat brain membranes.
Inhibition of NMDA-induced depolarizations in rat cortical slices.
Cyclopentane-1,3-diones
Related to tetronic and tetramic acids, cyclopentane-1,3-diones (e.g., 74; Scheme 7) exist as rapidly exchanging enol–ketone tautomers.[73] As such, consistent with the principle of vinylogy,[61] 1,3-diones exhibit pKa values that fall within the range of carboxylic acids.[73] The characterization of the cyclopentane-1,3-diones as potential bioisosteres of the carboxylic acid moiety was recently demonstrated for the first time in our laboratory.[74] Evaluation of physicochemical properties revealed that, in addition to the intrinsic acidity, the cyclopentane-1,3-dione unit, like carboxylic acids, can form relatively stable 1:1 salt complexes with amidines.[74] Furthermore, a series of derivatives of the known thromboxane A2 (TP) receptor antagonist, 75,[75] in which the carboxylic acid moiety is replaced by a mono or di-substituted cyclopentabe-1,3-dione unit, resulted in TP receptor antagonists with IC50 values in the nanomolar range against human and mouse TP receptors (e.g., compounds 76, 78, and 79; Table 13).[74] A unique aspect of the cyclopentane-1,3-dione unit is that it presents multiple points of attachment, which can be exploited to impart alternative orientations of the bioisostere (cf., 76 and 77; Table 13) and/or to diversify the structure of the acidic moiety.
Scheme 7.

Enol-ketone tautomers of cyclopentane-1,3-dione 74.
Table 13.
Thromboxane A2 (TP) receptor antagonist activity of cyclopentane-1,3-dione derivatives.[74]
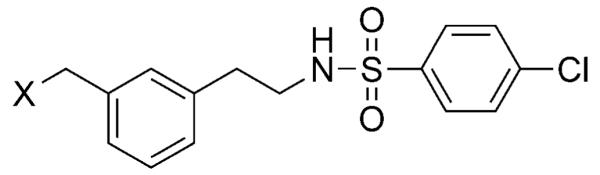
| |||
|---|---|---|---|
| Compd | X | IC50[a] [nm] | |
| mouse | human | ||
| 75 | CH2COOH | 27 | 61 |
| 76 |

|
52 | 250 |
| 77 |
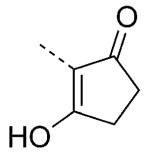
|
>10000 | >10000 |
| 78 |
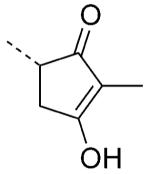
|
3.1 | 131 |
| 79 |
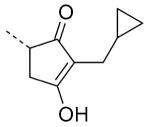
|
5.8 | 41 |
Results of an IP1 assay using mouse or human TP receptor.
Other Potential Bioisosteres
In addition to the isosteres discussed above, several other cyclic and acyclic structures, including boronic acids, mercaptoazoles, and sulfonimidamides, might be useful bioisosteres of the carboxylic acid moiety. The boronic acid moiety, for example, which is found in a number of biologically active compounds,[76] is likely to establish hydrogen bonds of similar geometries as carboxylic acids despite the limited acidity of this functional group (pKa= ~9–10). Likewise, mercaptoazoles (e.g., 80–91; Table 14),[77] which, depending on the oxidation state of the sulfur atom, can exhibit relatively wide ranges of acidity, might also be useful in drug design. Finally, cyclic sufonimidamides (e.g., 92; Scheme 8) have recently been proposed by scientists at AstraZeneca as potential bioisosteres of the carboxylic acid moiety.[78] The sufonimidamide heterocycle exhibits pKa values of 5–6, and although they can exist in four possible tautomers (Scheme 8), computational studies and X-ray crystal structure data suggest that the most stable tautomer has the acidic proton on the nitrogen in position 2 (92A). The most distinctive feature of this heterocycle is the presence of a stereogenic tetrahedral sulfur atom, as well as an sp3 carbon that can permit structural differentiation of the acidic moiety. Although there have been no reports of this particular heterocycle being applied successfully to the synthesis of biologically active compounds, the structural and physicochemical properties of this class of heterocycles make them potentially promising carboxylic acid bioisosteres.
Table 14.
Acidity of a series of mercaptoazoles.[77]

| |||||
|---|---|---|---|---|---|
| Compd | n | X | Y | Z | pKa |
| 80 | 0 | CH | CH | N | 11.6 |
| 81 | 1 | CH | CH | N | 9.8 |
| 82 | 2 | CH | CH | N | 8.67 |
| 83 | 0 | N | CH | N | 9.5 |
| 84 | 1 | N | CH | N | 7.38 |
| 85 | 2 | N | CH | N | 6.23 |
| 86 | 0 | N | N | CH | 8.2 |
| 87 | 1 | N | N | CH | 6.09 |
| 88 | 2 | N | N | CH | 5.15 |
| 89 | 0 | CO | NH | N | 7.85 |
| 90 | 1 | CO | NH | N | 5.2 |
| 91 | 2 | CO | NH | N | 4.79 |
Scheme 8.

The four possible tautomers (A–D) of sulfonimidamide 92.
Outlook
As highlighted in this Minireview, a diverse set of carboxylic acid bioisosteres have been identified, which exhibit various degrees of similarities with the carboxylic acid moiety in terms of size, geometry, charge distribution, acidity, and lipophilicity. However, as the outcome of any isosteric replacement is typically dependent upon the particular context defined by the physicochemical properties of the isostere and the corresponding binding counterpart(s) in the biological target(s), the availability of an increasingly larger set of potential carboxylic acid surrogates is ultimately critical to the success of the isosteric replacement strategy. Thus, the investigation of new and alternative structures that could complement the existing palette of carboxylic acid surrogates remains an important area of research.
Acknowledgements
The Authors are grateful to Prof. E. Amin and Dr. T.-L. Chiu (University of Minnesota, USA) for providing calculated logD values. Financial support for this work has been provided by the US National Institutes of Health (NIH)/National Institute on Aging (NIA) (grant no. AG034140).
References
- [1].Hajduk PJ, Bures M, Praestgaard J, Fesik SW. J. Med. Chem. 2000;43:3443–3447. doi: 10.1021/jm000164q. [DOI] [PubMed] [Google Scholar]
- [2].Pajouhesh H, Lenz GR. NeuroRx. 2005;2:541–553. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gardner I, Obach RS, Smith DA, Miao Z, Alex AA, Beaumont K, Kalgutkar A, Walker D, Dalvie D, Prakash C, Alf V. In: Metabolism, Pharmacokinetics and Toxicity of Functional Groups: Impact of Chemical Building Blocks on ADMET. Smith DA, editor. RSC Publishing; Cambridge: 2010. [Google Scholar]
- [4].Fung M, Thornton A, Mybeck K. Drug Inf. J. 2001;35:293–317. [Google Scholar]
- [5].Patani GA, LaVoie EJ. Chem. Rev. 1996;96:3147–3176. doi: 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]; Thornber CW. Chem. Soc. Rev. 1979;8:563. [Google Scholar]
- [6].Ventura ON, Rama JB, Turi L, Dannenberg JJ. J. Am. Chem. Soc. 1993;115:5754–5761. [Google Scholar]
- [7].Mai A. In: The Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids. Rappoport Z, Liebman JF, editors. Vol. 2. Wiley; Hoboken: 2009. pp. 731–806. Part 1, (Patai’s Chemistry of Functional Groups Series) Chapter 12. [Google Scholar]
- [8].Emery T. In: Advances in Enzymology and Related Areas of Molecular Biology. Meister A, editor. Vol. 35. Wiley; Hoboken: 1971. pp. 135–185. [DOI] [PubMed] [Google Scholar]
- [9].Muri EMF, Nieto MJ, Sindelar RD, Williamson JS. Curr. Med. Chem. 2002;9:1631–1653. doi: 10.2174/0929867023369402. [DOI] [PubMed] [Google Scholar]
- [10].Bauer L, Exner O. Angew. Chem. 1974;86:419–428. [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1974;13:376–384. [Google Scholar]
- [11].Bçhm S, Exner O. Org. Biomol. Chem. 2003;1:1176–1180. doi: 10.1039/b212298g. [DOI] [PubMed] [Google Scholar]
- [12].Summers JB, Mazdiyasni H, Holms JH, Ratajczyk JD, Dyer RD, Carter GW. J. Med. Chem. 1987;30:574–580. doi: 10.1021/jm00386a022. [DOI] [PubMed] [Google Scholar]
- [13].Summers JB, Gunn BP, Martin JG, Mazdiyasni H, Stewart AO, Young PR, Goetze AM, Bouska JB, Dyer RD. J. Med. Chem. 1988;31:3–5. doi: 10.1021/jm00396a002. [DOI] [PubMed] [Google Scholar]
- [14].Mulder GJ, Meerman JH. Environ. Health Perspect. 1983;49:27–32. doi: 10.1289/ehp.834927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Barrett SD, Bridges AJ, Dudley DT, Saltiel AR, Fergus JH, Flamme CM, Delaney AM, Kaufman M, LePage S, Leopold WR, Przybranowski SA, Sebolt-Leopold J, Van Becelaere K, Doherty AM, Kennedy RM, Marston D, Howard WA, Jr., Smith Y, Warmus JS, Tecle H. Bioorg. Med. Chem. Lett. 2008;18:6501–6504. doi: 10.1016/j.bmcl.2008.10.054. [DOI] [PubMed] [Google Scholar]
- [16].Froestl W, Mickel SJ, Hall RG, von Sprecher G, Strub D, Baumann PA, Brugger F, Gentsch C, Jaekel J. J. Med. Chem. 1995;38:3297–3312. doi: 10.1021/jm00017a015. [DOI] [PubMed] [Google Scholar]
- [17].Franz RG. AAPS PharmSci. 2001;3:1–13. [Google Scholar]
- [18].Watkins JC, Krogsgaard-Larsen P, Honor T. Trends Pharmacol. Sci. 1990;11:25–33. doi: 10.1016/0165-6147(90)90038-a. [DOI] [PubMed] [Google Scholar]
- [19].Kerr DIB, Ong J, Prager RH, Gynther BD, Curtis DR. Brain Res. 1987;405:150–154. doi: 10.1016/0006-8993(87)90999-1. [DOI] [PubMed] [Google Scholar]
- [20].Curtis DR, Watkins JC. J. Physiol. 1963;166:1–14. doi: 10.1113/jphysiol.1963.sp007087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kerr DIB, Ong J. Med. Res. Rev. 1992;12:593–636. doi: 10.1002/med.2610120604. [DOI] [PubMed] [Google Scholar]
- [22].Drysdale MJ, Pritchard MC, Horwell DC. J. Med. Chem. 1992;35:2573–2581. doi: 10.1021/jm00092a007. [DOI] [PubMed] [Google Scholar]
- [23].Woods DD, Fildes P. Chem. Ind. 1940;59:133. [Google Scholar]; Woods DD. Br. J. Exp. Pathol. 1940;21:74–90. [Google Scholar]
- [24].Bell PH, Roblin RO. J. Am. Chem. Soc. 1942;64:2905–2917. [Google Scholar]
- [25].King JF. In: The Chemistry of Sulphonic Acids, Esters and their Derivatives. Patai S, Rappoport Z, editors. Wiley; Chichester: 1991. pp. 249–259. Patai’s Chemistry of Functional Groups Series. [Google Scholar]
- [26].Yee YK, Bernstein PR, Adams EJ, Brown FJ, Cronk LA, Hebbel KC, Vacek EP, Krell RD, Snyder DW. J. Med. Chem. 1990;33:2437–2451. doi: 10.1021/jm00171a018. [DOI] [PubMed] [Google Scholar]
- [27].Uehling DE, Donaldson KH, Deaton DN, Hyman CE, Sugg EE, Barrett DG, Hughes RG, Reitter B, Adkison KK, Lancaster ME, Lee F, Hart R, Paulik MA, Sherman BW, True T, Cowan C. J. Med. Chem. 2002;45:567–583. doi: 10.1021/jm0101500. [DOI] [PubMed] [Google Scholar]
- [28].Ducharme Y, Blouin M, Carriere MC, Chateauneuf A, Cote B, Denis D, Frenette R, Greig G, Kargman S, Lamontagne S, Martins E, Nantel F, O’Neill G, Sawyer N, Metters KM, Friesen RW. Bioorg. Med. Chem. Lett. 2005;15:1155–1160. doi: 10.1016/j.bmcl.2004.12.005. [DOI] [PubMed] [Google Scholar]
- [29].Herr RJ. Bioorg. Med. Chem. 2002;10:3379–3393. doi: 10.1016/s0968-0896(02)00239-0. [DOI] [PubMed] [Google Scholar]
- [30].Myznikov L, Hrabalek A, Koldobskii G. Chem. Heterocycl. Compd. 2007;43:1–9. [Google Scholar]
- [31].McManus JM, Herbst RM. J. Org. Chem. 1959;24:1643–1649. [Google Scholar]
- [32].Costantino G, Maltoni K, Marinozzi M, Camaioni E, Prezeau L, Pin J-P, Pellicciari R. Bioorg. Med. Chem. 2001;9:221–227. doi: 10.1016/s0968-0896(00)00270-4. [DOI] [PubMed] [Google Scholar]
- [33].Allen FH, Groom CR, Liebeschuetz JW, Bardwell DA, Olsson TSG, Wood PA. J. Chem. Inf. Model. 2012;52:857–866. doi: 10.1021/ci200521k. [DOI] [PubMed] [Google Scholar]
- [34].Hansch C, Leo A. Exploring QSAR: Fundamentals and Applications in Chemistry and Biology. American Chemical Society; Washington DC: 1995. [Google Scholar]
- [35].Kenny PW. J. Chem. Inf. Mod. 2009;49:1234–1244. doi: 10.1021/ci9000234. [DOI] [PubMed] [Google Scholar]
- [36].Tominey AF, Docherty PH, Rosair GM, Quenardelle R, Kraft A. Org. Lett. 2006;8:1279–1282. doi: 10.1021/ol053072+. [DOI] [PubMed] [Google Scholar]; Peters L, Frohlich R, Boyd ASF, Kraft A. J. Org. Chem. 2001;66:3291–3298. doi: 10.1021/jo005632i. [DOI] [PubMed] [Google Scholar]
- [37].Timmermans PB, Wong PC, Chiu AT, Herblin WF, Benfield P, Carini DJ, Lee RJ, Wexler RR, Saye JA, Smith RD. Pharmacol. Rev. 1993;45:205–251. [PubMed] [Google Scholar]
- [38].Murphy TJ, Alexander RW, Griendling KK, Runge MS, Bernstein KE. Nature. 1991;351:233–236. doi: 10.1038/351233a0. [DOI] [PubMed] [Google Scholar]
- [39].Carini DJ, Duncia JV, Aldrich PE, Chiu AT, Johnson AL, Pierce ME, Price WA, Santella JB, Wells GJ. J. Med. Chem. 1991;34:2525–2547. doi: 10.1021/jm00112a031. [DOI] [PubMed] [Google Scholar]
- [40].Noda K, Saad Y, Kinoshita A, Boyle TP, Graham RM, Husain A, Karnik SS. J. Biol. Chem. 1995;270:2284–2289. doi: 10.1074/jbc.270.5.2284. [DOI] [PubMed] [Google Scholar]
- [41].Wexler RR, Greenlee WJ, Irvin JD, Goldberg MR, Prendergast K, Smith RD, Timmermans PBMWM. J. Med. Chem. 1996;39:625–656. doi: 10.1021/jm9504722. [DOI] [PubMed] [Google Scholar]
- [42].Naka T, Kubo K. Curr. Pharm. Des. 1999;5:453–472. [PubMed] [Google Scholar]
- [43].Kohara Y, Kubo K, Imamiya E, Wada T, Inada Y, Naka T. J. Med. Chem. 1996;39:5228–5235. doi: 10.1021/jm960547h. [DOI] [PubMed] [Google Scholar]
- [44].Lipinski CA, Fiese EF, Korst RJ. Quant. Struct.-Act. Relat. 1991;10:109–117. [Google Scholar]
- [45].Saltiel AR, Olefsky JM. Diabetes. 1996;45:1661–1669. doi: 10.2337/diab.45.12.1661. [DOI] [PubMed] [Google Scholar]
- [46].Henke BR, Blanchard SG, Brackeen MF, Brown KK, Cobb JE, Collins JL, Harrington WW, Hashim MA, Hull-Ryde EA, Kaldor I, Kliewer SA, Lake DH, Leesnitzer LM, Lehmann JM, Lenhard JM, Orband-Miller LA, Miller JF, Mook RA, Noble SA, Oliver W, Parks DJ, Plunket KD, Szewczyk JR, Willson TM. J. Med. Chem. 1998;41:5020–5036. doi: 10.1021/jm9804127. [DOI] [PubMed] [Google Scholar]
- [47].Zhou C, Tang C, Chang E, Ge M, Lin S, Cline E, Tan CP, Feng Y, Zhou Y-P, Eiermann GJ, Petrov A, Salituro G, Meinke P, Mosley R, Akiyama TE, Einstein M, Kumar S, Berger J, Howard AD, Thornberry N, Mills SG, Yang L. Bioorg. Med. Chem. Lett. 2010;20:1298–1301. doi: 10.1016/j.bmcl.2009.10.052. [DOI] [PubMed] [Google Scholar]; Medina JC, Houze JB. Annu. Rep. Med. Chem. 2008;43:75–85. [Google Scholar]
- [48].Blad CC, Tang C, Offermanns S. Nat. Rev. Drug Discovery. 2012;11:603–619. doi: 10.1038/nrd3777. [DOI] [PubMed] [Google Scholar]
- [49].Sasaki S, Kitamura S, Negoro N, Suzuki M, Tsujihata Y, Suzuki N, Santou T, Kanzaki N, Harada M, Tanaka Y, Kobayashi M, Tada N, Funami M, Tanaka T, Yamamoto Y, Fukatsu K, Yasuma T, Momose Y. J. Med. Chem. 2011;54:1365–1378. doi: 10.1021/jm101405t. [DOI] [PubMed] [Google Scholar]; Mikami S, Kitamura S, Negoro N, Sasaki S, Suzuki M, Tsujihata Y, Miyazaki T, Ito R, Suzuki N, Miyazaki J, Santou T, Kanzaki N, Funami M, Tanaka T, Yasuma T, Momose Y. J. Med. Chem. 2012;55:3756–3776. doi: 10.1021/jm2016123. [DOI] [PubMed] [Google Scholar]
- [50].Alvarez-Sánchez R, Montavon F, Hartung T, Pähler A. Chem. Res. Toxicol. 2006;19:1106–1116. doi: 10.1021/tx050353h. [DOI] [PubMed] [Google Scholar]
- [51].Bräuner-Osborne H, Egebjerg J, Nielsen EØ, Madsen U, Krogsgaard-Larsen P. J. Med. Chem. 2000;43:2609–2645. doi: 10.1021/jm000007r. [DOI] [PubMed] [Google Scholar]
- [52].Krogsgaard-Larsen P, Johnston GAR, Curtis DR, Game CJA, McCulloch RM. J. Neurochem. 1975;25:803–809. doi: 10.1111/j.1471-4159.1975.tb04411.x. [DOI] [PubMed] [Google Scholar]
- [53].Krogsgaard-Larsen P, Johnston GA, Lodge D, Curtis DR. Nature. 1977;268:53–55. doi: 10.1038/268053a0. [DOI] [PubMed] [Google Scholar]
- [54].Frølund B, Jørgensen AT, Tagmose L, Stensbøl TB, Vestergaard HT, Engblom C, Kristiansen U, Sanchez C, Krogsgaard-Larsen P, Liljefors T. J. Med. Chem. 2002;45:2454–2468. doi: 10.1021/jm020027o. [DOI] [PubMed] [Google Scholar]; Frølund B, Jensen LS, Guandalini L, Canillo C, Vestergaard HT, Kristiansen U, Nielsen B, Stensbøl TB, Madsen C, Krogsgaard-Larsen P, Liljefors T. J. Med. Chem. 2005;48:427–439. doi: 10.1021/jm049256w. [DOI] [PubMed] [Google Scholar]
- [55].Jørgensen CG, Clausen RP, Hansen KB, Bräuner-Osborne H, Nielsen B, Bjorn M, Kehler J, Krogsgaard-Larsen P, Madsen U. Org. Biomol. Chem. 2007;5:463–471. doi: 10.1039/b615162k. [DOI] [PubMed] [Google Scholar]; Bunch L, Krogsgaard-Larsen P, Madsen U. J. Org. Chem. 2002;67:2375–2377. doi: 10.1021/jo0162134. [DOI] [PubMed] [Google Scholar]; Hermit MB, Greenwood JR, Nielsen B, Bunch L, Jørgensen CG, Vestergaard HT, Stensbøl TB, Sanchez C, Krogsgaard-Larsen P, Madsen U, Bräuner-Osborne H. Eur. J. Pharmacol. 2004;486:241–250. doi: 10.1016/j.ejphar.2003.12.033. [DOI] [PubMed] [Google Scholar]
- [56].Perez GV, Perez AL. J. Chem. Educ. 2000;77:910. [Google Scholar]
- [57].Stefanidis D, Cho S, Dhe-Paganon S, Jencks WP. J. Am. Chem. Soc. 1993;115:1650–1656. [Google Scholar]
- [58].Qiu J, Stevenson SH, O’Beirn MJ, Silverman RB. J. Med. Chem. 1999;42:329–332. doi: 10.1021/jm980435l. [DOI] [PubMed] [Google Scholar]
- [59].Nicolaou I, Zika C, Demopoulos VJ. J. Med. Chem. 2004;47:2706–2709. doi: 10.1021/jm031060t. [DOI] [PubMed] [Google Scholar]
- [60].Avila J, Lucas JJ, Perez MAR, Hernandez F. Physiol. Rev. 2004;84:361–384. doi: 10.1152/physrev.00024.2003. [DOI] [PubMed] [Google Scholar]
- [61].Claisen L. Justus Liebigs Ann. Chem. 1894;281:306–313. [Google Scholar]; Claisen L. Justus Liebigs Ann. Chem. 1896;291:25–137. [Google Scholar]
- [62].Ito M, West R. J. Am. Chem. Soc. 1963;85:2580–2584. [Google Scholar]
- [63].Elliott TS, Slowey A, Ye Y, Conway SJ. Med. Chem. Commun. 2012;3:735–751. [Google Scholar]
- [64].Kinney WA, Lee NE, Garrison DT, Podlesny EJ, Simmonds JT, Bramlett D, Notvest RR, Kowal DM, Tasse RP. J. Med. Chem. 1992;35:4720–4726. doi: 10.1021/jm00103a010. [DOI] [PubMed] [Google Scholar]
- [65].Kinney WA, Abou-Gharbia M, Garrison DT, Schmid J, Kowal DM, Bramlett DR, Miller TL, Tasse RP, Zaleska MM, Moyer JA. J. Med. Chem. 1998;41:236–246. doi: 10.1021/jm970504g. [DOI] [PubMed] [Google Scholar]
- [66].Kulagowski JJ, Baker R, Curtis NR, Mawer IM, Moseley AM, Ridgill MP, Rowley M, Stansfield I, Leeson PD. J. Med. Chem. 1994;37:1402–1405. doi: 10.1021/jm00036a002. [DOI] [PubMed] [Google Scholar]
- [67].Rowley M, Leeson PD, Stevenson GI, Moseley AM, Stansfield I, Sanderson I, Robinson L, Baker R, Kemp JA. J. Med. Chem. 1993;36:3386–3396. doi: 10.1021/jm00074a020. [DOI] [PubMed] [Google Scholar]
- [68].Duplantier AJ, Becker SL, Bohanon MJ, Borzilleri KA, Chrunyk BA, Downs JT, Hu L-Y, El-Kattan A, James LC, Liu S, Lu J, Maklad N, Mansour MN, Mente S, Piotrowski MA, Sakya SM, Sheehan S, Steyn SJ, Strick CA, Williams VA, Zhang L. J. Med. Chem. 2009;52:3576–3585. doi: 10.1021/jm900128w. [DOI] [PubMed] [Google Scholar]
- [69].Mawer IM, Kulagowski JJ, Leeson PD, Grimwood S, Marshall GR. Bioorg. Med. Chem. Lett. 1995;5:2643–2648. [Google Scholar]
- [70].Royles BJL. Chem. Rev. 1995;95:1981–2001. [Google Scholar]; Tejedor D, Garcia-Tellado F. Org. Prep. Proc. Int. 2004;36:33. [Google Scholar]
- [71].Mulholland TPC, Foster R, Haydock DB. J. Chem. Soc. Perkin Trans. 1. 1972:2121–2128. [Google Scholar]
- [72].Zall A, Kieser D, Hçttecke N, Naumann EC, Thomaszewski B, Schneider K, Steinbacher DT, Schubenel R, Masur S, Baumann K, Schmidt B. Bioorg. Med. Chem. 2011;19:4903–4909. doi: 10.1016/j.bmc.2011.06.062. [DOI] [PubMed] [Google Scholar]
- [73].Hiraga K. Chem. Pharm. Bull. 1965;13:1300–1306. doi: 10.1248/cpb.13.1300. [DOI] [PubMed] [Google Scholar]
- [74].Ballatore C, Soper JH, Piscitelli F, James M, Huang L, Atasoylu O, Huryn DM, Trojanowski JQ, Lee VM, Brunden KR, Smith AB. J. Med. Chem. 2011;54:6969–6983. doi: 10.1021/jm200980u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Dickinson RP, Dack KN, Long CJ, Steele J. J. Med. Chem. 1997;40:3442–3452. doi: 10.1021/jm9702793. [DOI] [PubMed] [Google Scholar]
- [76].Yang WQ, Gao XM, Wang BH. Med. Res. Rev. 2003;23:346–368. doi: 10.1002/med.10043. [DOI] [PubMed] [Google Scholar]
- [77].Chenard BL, Lipinski CA, Dominy BW, Mena EE, Ronau RT, Butterfield GC, Marinovic LC, Pagnozzi M, Butler TW, Tsang T. J. Med. Chem. 1990;33:1077–1083. doi: 10.1021/jm00165a030. [DOI] [PubMed] [Google Scholar]; Lipinski CA, Chenard BL. Pestic. Sci. 1990;29:227–240. [Google Scholar]
- [78].Pemberton N, Graden H, Evertsson E, Bratt E, Lepistç M, Johannesson P, Svensson PH. ACS Med. Chem. Lett. 2012;3:574–578. doi: 10.1021/ml3000935. [DOI] [PMC free article] [PubMed] [Google Scholar]