

Abstract
Depressive symptoms are associated with an increased risk of Alzheimer’s Disease (AD) but the mechanism(s) involved has not been well established. In a convenience sample of participants in the University of Kansas’ Brain Aging Project, we use structural equation modeling (SEM) to explicitly distinguish depressive symptom-related variance in cognitive task performance (i.e., “DEPCOG”) from that which is unrelated to a depressive symptoms. DEPCOG is strongly associated with the cognitive correlates of functional status (δ), which we previously associated with elements of the Default Mode Network (DMN). Both δ and DEPCOG map to a Posterior Cingulate seeded network that has recently been associated with β-amyloid deposition and includes elements of the DMN. Both contribute significantly to clinical dementia status and dementia severity, as measured by the Clinical Dementia Rating Scale Sum of Boxes (CDR-SB). These findings suggest that the cognitive correlates of depressive symptoms, even in the absence of a major depressive episode, may contribute to dementia in their own right, and could be responsible for some cases of incident clinical “AD”. This conclusion suggests new opportunities for the latter’s diagnosis, prevention and treatment.
Keywords: Aging, Cognition, Dementia, Depression, g, Functional Status
Introduction
It has been suggested that depressive symptoms in AD may reflect a specific syndrome of “depression in AD” (dAD) [1]. Depression in non-demented persons has been identified as a possible risk factor for incident Alzheimer’s disease (AD) [2–4]. In a recent meta-analysis, depression appeared to double the risk of AD [5]. Depressive symptoms are common in Mild Cognitive Impairment (MCI) [6] and appear to hasten conversion from MCI to clinical AD [7]. Even subclinical depressive symptoms may be sufficient to convey this risk [8].
The mechanism(s) by which depression and depressive symptoms might effect these risks has not been well established. However, we have recently shown that depressive symptoms are associated with incident changes in executive control, not memory [9], and that depressive symptom-related cognitive change is not mediated through AD pathology [10]. Similarly, a history of past depressive episodes is not associated with the distribution of [(11)C] Pittsbugh Compound B (PiB) binding [11] and depressive symptoms are not associated with apolipoprotein e4 (ApoE4) in MCI [12]. These findings suggest that depression’s effects on dementia status are independent of the AD process. Therefore, depression may itself be dementing. Moreover, because none of these studies involved clinically depressed persons, sub-syndromal depressive symptoms may indicate an independent dementing process.
We have recently developed an analytical technique that can address this issue. We use structural equation modeling (SEM) to explicitly distinguish dementia-relevant variance in cognitive task performance (i.e., “δ”) from that which is unrelated to a dementing process (i.e., “g′”) [13–14]. Together, g′ and δ effectively comprise Spearman’s “g” (i.e., “general intelligence”) [15]. We recently validated δ in the Texas Alzheimer’s Research and Care Consortium (TARCC), a well characterized cohort of Alzheimer’s disease (AD) cases and controls [14].
One of the advantages of our approach is that δ’s factor scores represent a continuously varying and arguably measurement error free dementia endophenotype. We have examined the bio-markers of this endophenotype, and recently co-localized δ specifically with grey matter atrophy in the Default Mode Network (DMN) [16]. The DMN is comprised of highly interconnected neocortical regions that are active during wakeful self-reflection and introspection, and inactive during task specific processing [17]. Its hubs include parts of the medial temporal lobe, the medial prefrontal cortex, the posterior cingulate, the precuneus, and the medial, lateral and inferior parietal cortex [18]. The DMN is abnormal in AD, but also in depression [19]. Depression-related atrophy in DMN-related regions [20] may provide an explanation for the disabling, and therefore intrinsically “dementing” nature of depressive illness.
In this analysis, we return to the same dataset (i.e., the Brain Aging Project (BAP) of the Univeristy of Kansas Department of Neurology’s Alzheimer’s Disease Center) to examine whether there is overlap between the cognitive correlates of depressive symptoms (DEPCOG) and the cognitive correlates of functional status (i.e., δ), and whether DEPCOG can also be specifically associated with structural changes in the DMN. If so, then sub-syndromal depression itself, independent of AD lesions, may be responsible for some cases of incident clinical “AD”, suggesting new opportunities for the latter’s diagnosis, prevention and treatment.
Methods
Sample
Participants were enrolled in the University of Kansas Brain Aging Project (BAP). Data used in these analyses were from individuals with early-stage Alzheimer’s disease (AD), defined by a Clinical Dementia Rating (CDR) scale score of 0.5 or 1.0, n = 70) or those without dementia (CDR = 0, n=76) aged 60 years and older [18]. Study exclusions have been reported previously [21] and briefly include baseline neurologic disease other than AD with the potential to impair cognition, current or past history of diabetes mellitus, recent history of cardiovascular disease, clinically significant depressive symptoms, and MRI exclusions among others. Portions of these data have been reported previously as part of a larger cohort [22–24]. Institutionally approved informed consent was obtained from all participants and their legal representative as appropriate before enrollment into the study.
Clinical assessment
The clinical assessment included a semi-structured interview with the participant and a collateral source knowledgeable about the participant. Medications, past medical history, education, demographic information and family history were collected from the collateral source. Dementia status of the participant was based on clinical evaluation [25]. Diagnostic criteria for AD require the gradual onset and progression of impairment in memory and at least one other cognitive and functional domain [26]. The CDR assesses function in multiple domains and was used to assess dementia severity, such that CDR 0.0 indicates no dementia, CDR 0.5 indicates very mild, and CDR 1.0 indicates mild dementia [27]. These methods have a diagnostic accuracy for AD of 93% and have been shown to be accurate in discriminating those with mild cognitive impairment (MCI) who have early stage AD [25, 28]. We encountered problems when trying to model three diagnostic classes, including the relatively small numbers of AD and MCI cases, and instead modeled “Diagnosis” as AD and MCI (N = 70) vs. controls (n = 76).
Depressive symptoms were assessed using the short Geriatric Depression Scale (GDS) [29–30]. Subjects were asked to self-report their depressive symptoms, while their caregivers were asked to assess the subjects’ dysphoria. GDS scores range from zero–15. Higher scores are worse. A cut-point of 6–7 best discriminates clinically depressed from non-depressed elderly.
Cognitive Assessment
A trained psychometrician administered a psychometric test battery that included common measures of memory (Wechsler Memory Scale [WMS]–Revised Logical Memory IA and IIA [31], Free and Cued Selective Reminding Task [32], working memory [WMS III Digit Span Forwards and Backwards [31], executive function [Verbal Fluency – Animals [33], and Stroop Color-Word Interference Test [34]. The Mini-Mental State Examination (MMSE) [35] was also administered.
Functional Assessment
We used the Alzheimer’s Disease Cooperative Study Activities of Daily Living Scale for Mild Cognitive Impairment (ADCS-ADL) with information collected from the informant. The ADCS-ADL is a well characterized measure of independence in activities of daily living [36]. The 18-item measure is heavily weighted towards instrumental activities of daily living (IADL) such as meal preparation, travel outside the home, shopping, and performing household chores. Tasks are scored by increasing level of independence with greater scores reflecting more independence in IADL.
Statistical approach
This analysis was performed using AMOS software [37]. All observed variables were adjusted for age, gender and education. Latent variables of interest were constructed from confirmatory factor analyses performed in a structural equation framework. The latent variables d and DEPCOG were uniquely indicated by IADL and GDS scores, respectively. Otherwise, they were derived from an identical cognitive battery that was both more circumscribed and had no overlap with that of our previously validated latent variable “δ” [16]. There was no overlap at all in the indicator variables used to construct DEPCOG and δ. Residual covariances were empirically modeled to optimize model fit. Model parameters were compared across models to ensure that model interpretation remained stable across alternative residual covariance structures.
Missing data
We used Full Information Maximum Likelihood (FIML) methods to address missing data. FIML uses the entire observed data matrix to estimate parameters with missing data. In contrast to listwise or pairwise deletion, FIML yields unbiased parameter estimates, preserves the overall power of the analysis, and is arguably superior to alternative methods, e.g., multiple imputation [38–39].
Fit Indices
The validity of structural models was assessed using three common test statistics. A non-significant chi-square signifies that the data are consistent with the mode [40]. The comparative fit index (CFI), with values ranging between 0 and 1, compares the specified model with a model of no change [41]. CFI values below 0.95 suggest model misspecification. Values of 0.95 or greater indicate adequate to excellent fit. A root mean square error of approximation (RMSEA) of 0.05 or less indicates a close fit to the data, with models below 0.05 considered “good” fit, and up to 0.08 as “acceptable” [42]. All three fit statistics should be simultaneously considered to assess the adequacy of the models to the data.
Neuroimaging
Baseline and follow-up whole brain structural MRI data were obtained using a Siemens 3.0 Tesla Allegra MRI Scanner. High-resolution T1 weighted anatomical images were acquired (magnetization-prepared rapid gradient echo [MPRAGE]; 1 × 1 × 1 mm3 voxels, repetition time [TR] =2500, echo time [TE] =4.38 ms, inversion time [TI] =1,100 ms, field of view=256 × 256, flip angle=8 degrees). Data analysis was performed using the VBM5 toolbox (http://dbm.neuro.uni-jena.de), an extension of the SPM5 algorithms (Wellcome Department of Cognitive Neurology, London, UK) running under MATLAB 7.1(The MathWorks, Natick, MA, USA).
Voxel-based morphometry (VBM) is a method for detecting differences in the volume of brain matter. Our structural image processing method for VBM is detailed elsewhere [22, 43]. Briefly, tissue classification, image registration and MRI inhomogeneity bias correction were performed as part of the unified segmentation approach implemented in SPM5 [44]. We used the Hidden Markov Field (HMRF) model on the estimated tissue maps (3 × 3 × 3 mm3). Estimated tissue probability maps were written without making use of the International Consortium for Brain Mapping tissue priors to avoid a segmentation bias [45]. Images were then modulated and saved using affine registration plus nonlinear spatial normalization [46]. The resulting gray matter volume maps were smoothed with a 10 mm FWHM Gaussian kernel before statistical analysis.
Imaging Statistics
We used a multiple regression model in SPM5 with age, education, and gender as covariates (age and education centered on the mean). DEPCOG scores are also implicitly adjusted for g′ factor scores. The absolute threshold masking was set at 0.10 to restrict each analysis to gray matter. Our primary interest was the relationship of DEPCOG factor scores with regional gray matter volume, independent of the remaining regressors. Results were considered significant at p<0.05 [family-wise error corrected (FWE)], with clusters exceeding 50 voxles. Peak voxels are reported with reference to the MNI standard space and anatomic labels are reported with reference to the computerized Talairach Daemon [47] within the Pickatlas [48]. DEPCOG’s regional gray matter volume correlates are compared in Figures 6 and 7 to those of d and our previously validated original model “δ” [16].
Figure 6.
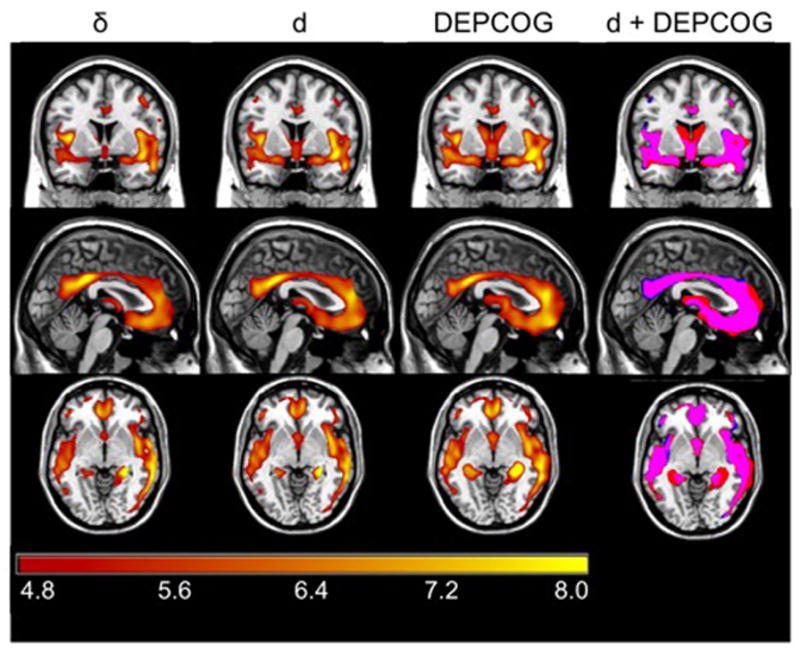
Regional Cortical Atrophy Associated Specifically with DEPCOG. Figure 6 shows regional cortical volume associated with δ (left column), d (middle left column), and DEPCOG (middle right column). Each analysis is adjusted for age, gender and education (and implicitly for g′). The colorbar represents the voxel-wise T statistic, only significant voxels are presented (FWE>0.05, k>50). The overlap of the maps can be seen in right column: d is shown in blue, DEPCOG is in red and the overlap is shown in magenta. Adjusted for age and gender (and implicitly for education and g′). Note overlap with elements of the Default Mode Network.
Figure 7.
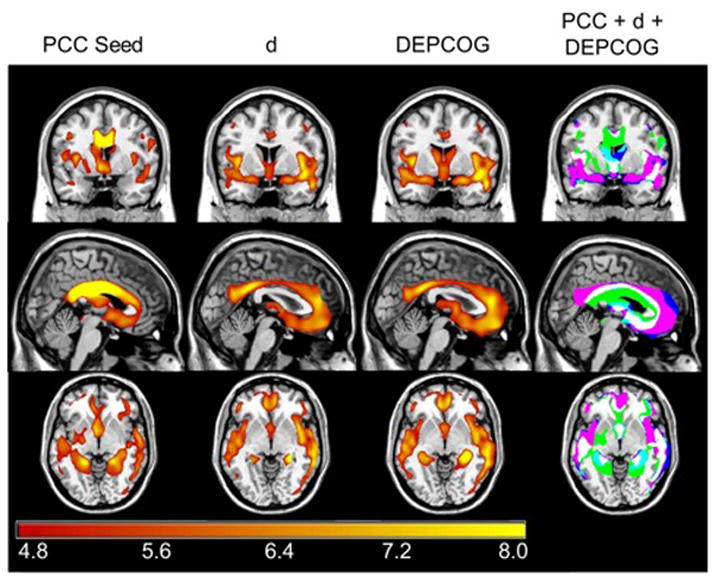
A PCC seed Replicates d and DEPCOG. Adjusted for age and gender (and implicitly for education and g′). PCC = Posterior Cingulate. Figure 7 shows regional cortical volume associated with volume in the posterior cingulate (PCC seed, left column), d (middle left column), and DEPCOG (middle right column). The colorbar represents the voxel-wise T statistic, only significant voxels are presented (FWE>0.05, k>50). The overlap of the maps can be seen in right column: DEPCOG alone = blue; PCC alone = green; d+DEPCOG = magenta; PCC + DEPCOG = aqua; all three = white.
Oh et al. [49] have demonstrated that Pittsburgh Compound B (PiB) burden in non-demented older persons is associated with atrophy in the posterior cingulate. When a seed was placed in that structure, an inter-related set of structures emerged. We tested whether the grey matter atrophy associated with our latent constructs is co-localized with the same structures. We used a multiple regression model in SPM5 with age, education, and gender as covariates (age and education centered on the mean). d and DEPCOG scores are also implicitly adjusted for g′ factor scores. The absolute threshold masking was set at 0.10 to restrict each analysis to gray matter. Our primary interest was the relationship of DEPCOG factor scores with regional gray matter volume, independent of the remaining regressors. We extracted relative brain volume from a bilateral posterior cingulate ROI (4 mm spheres at −10, −38, 30 and 10, −38, 30) [49] and regressed the relative gray matter volume corrected for age, sex and education as has been done recently by Monembault et al. [50].
Results
Sample demographics are presented in Table 1. Clinical assessment means are presented in Table 2. All groups had sub-clinical mean GDSs and GDSc scores. However, there were significant cross-group differences by both measures (by MANOVA, adjusted for age, education and gender). MCI cases exhibited significantly more depressive symptoms by both measures than either AD or controls. There were no significant differences between AD cases and controls on either measure. Although AD and MCI cases were significantly more likely to report a past history of depression than controls, MCI cases were no less likely to report a past history of depression than AD cases. There were no group differences with regard to the current use of either benzodiazepines or serononin-selective reuptake inhibitors (SSRI). AD cases were more likely than either MCI cases or controls to use other psychotropics (all group comparisons by post hoc Honest Significant Difference test for uneaqual N’s).
Table 1.
Subject Characteristics
| Variable | Control Mean (SD) (N = 76) |
MCI Mean (SD) (N = 47) |
AD Mean (SD) (N = 23) |
Total Mean (SD) (N = 146) |
|---|---|---|---|---|
| AGE (years) | 74.2 (7.2) | 75.9 (6.5) | 73.2 (5.8) | 74.6 (6.8) |
| % Female | 58% | 62% | 70% | 61% |
| Education (years) | 16.3 (2.7) | 14.9 (3.2) | 15.2 (3.0) | 15.7 (3.0) |
| MMSE | 29.4 (0.8) | 28.1 (1.3) | 22.1 (3.1) | 27.8 (3.0) |
| CDR-SB | 0 (0.1) | 2.7 (1.1) | 4.2 (1.2) | 1.5 (1.8) |
CDR-SB = CDR Sum of Boxes; MMSE = Mini-Mental State Examination
Table 2.
Raw Clinical Means
| Variable | Control Mean (SD) (N = 76) |
MCI Mean (SD) (N = 47) |
AD Mean (SD) (N = 23) |
Total Mean (SD) (N = 146) |
|---|---|---|---|---|
| Boston | 14.2 (1.0) | 12.7 (2.7) | 9.4 (3.6) | 13.0 (2.8) |
| LMIIA | 10.8 (4.6) | 4.6 (4.8) | 1.1 (2.0) | 7.3 (5.8) |
| SRTFR | 28.3 (6.3) | 17.5 (8.8) | 6.0 (5.0) | 21.3 (10.8) |
| Animals | 18.6 (4.3) | 15.5 (4.5) | 9.2 (3.9) | 16.1 (5.4) |
| StrI | 35.9 (8.7) | 27.0 (7.8) | 14.5 (8.8) | 29.7 (11.4) |
| MCI-ADL | 49.4 (2.3) | 43.1 (5.4) | 36.4 (10.2) | 45.1 (7.1) |
| GDSs | 0.8 (0.9) | 1.9 (1.6) | 1.7 (1.4) | 1.3 (1.4) |
| GDSc | 0.6 (1.0) | 3.2 (2.8) | 4.0 (3.3) | 2.0 (2.6) |
| % report a h/o “Depression” | 7.9 | 29.8 | 34.8 | 19.2 |
Animals = Category Fluency: Animals; Boston = Boston Naming Test (15 item); GDSc = Geriatric Depression Scale Caregiver rated; GDSs = Geriatric Depression Scale Subject rated; LMIIA = Wechsler Memory Scale–Revised Logical Memory Story A Delayed; MCI-ADL = Alzheimer’s Disease Cooperative Study Activities of Daily Living Scale for Mild Cognitive Impairment; SRTFR = Selective Reminding Task Free Recall Total; StrI = Stroop Color Task Color-Word Interference Task
First, we replicated δ from a more circumscribed cognitive and clinical assessment (Figure 1). As in previous analyses, the new latent construct “d” was a strong independent predictor of CDR-SB (r = −0.94, p < 0.001), and was more strongly labeled by IADL (i.e., MCI-ADL; r = 0.77, p < 0.001) than by any cognitive measure (range r = 0.52, Animals; −0.75, SRTFR; all p <0.001). This model had excellent fit (Table 3).
Figure 1.
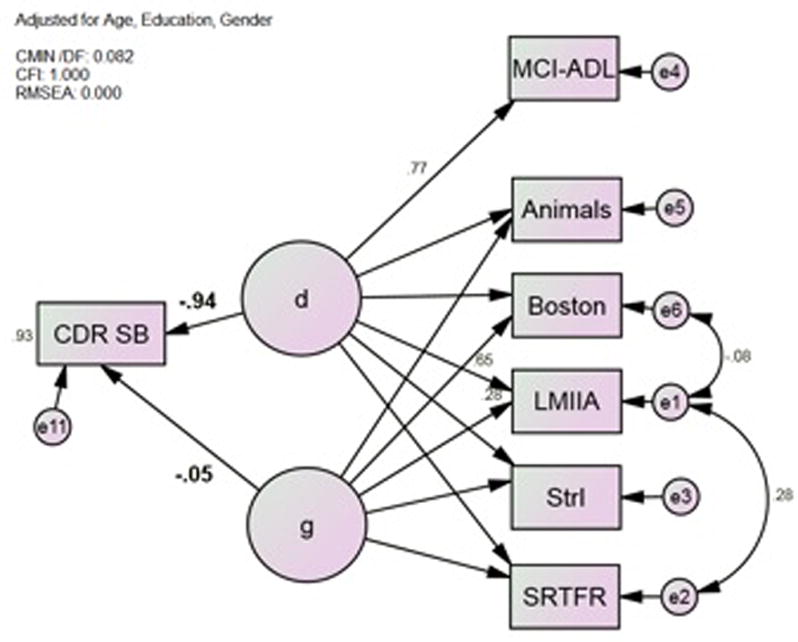
Model 1*; d Correlates Strongly with CDR-SB. Animals = Category Fluency: Animals; Boston = Boston Naming Test (15 item); CDR-SB = Clinical Dementia Rating Scale Sum of Boxes; LMIIA = Wechsler Memory Scale–Revised Logical Memory Story A Delayed; MCI-ADL = Alzheimer’s Disease Cooperative Study Activities of Daily Living Scale for Mild Cognitive Impairment; SRTFR = Selective Reminding Task Free Recall Total; StrI = Stroop Color Task Color-Word Interference Task *All observed variables adjusted for age, gender and education (not shown).
Table 3.
Model Fit
| Model | X2:df, p | CMIN | RMSEA | CFI |
|---|---|---|---|---|
| 1 | 7.4:9, p = 0.60 | 0.08 | 0.000 | 1.000 |
| 2 | 9.9:9, p = 0.36 | 1.10 | 0.023 | 0.986 |
| 3 | 7.6:8, p = 0.47 | 0.95 | 0.000 | 1.000 |
| 4 | 15.7:15, p = 0.41 | 1.04 | 0.017 | 0.999 |
Next, we constructed DEPCOG from the same cognitive battery (Figure 2). DEPCOG represents the shared variance between these cognitive measures and self-rated GDS scores. Aside from the fact that MCI-ADL has been replaced by GDSs, Model 2 is identical to Model 1.
Figure 2.
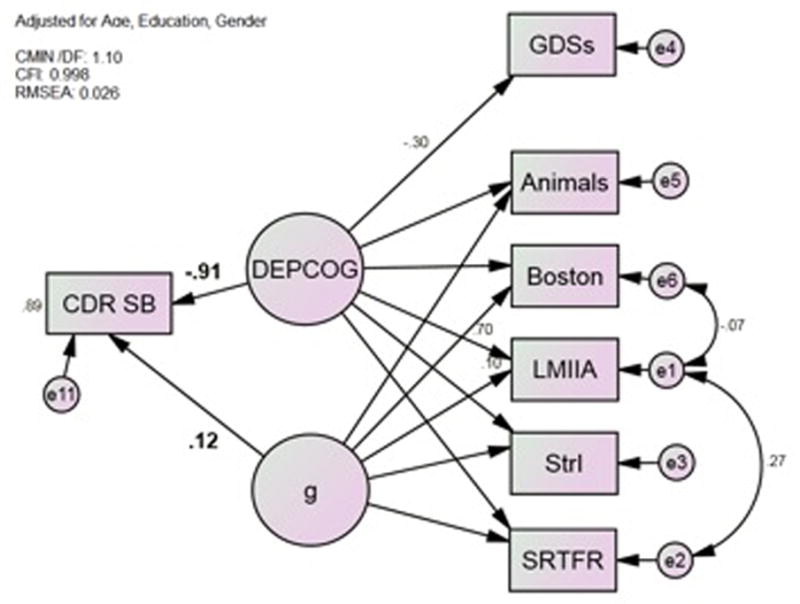
Model 2*; DEPCOG Correlates Strongly with CDR-SB. Animals = Category Fluency: Animals; Boston = Boston Naming Test (15 item); CDR-SB = Clinical Dementia Rating Scale Sum of Boxes; GDSs = Geriatric Depression Scale Subject rated; LMIIA = Wechsler Memory Scale–Revised Logical Memory Story A Delayed; MCI-ADL = Alzheimer’s Disease Cooperative Study Activities of Daily Living Scale for Mild Cognitive Impairment; SRTFR = Selective Reminding Task Free Recall Total; StrI = Stroop Color Task Color-Word Interference Task *All observed variables adjusted for age, gender and education (not shown).
As was true for d, DEPCOG was a strong independent predictor of CDR-SB (r = −0.91, p < 0.001). DEPCOG is labelled moderately strongly by GDSs (r = −0.30, p < 0.001) and more strongly by the cognitive measures (range r = 0.61, StrI; −0.81, SRTFR; all p <0.001). This model again has excellent fit, and fit marginally less well than Model 1 (Table 3).
We compared d, g′ and DEPCOG as predictors of a wide range of clinical outcomes, bedsides CDR-SB. The latent variable d and DEPCOG were comprably strong predictors of categorical BAP clinical diagnoses, and of our previously reported “δ” dementia endophenotype, constructed from an overlapping but more extensive psychometric battery, and localized to the DMN [14] (Table 4). DEPCOG is significantly associated with the subject’s history of depressive illness (r = −0.51, p < 0.001) as is d (r = −0.30, p < 0.001). The latent variable g′ is not, in either model (both p > 0.05). DEPCOG is strongly associated with ADL-MCI scores (r = 0.69, p < 0.001), but slightly less strongly than that measure’s loading on d (r = 0.77, Figure 1).
Table 4.
Latent Variable Partial Correlations with Clinical Outcomes
| Predictor | Outcome | r* | p |
|---|---|---|---|
| d | CDR-SB | −0.94 | ≤ 0.001 |
| DEPCOG | CDR-SB | −0.91 | ≤ 0.001 |
| g′** | CDR-SB | 0.12 | 0.745 |
| d | Diagnosis | −0.81 | ≤ 0.001 |
| DEPCOG | Diagnosis | −0.99 | ≤ 0.001 |
| g′ | Diagnosis | 0.22 | 0.627 |
| d | δ† | 0.94 | ≤ 0.001 |
| DEPCOG | δ | 0.86 | ≤ 0.001 |
| g′ | δ | −0.15 | 0.736 |
| DEPCOG | ADL-MCI | 0.69 | ≤ 0.001 |
| g′ | ADL-MCI | −0.24 | 0.655 |
| DEPCOG | Depression | −0.51 | 0.016 |
| d | Depression | −0.30 | 0.001 |
| g′ | Depression | 0.23 | 0.425 |
Next we examined d and DEPCOG’s independent multivariate associations with BAP clinical diagnoses (Figure 3). In this model, “d” and “DEPCOG” are orthogonal and are obviously not identical to their unadjusted analogs in Models 1 and 2 (respectively). Adjusting these constructs is desirable in order to demonstrate any residual independent effect of DEPCOG on AD’s diagnosis. Eighty percent of the variance was explained by DEPCOG, d, g′ and covariates, but only DEPCOG (r = −0.44, p = 0.044), d (r = −0.73, p < 0.001) and education (r = − 0.21, p = 0.011) made significant independent contributions. DEPCOG and d attenuate each other relative to their mutually unadjusted associations in Table 4.
Figure 3.
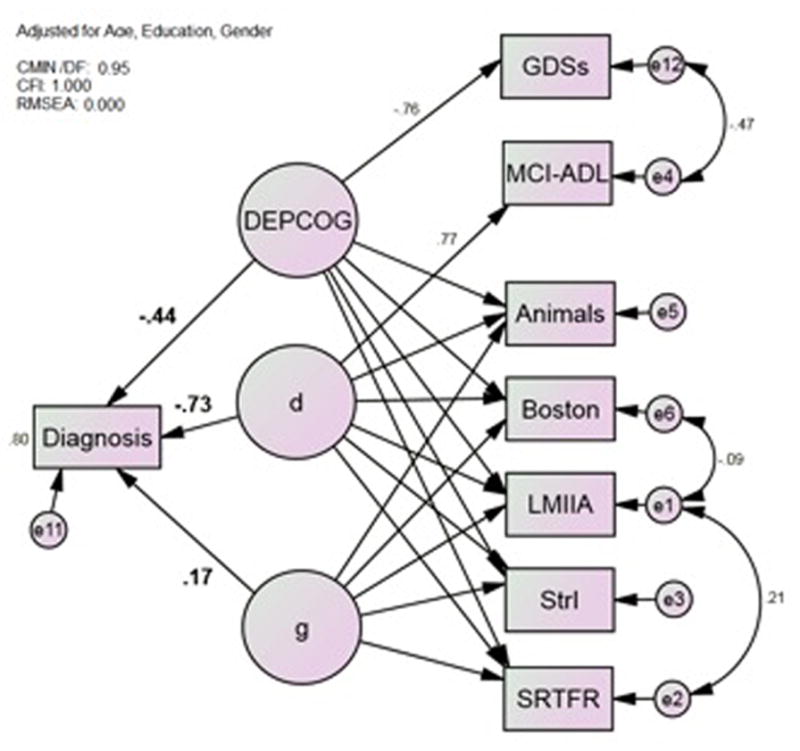
Model 3*; The Latent Variables d and DEPCOG Contribute Independently to Diagnosis. Animals = Category Fluency: Animals; Boston = Boston Naming Test (15 item); CDR-SB = Clinical Dementia Rating Scale Sum of Boxes; GDSc = Geriatric Depression Scale Caregiver rated; LMIIA = Wechsler Memory Scale–Revised Logical Memory Story A Delayed; MCI-ADL = Alzheimer’s Disease Cooperative Study Activities of Daily Living Scale for Mild Cognitive Impairment; SRTFR = Selective Reminding Task Free Recall Total; StrI = Stroop Color Task Color-Word Interference Task *All observed variables adjusted for age, gender and education (not shown).
Because the DMN is activated by self-referential cognitive tasks, we considered the possibility that DEPCOG’s association with dementia status could be mediated through the self-reported nature of depressive symptoms surveys, independent of their symptom content. However, GDSs’ loading on DEPCOG is completely mediated by caregiver-rated GDS scores (GDSc), while DEPCOG’s association with CDR-SB is unaffected. Therefore, the cognitive correlates of self-reported depression ratings are mediated by their depressive symptom content. This model has excellent fit (Table 3).
Next, we constructed d and DEPCOG endophenotypes from their age, education and gender adjusted (but not mutually adjusted) factor loadings. DEPCOG factor scores correlated strongly with d’s (r = 0.93, p < 0.001) (Figure 5). A significant fraction of the cases are disproportionately affected by the cognitive correlates of depressive symptoms. This presentation appears most common among MCI cases.
Figure 5.
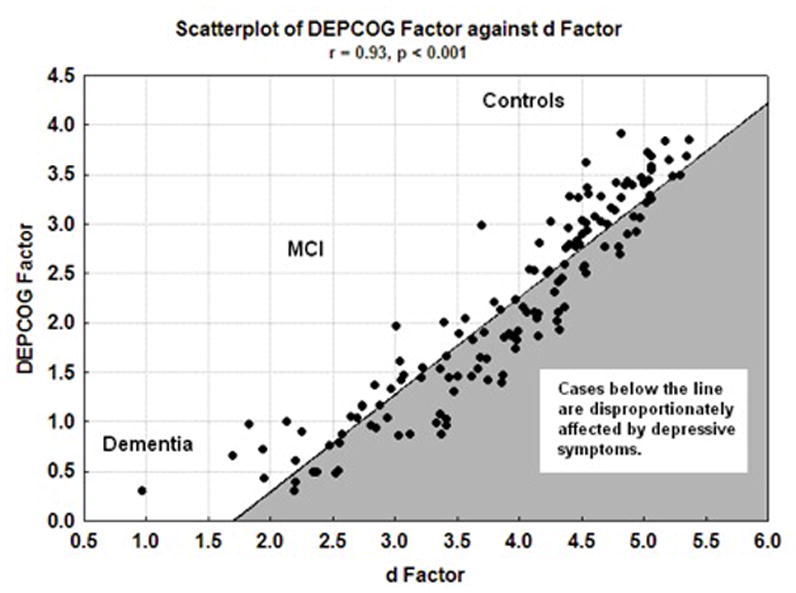
Scatter plot of DEPCOG Factor against d Factor. Cases in the gray space are disproportionately affected by depressive symptoms.
Figure 6 and Table 5 present the locations of peak correlation of gray matter volume with DEPCOG. All associations are adjusted for age, education and gender (and implicitly g′). Visual inspection of Figure 6 reveals a strong overlap with elements of the DMN we previously associated with d, notably bilateral medial frontal and anterior cingulate gyri, bilateral posterior cingulate and precuneus, and bilateral superior temporal lobe (Table 5).
Table 5.
Locations of peak correlation of gray matter volume with DEPCOG*
| Region | BA | x | y | z | cluster size | T | Z |
|---|---|---|---|---|---|---|---|
| Medial Frontal and Anterior Cingulate Gyri | 10 & 32 | −7 | 45 | 16 | 17101 | 9.4 | > 8 |
| 5 | 47 | −12 | 8.5 | 7.62 | |||
| 6 | 42 | 16 | 8.47 | 7.6 | |||
| Middle and Posterior Cingulate Gyrus | 31 | 3 | −45 | 30 | 5238 | 8.32 | 7.49 |
| 5 | −61 | 21 | 7.56 | 6.92 | |||
| 8 | −38 | 39 | 7.53 | 6.89 | |||
| Transverse and Superior Temporal Gyri, & Insula | 38 & 13 | 33 | −30 | −11 | 39434 | 10.06 | > 8 |
| 45 | 1 | −12 | 9.87 | > 8 | |||
| 40 | −5 | 11 | 9.85 | > 8 | |||
| Superior Temporal Gyrus | 42 | 62 | −34 | 18 | 283 | 7.07 | 6.53 |
| Middle and Superior Temporal Gyri | 22 | −56 | −8 | −11 | 5313 | 7.71 | 7.03 |
| −57 | −17 | −8 | 7.65 | 6.98 | |||
| −47 | −12 | −6 | 7.11 | 6.56 | |||
| Insula | −38 | 1 | 6 | 2899 | 8.03 | 7.28 | |
| −36 | 10 | 4 | 7.98 | 7.24 | |||
| −38 | −11 | 13 | 7.58 | 6.93 | |||
| Hippocampus/Parahippocampal Gyrus | −23 | −13 | −21 | 4507 | 8.65 | 7.73 | |
| −29 | −35 | −8 | 7.57 | 6.92 | |||
| Parahippocampal Gyrus | −21 | 2 | −16 | 575 | 7.57 | 6.92 | |
| Middle Frontal Gyrus | 23 | 33 | −16 | 462 | 7.41 | 6.8 | |
| 30 | 52 | 12 | 289 | 7.18 | 6.61 | ||
| Inferior Frontal Gyrus | 46 | −44 | 40 | 2 | 221 | 7.00 | 6.47 |
| 44 | 41 | 11 | 306 | 6.98 | 6.45 | ||
| Thalamus | −7 | 1 | 8 | 389 | 6.84 | 6.34 | |
| −1 | −2 | 3 | 6.72 | 6.25 |
Adjusted for age, gender, education (and implicitly for g′). Higher d scores are associated with greater gray matter volume in these regions.
To more precisely define regions of peak association, a higher threshold is used for reporting (FWE 0.001, k>200).
Finally, we examined whether the grey matter atrophy associated with d and DEPCOG is co-localized with PCC-related structures. After placing a seed in that ROI, a network of inter-correlated structures emerged that overlaps with those associated with d and DEPCOG (Figure 7). All three are co-localized within a subset of the PCC seeded network. Their region of overlap does not include the PCC’s thalamic or periventricular corpus callosum connections. Nor does it include d and DEPCOG’s hippocampal insular, or precuneus and inferior medio-frontal overlap. Instead, these three networks overlap primarily in DMN-related structures, including again the bilateral anterior cingulate gyri, bilateral posterior cingulate, and bilateral superior temporal lobe.
Discussion
We have proposed to re-conceptualize dementing processes as the set of disorders affecting the cognitive correlates of functional status (i.e., “δ”). In this paper, we have replicated δ in a more circumscribed assessment, and demonstrated that the resulting latent variable (d) is again strongly associated with CDR-SB, clinical categorical dementia diagnoses, and δ itself (r = 0.94) (Table 4). Moreover, both latent variables can be specifically associated with elements of the DMN. The homology between δ and d is consistent with Spearman’s description of the latent variable g’s “indifference to its indicators”.
However, these characteristics are also generally true of the cognitive correlates of self-reported depressive symptoms (DEPCOG). This latent variable’s association with dementia severity does not appear to be an artifact of the DMN’s involvement in self-reflection and autobiographical cognitive tasks [51]. Intead, DEPCOG’s association with dementia severity is mediated via the shared variance in cognitive performance and the content of depression symptom ratings. Because DEPCOG’s association with categorical dementia diagnoses is attenuated by d, we can conclude that there is some overlap between DEPCOG and d and therefore, that the cognitive correlates of depressive symptoms are dementing.
Technically, the latent variables labeled “d”, “g”, and “DEPCOG” may not be identical across models 1–4. For example, the replacement of d by DEPCOG in Model 2 should alter “g” relative to Model 1 by changing the DEPCOG adjusted residual variance in g’s cognitive indicators, and therefore their loadings on g itself. However, the practical consequences of this are trivial. The latent variable g is weakly related to CDR-SB in Models 1, 2, and 4, while d and DEPCOG are both strongly related to that outcome. In all three models, the amount of explained CDR-SB variance is essentially unchanged.
A more significant limitation is that the mean GDS score in this sample is in a “sub-syndromal” range. Thus, our results do not speak to categorical diagnoses of “Major Depression”, particularly as operationalized by Diagnostic and Statistical Manual -IVR (DSM) [52] or the Structured Clinical Interview for DSM Disorders (SCID) [53]. However, even sub-syndromal depressive symptoms are relevant to dementia case-finding. Sub-syndromal depressive symptoms are significant risks factors for incident AD [54–56]. Our group has previously shown, in three separate datasets, including a diverse range of ethnic community dwelling elders, that sub-syndromal depressive symptoms are associated with longitudinal cognitive decline, functional status, and mortality risk [9–10; 57]. Moreover, we are not modeling GDS scores here, but instead the fraction of observed variance in GDS scores, no more than 20–30% of the total, that is specifically related to cognitive performance. DEPCOG should be considered a cognitive performance measure first, and only secondarily as a measure of affect.
Epidemiological studies have previously detected an atypical mild depressive syndrome, which is both more common among the elderly and associated with cognitive impairment [58–59]. However, if sub-syndromal GDS scores reflected endemic non-specific mood symptoms, there would be no apriori reason why they would particularly be associated with cognitive performance measures, much less the specific fraction of such variance that is also strongly associated with IADL through d. Thus, our major finding is that even in the absence of major depression, the relatively small fraction of cognitive variance that is associated with self-reported mood is disabling, and significantly influences “AD”’s clinical diagnosis.
In any case, the cognitive correlates of sub-syndromal depressive symptoms appear to map to the very same network as the cognitive correlates of functional status. Their associations with co-localized brain atrophy helps explain the high ranking of depression-related illnesses among the World Health Organisation’s list of disabling conditions [60]. It remains to be determined whether this potential “dementia of sub-syndromal depressive symptoms” is mediated through AD pathology, or if it would be still be disabling in the latter’s absence. Future studies can pursue this issue in a non-AD sample. However, in the current highly selected sample, AD is strongly suspected to be the dementing process reflected by δ/d, and DEPCOG.
Surprisingly, this conclusion conflicts with our recent work in another cohort, which suggests that depressive symptoms and AD pathology are in fact independent of each other [10]. Similarly, a latent d homolog has previously been shown not to be associated with the apolipoprotein (APOE) e4 allele [61]. Moreover, APOE’s effect on whole brain atrophy does not resemble d’s [62], neither does APOE e4 status affect the association between depressive symtpoms and MCI [12]. These observations appear to argue against DEPCOG’s association with AD.
On the other hand, both d and DEPCOG appear to map to portions of a PCC seeded network. This network has previously been associated with β-amyloid deposition by PiB PET [49]. β-amyloid has recently been demonstrated to correlate strongly voxelwise with DMN connectivity [63], and both d and DEPCOG appear to be co-localized with PCC’s DMN-related hubs, notably the cingulate and superior temporal gyri. DEPCOG’s apparent co-localization with β-amyloid-affected structures argues for AD’s mediation of DEPCOG scores.
It may be possible to reconcile these descrepancies. β-amyloid’s association with DMN connectivity is positively inflected [49]. Regions with the highest β-amyloid burdens will therefore have the highest connectivity. DMN connectivity in PCC is similarly increased in cognitively normal persons in the presence of β-amyloid [64]. DMN connectivity is also increased in depressed cases, both young and late onset [65]. The positive associations between depression, β-amyloid burden and DMN connectivity suggest that sub-syndromal depressive symptoms may be an emergent consequence of AD-related β-amyloid-mediated changes in DMN connectivity. Because β-amyloid is first deposited in preclinical AD cases, this might also explain MCI’s disproportionate DEPCOG burden in Figure 5, and the association between sub-syndromal depressive symptoms and MCI conversion risk.
It is also striking that the atrophy related to the latent variables d and DEPCOG has relatively little neocortical extent. These variables’ lack of association with atrophy in the neocortex may explain why so little of the variance in cognitive performance can be related to them [13–14], why observed cognitive performance explains so little variance in IADL’s [66], and the weak to moderate correlations between whole brain averaged Pib PET burden and IADL [67].
Interestingly, neocortical AD neuropathology peforms better as a predictor of Basic Acitivties of Daily Living (BADL) [68], which we have shown not to load strongly on δ [14, 16], and also not to predict CDR-SB independently of δ. In contrast, d and DEPCOG are largely limited to midline paralimbic structures (Figure 6). Midline paralimbic structures are increasingly implicated in the processing of self-referential information [69].
It remains to be seen if anti-depressant treatment can affect DEPCOG scores, but some anti-depressant treatments are known to have saultatory effects on cognition in elderly persons. The most responsive cognitive measures load heavily on δ and presumably DEPCOG (e.g., Digit Symbol Substitution) [70]. This observation is important because the effect of sub-syndromal depressive symptoms on prospective cognitive decline can be shown to be comparable to that of β-amyloid lesions, and yet independent of that neuropathology [10]. In the absence of effective anti-amyloid interventions, anti-depressant treatment of MCI might offer another path towards dementia’s prevention.
Moreover, sub-syndromal depressive symptoms are significantly associated with clinical diagnoses of AD and MCI independently of d (Figure 3). We can conclude then that sub-syndromal depressive symptoms partially contribute to clinicians’ estimates of dementia status via non-disabling (i.e., non-dementing) effects on cognitive performance. This situation is most easily interpreted as a case selection bias. Despite, and possibly because of the fact that these subjects do not meet criteria for a major depressive syndrome, their depressive symptom-related cognitive and cognitively mediated functional impairment(s) risk to be misattributed to “AD”. Thus, effective pharmacotherapy against sub-syndromal depressive symptoms may again have potential to prevent or delay MCI conversion to “AD”, regardless of whether it acts through d or the non-disabling fraction of cognitive performance (i.e., g).
In summary, this analysis indicates that the cognitive correlates of depressive symptoms are empirically highly correlated with d/δ, disabling, and therefore “dementing” in their own right. They cannot be dismissed as “pseudodementia”. This analysis cannot determine whether the latent variables d and DEPCOG are explained by a common dementing process, but AD is suspected in this highly enriched sample. Although our previous work in other cohorts suggests that the effect of depressive symptoms on cognitive decline is not mediated by the better studied AD lesions, other AD-related pathology may be responsible. If so, then that pathology may also be responsible for d’s variability, and thus the dementing character of AD itself.
Our analyses also demonstrate that the cognitive correlates of depressive symptoms affect clinicians’ dementia diagnoses despite, and perhaps even because of, sub-syndromal symptom burdens. Future studies should address DEPCOG’s association with d in non-AD cases, and its relationship to clinical levels of depression. As in cognition, the disability-relevant fraction of depressive symptoms may be small and insuffient to warrant a diagnosis of “Major Depression”, despite its inherently disabling character.
Independent of their associations with d and atrophy in the DMN, depressive symptoms also impart a non-disabling effect on cognitive performance. Clinicians may underappreciate both contributions to their patients’ overall cognitive burden. In the absence of disease modifying agents, intervention on depressive symptoms may yet offer the potential to prevent or reverse MCI’s conversion to “clinical AD.”
Figure 4.
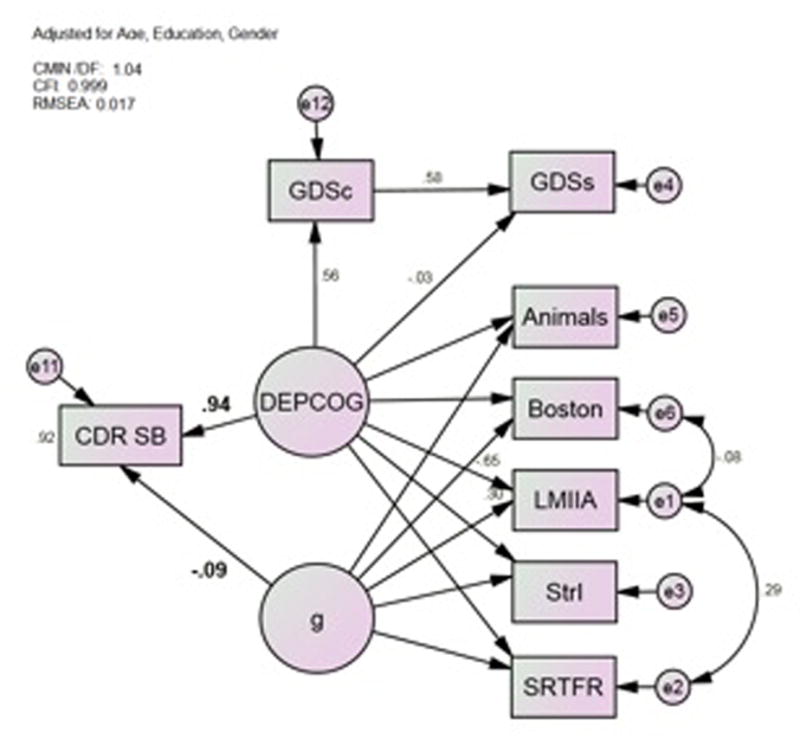
Model 4*; Symptom Content Mediates the GDS’ Effect. Animals = Category Fluency: Animals; Boston = Boston Naming Test (15 item); CDR-SB = Clinical Dementia Rating Scale Sum of Boxes; GDSc = Geriatric Depression Scale Caregiver rated; LMIIA = Wechsler Memory Scale–Revised Logical Memory Story A Delayed; MCI-ADL = Alzheimer’s Disease Cooperative Study Activities of Daily Living Scale for Mild Cognitive Impairment; SRTFR = Selective Reminding Task Free Recall Total; StrI = Stroop Color Task Color-Word Interference Task *All observed variables adjusted for age, gender and education (not shown)
Acknowledgments
This study was made possible by the Julia and Van Buren Parr endowment for the study of Alzheimer’s Disease. The sponsor had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The Brain Aging Project is supported by grants R03AG026374 and R21AG029615 from the National Institutes of Aging, grant K23NS058252 from the National Institute on Neurological Disorders and Stroke. The University of Kansas General Clinical Research Center (M01RR023940) provided essential space, expertise, and nursing support. Dr. Burns and Vidoni are supported by the University of Kansas Alzheimer’s Disease Center (P30AG035982). Dr. Burns is also supported by R01AG034614 and R01AG033673. Dr. Vidoni is also supported by the Heartland Institute for Clinical & Translational Research, University of Kansas Medical Center’s CTSA, KL2 TR000119 & UL1 TR00001. Dr. Honea is supported by K01AG035042. The extraction of these latent variables and their biomarkers is covered under provisional patents 61/603,226 and 61/671,858.
Contributor Information
Donald R. Royall, Email: royall@uthscsa.edu.
Raymond F. Palmer, Email: palmerr@uthscsa.edu.
Eric D. Vidoni, Email: evidoni@kumc.edu.
Robyn A. Honea, Email: rhonea@kumc.edu.
References
- 1.Vilalta-Franch J, Garre-Olmo J, Lopez-Pousa S, Turon-Estrada A, Lozano-Gallego M, Hernandez-Ferrandiz M, Pericot-Nierga I, Feijoo-Lorza R. Comparison of different clinical diagnostic criteria for depression in Alzheimer disease. Am J Geriatr Psychiatry. 2006;14:589–597. doi: 10.1097/01.JGP.0000209396.15788.9d. [DOI] [PubMed] [Google Scholar]
- 2.Speck CE, Kukull WA, Brenner DE, Bowen JD, McCormick WC, Teri L, Pfanschmidt ML, Thompson JD, Larson EB. History of depression as a risk factor for Alzheimer’s disease. Epidemiol. 1995;6:366–369. doi: 10.1097/00001648-199507000-00006. [DOI] [PubMed] [Google Scholar]
- 3.Green RC, Cupples LA, Kurz A, Auerbach S, Go R, Sadovnick D, Duara R, Kukull WA, Chui H, Edeki T, Griffith PA, Friedland RP, Bachman D, Farrer L. Depression as a risk factor for Alzheimer disease: the MIRAGE Study. Arch Neurol. 2003;60:753–759. doi: 10.1001/archneur.60.5.753. [DOI] [PubMed] [Google Scholar]
- 4.Steenland K, Karnes C, Seals R, Carnevale C, Hermida A, Levey A. Late-Life Depression as a Risk Factor for Mild Cognitive Impairment or Alzheimer’s Disease in 30 US Alzheimer’s Disease Centers. J Alzheimers Dis. 2012;31:265–275. doi: 10.3233/JAD-2012-111922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ownby RL, Crocco E, Acevedo A, John V, Loewenstein D. Depression and risk for Alzheimer disease: systematic review, meta-analysis, and metaregression analysis. Arch Gen Psychiatry. 2006;63:530–538. doi: 10.1001/archpsyc.63.5.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee JS, Potter GG, Wagner HR, Welsh-Bohmer KA, Steffens DC. Persistent mild cognitive impairment in geriatric depression. Int Psychogeriatr. 2007;19:125–135. doi: 10.1017/S1041610206003607. [DOI] [PubMed] [Google Scholar]
- 7.Gabryelewicz T, Styczynska M, Luczywek E, Barczak A, Pfeffer A, Androsiuk W, Chodakowska-Zebrowska M, Wasiak B, Peplonska B, Barcikowska M. The rate of conversion of mild cognitive impairment to dementia: predictive role of depression. Int J Geriatr Psychiatry. 2007;22:563–567. doi: 10.1002/gps.1716. [DOI] [PubMed] [Google Scholar]
- 8.Rosenberg PB, Mielke MM, Appleby BS, Oh ES, Geda YE, Lyketsos CG. The association of neuropsychiatric symptoms in MCI with incident dementia and Alzheimer Disease. Am J Geriatr Psychiatry. 2012 doi: 10.1016/j.jagp.2013.01.006. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Royall DR, Palmer R, Chiodo LK, Polk M. Depressive symptoms predict longitudinal change in executive control but not memory. Int J Geriatr Psychiatry. 2012;27:89–96. doi: 10.1002/gps.2697. [DOI] [PubMed] [Google Scholar]
- 10.Royall DR, Palmer RF. Alzheimer pathology does not mediate the association between depressive symptoms and subsequent cognitive decline. Alz Dis & Dementia. doi: 10.1016/j.jalz.2011.11.009. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Madsen K, Hasselbalch BJ, Frederiksen KS, Haahr ME, Gade A, Law I, Price JC, Knudsen GM, Kessing LV, Hasselbalch SG. Lack of association between prior depressive episodes and cerebral [(11)C] PiB binding. Neurobiol Aging. 2012;33:2334–2342. doi: 10.1016/j.neurobiolaging.2011.11.021. [DOI] [PubMed] [Google Scholar]
- 12.Nose M, Kodama C, Ikejima C, Mizukami K, Matsuzaki A, Tanaka S, Yoshimura Aasumo F, Asada T. ApoE4 is not associated with depressive symptoms when mild cognitive impairment is considered. Int J Geriatr Psychiatry. doi: 10.1002/gps.3803. in press. [DOI] [PubMed] [Google Scholar]
- 13.Royall DR, Palmer RF. Getting Past “g”: Testing a new model of dementing processes in non-demented persons. J Neuropsychiatry Clin Neurosci. 2012;24:37–46. doi: 10.1176/appi.neuropsych.11040078. [DOI] [PubMed] [Google Scholar]
- 14.Royall DR, Palmer RF, O’Bryant S. Validation of a latent variable representing the dementing process. J Alzheimer’s Dis. 2012;30:639–649. doi: 10.3233/JAD-2012-120055. [DOI] [PubMed] [Google Scholar]
- 15.Spearman C. General intelligence, objectively determined and measured. Am J Psychol. 1904;15:201–293. [Google Scholar]
- 16.Royall DR, Palmer RF, Vidoni ED, Honea RA, Burns JM. The Default Mode Network and related right hemisphere structures may be the key substrates of dementia. J Alzheimer’s Dis. 2012;32:467–478. doi: 10.3233/JAD-2012-120424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uddin LQ, Kelly AM, Biswal BB, Xavier Castellanos F, Milham MP. Functional connectivity of default mode network components: correlation, anticorrelation, and causality. Hum Brain Mapp. 2009;30:625–637. doi: 10.1002/hbm.20531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buckner RL, Andrews-Hanna JR, Schacter DL. The Brain’s Default Network: Anatomy, Function, and Relevance to Disease. Ann NY Acad Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- 19.Sheline YI, Barch DM, Price JL, Rundle MM, Vaishnavi SN, Snyder AZ, Mintun MA, Wang S, Coalson RS, Raichle ME. The default mode network and self-referential processes in depression. Proc Natl Acad Sci. 2009;106:1942–1947. doi: 10.1073/pnas.0812686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goveas JS, Espeland MA, Hogan P, Dotson V, Tarima S, Coker LH, Okene J, Brunner R, Woods NF, Wassertheil-Smoller S, Kotchen JM, Resnick S. Depressive symptoms, brain volumes and subclinical cerebrovascular disease in postmenopausal women: the Women’s Health Initiative MRI Study. J Affect Disord. 2011;132:275–284. doi: 10.1016/j.jad.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry. 1982;140:566–572. doi: 10.1192/bjp.140.6.566. [DOI] [PubMed] [Google Scholar]
- 22.Burns JM, Cronk BB, et al. Cardiorespiratory fitness and brain atrophy in early Alzheimer disease. Neurol. 2008;71:210–216. doi: 10.1212/01.wnl.0000317094.86209.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Honea R, Crow TJ, et al. Regional deficits in brain volume in schizophrenia: a meta-analysis of voxel-based morphometry studies. Am J Psychiatry. 2005;162:2233–2245. doi: 10.1176/appi.ajp.162.12.2233. [DOI] [PubMed] [Google Scholar]
- 24.Vidoni ED, Honea RA, Billinger SA, Swerdlow RH, Burns JM. Cardiorespiratory fitness is associated with atrophy in Alzheimer’s and aging over 2 years. Neurobiol Aging. 2012;33:1624–1632. doi: 10.1016/j.neurobiolaging.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morris JC, Storandt M, et al. Mild Cognitive Impairment Represents Early-Stage Alzheimer Disease. Arch Neurol. 2001;58:397–405. doi: 10.1001/archneur.58.3.397. [DOI] [PubMed] [Google Scholar]
- 26.McKhann G, Drachman D, et al. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 27.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 28.Berg LD, McKeel W, Jr, et al. Clinicopathologic Studies in Cognitively Healthy Aging and Alzheimer Disease: Relation of Histologic Markers to Dementia Severity, Age, Sex, and Apolipoprotein E Genotype. Arch Neurol. 1998;55:326–335. doi: 10.1001/archneur.55.3.326. [DOI] [PubMed] [Google Scholar]
- 29.Sheikh JI, Yesavage JA. Geriatric Depression Scale (GDS): Recent evidence and development of a shorter version. Clin Gerontologist. 1986;5:165–173. [Google Scholar]
- 30.Logsdon RG, Teri L. Depression in Alzheimer’s Disease patients: Caregivers as surrogate reporters. J Am Geriatr Soc. 1995;43:150–155. doi: 10.1111/j.1532-5415.1995.tb06380.x. [DOI] [PubMed] [Google Scholar]
- 31.Wechsler D, Stone CP. Manual: Wechsler Memory Scale. New York: Psychological Corporation; 1973. [Google Scholar]
- 32.Grober E, Buschke H, et al. Screening for dementia by memory testing. Neurology. 1988;38:900–903. doi: 10.1212/wnl.38.6.900. [DOI] [PubMed] [Google Scholar]
- 33.Hanninen T, Reinikainen KJ, et al. Subjective memory complaints and personality traits in normal elderly subjects. J Am Geriatr Soc. 1994;42:1–4. doi: 10.1111/j.1532-5415.1994.tb06064.x. [DOI] [PubMed] [Google Scholar]
- 34.Stroop J. Studies of interference in serial verbal reactions. J Exp Psychol. 1935;18:643–662. [Google Scholar]
- 35.Folstein MF, Folstein SE, McHugh PR. Mini-mental state: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:198–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 36.Galasko D, Bennett D, et al. An inventory to assess activities of daily living for clinical trials in Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. Alz Dis & Assoc Disord. 1997;11(Suppl 2):S33–39. [PubMed] [Google Scholar]
- 37.Arbuckle JL. Analysis of Moment Structures-AMOS (Version 7.0) [Computer Program] Chicago: SPSS; 2006. [Google Scholar]
- 38.Schafer JL, Graham JW. Missing data: Our view of the state of the art. Psychol Methods. 2002;7:147–177. [PubMed] [Google Scholar]
- 39.Graham JW. Missing Data Analysis: Making it work in the real world. Ann Rev Psychol. 2009;6:549–576. doi: 10.1146/annurev.psych.58.110405.085530. [DOI] [PubMed] [Google Scholar]
- 40.Bollen KA, Long JS. Testing Structural Equation Models. Sage Publications; Thousand Oaks, CA: 1993. [Google Scholar]
- 41.Bentler PM. Comparative fit indexes in structural models. Psychol Bull. 1990;107:238–246. doi: 10.1037/0033-2909.107.2.238. [DOI] [PubMed] [Google Scholar]
- 42.Browne M, Cudeck R. Alternative ways of assessing model fit. In: Bollen KA, Long JS, editors. Testing structural equation models. Sage Publications; Thousand Oaks, CA: 1993. pp. 136–162. [Google Scholar]
- 43.Honea RA, Thomas GP, et al. Cardiorespiratory fitness and preserved medial temporal lobe volume in Alzheimer disease. Alzheimer Dis Assoc Disord. 2009;23:188–197. doi: 10.1097/WAD.0b013e31819cb8a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ashburner J, Friston KJ. Unified segmentation. Neuroimage. 2005;26:839–851. doi: 10.1016/j.neuroimage.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 45.Gaser C, Altaye M, et al. Unified segmentation without tissue priors. Neuroimage. 2007;36(Suppl 1):S68. [Google Scholar]
- 46.Wilke M, Holland SK, et al. Template-O-Matic: a toolbox for creating customized pediatric templates. Neuroimage. 2008;41:903–913. doi: 10.1016/j.neuroimage.2008.02.056. [DOI] [PubMed] [Google Scholar]
- 47.Lancaster JL, Rainey LH, et al. Automated labeling of the human brain: A preliminary report on the development and evaluation of a forward-transform method. Hum Brain Mapp. 1997;5:238–242. doi: 10.1002/(SICI)1097-0193(1997)5:4<238::AID-HBM6>3.0.CO;2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maldjian JA, Laurienti PJ, et al. An automated method for neuroanatomic and cytoarchitectonic atlas-based interrogation of fMRI data sets. Neuroimage. 2003;19:1233–1239. doi: 10.1016/s1053-8119(03)00169-1. [DOI] [PubMed] [Google Scholar]
- 49.Oh H, Mormino EC, Madison C, Hayenga A, Smiljic A, Jagust WJ. β-Amyloid affects frontal and posterior brain networks in normal aging. Neuroimage. 2011;54:187–195. doi: 10.1016/j.neuroimage.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Montembeault M, Joubert S, Doyon J, Carrier J, Gagnon JF, Monchi O, Lungu O, Belleville S, Brambati SM. The impact of aging on gray matter structural covariance networks. Neuroimage. 2012;63:754–759. doi: 10.1016/j.neuroimage.2012.06.052. [DOI] [PubMed] [Google Scholar]
- 51.Sperleng RN, Mar RA, Kim AS. The common neural basis of autobiographical memory, prospection, navigation, theory of mind, and the default mode: a quantitative meta-analysis. J Cogn Neurosci. 2009;21:489–510. doi: 10.1162/jocn.2008.21029. [DOI] [PubMed] [Google Scholar]
- 52.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. Washington: American Psychiatric Association; 1994. [Google Scholar]
- 53.First MB, Spitzer RL, Gibbon M, Williams JBW. Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version, Non-patient Edition (SCID-I/NP) New York: Biometrics Research, New York State Psychiatric Institute; 2002. [Google Scholar]
- 54.Devanand DP, Sano M, Tang MX, Taylor S, Gurland BJ, Wilder D, Stern Y, Mayeux R. Depressed mood and the incidence of Alzheimer’s disease in the elderly living in the community. Arch Gen Psychiatry. 1996;53:175–182. doi: 10.1001/archpsyc.1996.01830020093011. [DOI] [PubMed] [Google Scholar]
- 55.Geerlings MI, Schmand B, Braam AW, Jonker C, Bouter LM, van Tilburg W. Depressive symptoms and risk of Alzheimer’s disease in more highly educated older people. J Am Geriatr Soc. 2000;48:1092–1097. doi: 10.1111/j.1532-5415.2000.tb04785.x. [DOI] [PubMed] [Google Scholar]
- 56.Wilson RS, Barnes LL, Mendes de Leon CF, Aggarwal NT, Schneider JS, Bach J, Pilat J, Beckett LA, Arnold SE, Evans DA, Bennett DA. Depressive symptoms, cognitive decline, and risk of AD in older persons. Neurology. 2002;59:364–370. doi: 10.1212/wnl.59.3.364. [DOI] [PubMed] [Google Scholar]
- 57.Royall DR, Palmer R, Chiodo LK, Polk MJ, Markides KS, Hazuda H. Clock-drawing potentially mediates depression’s effect on mortality: Replication in three cohorts. Int J Geriatr Psychiatry. 2008;23:821–829. doi: 10.1002/gps.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davidson J, Woodbury MA, Pelton S, Krishnan R. A study of depressive typologies using grade of membership analysis. Psychol Med. 1988;18:179–189. doi: 10.1017/s0033291700002002. [DOI] [PubMed] [Google Scholar]
- 59.Davidson JR, Woodbury MA, Zisook S, Giller EL., Jr Classification of depression by grade of membership: a confirmation study. Psychol Med. 1989;19:987–998. doi: 10.1017/s0033291700005717. [DOI] [PubMed] [Google Scholar]
- 60.Pollock B, Reynolds C. Depression in late life. Harvard Mental Health Letter. 2000;17:3–5. [PubMed] [Google Scholar]
- 61.Bogner HR, Richie MB, de Vries HF, Morales KH. Depression, cognition, apolipoprotein e genotype: latent class approach to identifying subtype. Am J Geriatr Psychiatry. 2009;17:344–352. doi: 10.1097/JGP.0b013e3181987730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pievani M, Rasser PE, Galluzzi S, Benussi L, Ghidoni R, Sabattoli F, Bonetti M, Binetti G, Thompson PM, Frisoni GB. Mapping the effect of APOE epsilon4 on gray matter loss in Alzheimer’s disease in vivo. Neuroimage. 2009;45:1090–1098. doi: 10.1016/j.neuroimage.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Buckner RL, Sepulcre J, Talukdar T, Krienen FM, Liu H, Hedden T, Andrews-Hanna JR, Sperling RA, Johnson KA. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer’s disease. J Neurosci. 2009;29:1860–1873. doi: 10.1523/JNEUROSCI.5062-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sheline YI, Raichle ME, Snyder AZ, Morris JC, Head D, Wang S, Mintun MA. Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol Psychiatry. 2010;67:584–587. doi: 10.1016/j.biopsych.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alexopoulos GS, Hoptman MJ, Kanellopoulos D, Murphy CF, Lim KO, Gunning FM. Functional connectivity in the cognitive control network and the default mode network in late-life depression. J Affect Disord. 2012;39:56–65. doi: 10.1016/j.jad.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Royall DR, Lauterbach EC, Kaufer DI, Malloy P, Coburn KL, Black KJ. The cognitive correlates of functional status: A review from the Committee on Research of the American Neuropsychiatric Association. J Neuropsychiatry Clin Neurosci. 2007;19:249–265. doi: 10.1176/jnp.2007.19.3.249. [DOI] [PubMed] [Google Scholar]
- 67.Marshall GA, Olson LE, Frey MT, Maye J, Becker JA, Rentz DM, Sperling RA, Johnson KA. Instrumental activities of daily living impairment is associated with increased amyloid burden. Dement Geriatr Cogn Disord. 2011;31:43–50. doi: 10.1159/000329543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marshall GA, Fairbanks LA, Tekin S, Vinters HV, Cummings JL. Neuropathologic correlates of activities of daily living in Alzheimer disease. Alzheimer Dis Assoc Disord. 2006;20:56–59. doi: 10.1097/01.wad.0000201852.60330.16. [DOI] [PubMed] [Google Scholar]
- 69.Qin P, Northoff G. How is our self related to midline regions and the default-mode network? NeuroImage. 2011;57:1221–1233. doi: 10.1016/j.neuroimage.2011.05.028. [DOI] [PubMed] [Google Scholar]
- 70.Doraiswamy PM, Krishnan KR, Oxman T, Jenkyn LR, Coffey DJ, Burt T, Clary CM. Does antidepressant therapy improve cognition in elderly depressed patients? J Gerontol A Biol Sci Med Sci. 2003;58:M1137–1144. doi: 10.1093/gerona/58.12.m1137. [DOI] [PubMed] [Google Scholar]