

SYNOPSIS
Abnormal smooth muscle cell proliferation is a hallmark of vascular disease. Although growth factors are known to contribute to cell hyperplasia, the changes in metabolism associated with this response, particularly mitochondrial respiration, remain unclear. Given the increased energy requirements for proliferation, we hypothesized that platelet-derived growth factor (PDGF) would stimulate glycolysis and mitochondrial respiration and that this elevated bioenergetic capacity is required for smooth muscle cell hyperplasia. To test this hypothesis, cell proliferation, glycolytic flux, and mitochondrial oxygen consumption were measured after treatment of primary rat aortic smooth muscle cells with PDGF. PDGF increased basal and maximal rates of glycolytic flux and mitochondrial oxygen consumption; enhancement of these bioenergetic pathways led to a substantial increase in the mitochondrial reserve capacity. Interventions with the PI3K inhibitor LY-294002 or the glycolysis inhibitor 2-deoxy-D-glucose abrogated PDGF-stimulated proliferation and prevented augmentation of glycolysis and mitochondrial reserve capacity. Similarly, when L-glucose was substituted for D-glucose, PDGF-dependent proliferation was abolished, as were changes in glycolysis and mitochondrial respiration. Interestingly, lactate dehydrogenase protein levels and activity were significantly increased after PDGF treatment. Moreover, L-lactate substitution for D-glucose was sufficient for increasing the mitochondrial reserve capacity and cell proliferation after treatment with PDGF; these effects were inhibited by the lactate dehydrogenase inhibitor, oxamate. These data suggest that glycolysis, by providing substrates that enhance the mitochondrial reserve capacity, plays an essential role in PDGF-induced cell proliferation, underscoring the integrated metabolic response required for proliferation of VSMC in the diseased vasculature.
Keywords: PI3K, vascular smooth muscle cells, proliferation, mitochondria, metabolic flux
INTRODUCTION
Vascular smooth muscle cell (VSMC) proliferation stimulated by platelet-derived growth factor (PDGF) is instrumental in the progression of atherosclerosis, angioplasty-dependent restenosis, neo-intimal hyperplasia and other complications associated with vascular disease [1–4]. Recent studies suggest that PDGF-dependent VSMC proliferation involves stimulation of glycolysis [5–7]. Other studies have reported that a major metabolite produced by proliferating VSMC is lactate [8], again supporting the concept that induction of glycolysis may be required for cell proliferation [5]. However, augmentation of glycolytic activity, supported by increased glycolytic protein expression and glucose utilization, has not been characterized further in the context of mitochondria-related metabolic changes. This is potentially important since the changes in metabolism associated with VSMC proliferation may contribute to the pathological changes associated with cell hyperplasia.
The increase in glycolytic flux due to growth factors has been shown to be critical in the bioenergetic shift that occurs during VSMC proliferation [9, 10]. Several signaling pathways, including the PI3K/Akt pathway, have been shown to regulate glucose metabolism, particularly through the glucose transporter-4 channel and activation of pathways that lead to increased glycolytic enzyme activity and expression [11–15]. Activation of the PI3K/Akt pathway in cells occurs rapidly upon PDGF exposure and has been shown to be a modulator of glycolytic activity. However, the role mitochondria play in this proliferative response remains unclear. Establishing the role of cellular bioenergetics in proliferating cells is therefore critical for identifying new cellular targets that may help prevent the uncontrolled proliferation of VSMC, yet spare normal functioning vascular cells.
We hypothesized that glycolysis serves to increase the availability of metabolic substrates for oxidative phosphorylation and thereby supports the energy required for proliferation in growth factor-stimulated cells. Using extracellular flux technology, we examined mitochondrial oxygen consumption as well as glycolytic flux in intact VSMC in response to PDGF. We show that PDGF increases both glycolysis and mitochondrial respiration in VSMC. Interruption of glycolysis via direct inhibition of PI3K, glycolysis, or lactate dehydrogenase decreases the capacity of the cells to mount the bioenergetic response required for proliferation in response to PDGF. In addition, these studies suggest that the lactate dehydrogenase pathway may play an important role in regulating the bioenergetic reserve of VSMC and therefore may be integral to the hyperplastic VSMC phenotype.
MATERIALS AND METHODS
Reagents and antibodies
Sodium pyruvate, D-glucose, L-glucose, sodium-L-lactate, NADH, 2-deoxy-D-glucose, 5,5′-dithiobis-(2-nitrobenzoic) acid (DTNB), lauryl maltoside, potassium ferricyanide, ascorbate, oxaloacetate, acetyl coenzyme A, cytochrome c, sodium oxamate, adenosine 5-triphosphate disodium salt (ATP) and adenosine 5-diphosphate disodium salt (ADP) were purchased from Sigma (St. Louis, MO). Chromatography grade tetrabutylammonium hydrogen sulfate (TBAHS) from Fluka Analytical was also obtained through Sigma. Analytical grade perchloric acid (69%), dipotassium hydrogen phosphate (K2HPO4), potassium dihydrogen phosphate (KH2PO4), methanol, sodium hydroxide (NaOH) and orthophosphoric acid (85%) were obtained from Fisher Scientific (Pittsburg, PA). LY-294002 was purchased from Calbiochem (San Diego, California). Akt, phospho-Akt (Ser473), LDH, GAPDH, phospho-retinoblastoma (Rb) protein (Ser807/811), cyclin D1 and β-actin antibodies were purchased from Cell Signaling Technologies (Danvers, MA). Phospho-PDH-E1-α (Ser293) antibody was purchased from Novus Biologicals (Littleton, Colorado). PDK-1 antibody was purchased from Assay Designs (Ann Arbor, Michigan). JC-1 dye (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide) was purchased from Invitrogen (Carlsbad, California). Complex I (39 kD), Complex II (70 kD), Complex III core 2, cytochrome c oxidase (complex IV) subunit I, PDH-E1α and VDAC antibodies were purchased from MitoSciences (Eugene, Oregon). Cytochrome c antibodies were purchased from BD Biosciences (San Jose, California). PDGF-BB was purchased from R&D Biosciences (San Diego, CA).
Cell culture and treatments
Rat aortic smooth muscle cells were harvested from descending thoracic aortas and maintained at 37°C in 5% CO2 in DMEM growth medium (Gibco, from Invitrogen) containing D-glucose (1 g/L), GLUTAMAX (4 mM), pyruvate (1 mM), sodium bicarbonate (3.7 g/L) and 10% fetal bovine serum (FBS) (Atlanta Biologicals; Lawrenceville, GA) with penicillin (100 U/ml) and streptomycin (100 ng/ml). Cells used in this study were between passages 4 to 12. Experiments were performed in complete serum media containing 10% FBS as described above unless specified. Glucose-free medium containing L-glutamate in place of GLUTAMAX was used for treatments with L-glucose (1 g/L) or L-lactate (18 mM). Sub-confluent cells grown in either 6-well, 12-well or Seahorse Bioscience V7 tissue culture plates were incubated with vehicle or inhibitor for 1 h prior to treatment with vehicle or PDGF for the indicated times.
Measurement of cellular bioenergetic function using the XF-24 Extracellular Flux Analyzer
The XF-24 Extracellular Flux Analyzer from Seahorse Biosciences (North Billerica, MA) was used to examine the effects of chronic PDGF treatment on glycolysis and mitochondrial function in rat aortic smooth muscle cells as described in [16–19].
A mitochondrial function assay based on inhibitors of the electron transport chain and uncoupling agents was used to identify changes in key aspects of respiratory function [20]. Briefly, three basal oxygen consumption rates (OCR) are measured to obtain a baseline OCR, followed by the sequential injection and measurement of OCR with oligomycin (1 μg/ml), FCCP (1 μM), and antimycin A (10 μM) after each intervention. In this study, experiments were designed to determine ECAR and OCR at the end of treatment without or with inhibitors and/or PDGF. The base media was changed prior to the bioenergetic measurements to serum-free unbuffered (without sodium bicarbonate) DMEM media base (Cellgro) supplemented with 4 mM L-glutamine, 5.5 mM D-glucose, and 1 mM sodium pyruvate at pH 7.4. In some experiments, L-glucose or L-lactate was substituted for D-glucose. Due to the effects of PDGF on vascular smooth muscle cell proliferation, total cellular protein was measured following each experiment using the Lowry method [21] and used to normalize mitochondrial function rates.
Cell counting
Cells treated in 6-well plates were washed twice with PBS followed by 3 min incubation with 300 μl of 0.25% trypsin-EDTA at 37°C. After trypsinization, cells were resuspended in 1 ml of 10% FBS-containing media and placed in Eppendorf tubes kept at 37°C. Cells were immediately counted using a hemacytometer. At least 5 fields per replicate were counted to obtain the average cell number for each treatment group.
LDH activity assay
LDH was measured in the cell lysates and media as both an indicator of cell viability and change in the specific activity of the enzyme. After the indicated treatments, the medium was collected, and lysates were prepared by scraping cells in 100 μl of PBS containing 0.1% Triton X-100. After centrifugation of the lysates and media, LDH activity was measured spectrophotometrically at 340 nm for 4 min at 37°C. Cytotoxicity was then estimated by dividing the change in absorbance of the medium by the values for LDH activity in the medium and the lysate combined. The values were then normalized to control, which represented 100% cell viability. Total protein in the cell lysates was measured using the Bradford method [22]. The extinction coefficient of NADH (6220 M−1 cm−1) was used to calculate the nanomoles of NADH oxidized per min per milligram of protein.
Western blot analysis
Cells were washed twice with PBS and lysed in protease inhibitor- and phosphatase-inhibitor-containing cell lysis buffer (20 mM HEPES pH 7.0, 1 mM DTPA, 1% NP-40, 0.1% SDS). Cell lysates were collected and centrifuged at 14,000 × g for 10 min at 4°C. Supernatants were collected and protein concentration was determined by the Lowry method using BSA as a standard. Samples were mixed with Laemmli sample buffer and boiled for 10 min. Equal amounts of protein were resolved by 10 or 12% SDS-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. Immunoblot analysis was performed using antibodies listed above.
Measurement of mitochondrial inner membrane potential (ΔψM)
The inner membrane ΔψM was assessed using JC-1 dye. Briefly, VSMC grown in 12-well culture plates were treated without or with PDGF for 24 h after which 7.4 μM JC-1 was added directly to cells in culture medium and incubated for 30 min. Next, cells were washed with PBS, and red/green fluorescence was measured using a fluorescence plate reader with excitation/emission filters suitable for rhodamine (560/595 nm) and fluorescein (484/535 nm), respectively. Data are expressed as the ratio of red to green fluorescence.
Citrate synthase activity assay
Citrate synthase was assayed by monitoring the conversion of oxaloacetate and acetyl coenzyme A into citrate and coenzyme A. The addition of 5,5′-dithiobis-(2-nitrobenzoic) acid (DTNB) promotes the chemical conversion of DTNB into 2-nitro-5-thiobenzoic acid (TNB) by coenzyme A. This reaction was monitored spectrophotometrically at 412 nm. VSMC treated without or with PDGF for 24 h were lysed in PBS containing 0.2% lauryl maltoside. At least 10 μg of whole cell lysate was added to a cuvette which contained buffer (100 mM Tris with 0.1% Triton X-100, pH 8.0), 100 μM acetyl coenzyme A, and 200 μM DTNB. The reaction was initiated by adding 200 μM oxaloacetate, and the absorbance at 412 nm was monitored for 4 min to obtain a rate of activity. The protein content was measured in cell lysates and used to normalize the rates of activity.
Complex IV activity assay
Complex IV activity was assessed by monitoring the rate of cytochrome c oxidation spectrophotometrically [23]. VSMC treated without or with PDGF for 24 h were lysed in PBS containing 0.2% lauryl maltoside. Purified reduced cytochrome c was used as a substrate in this assay, and the concentration was determined using the extinction coefficient (19200 M−1 cm−1) at 550 nm. A reference cuvette containing 50 μM purified oxidized cytochrome c (by addition of 1 mM ferricyanide) in 10 mM phosphate buffer at pH 7.0 was used to subtract background absorbance. At least 5 μg of cell lysate was added to a cuvette containing 10 mM phosphate buffer, pH 7.0 and 50 μM cytochrome c. The rate of cytochrome c oxidation was then measured. Complex IV activity was calculated by plotting the natural log of the change in absorbance at 550 nm versus time. The rate was then calculated and normalized to protein concentration.
Nucleotide Extraction
Briefly, an aliquot of protein-precipitated lysate was obtained from VSMC after the indicated treatments by scraping the cells after addition of 5% perchloric acid. This lysate was centrifuged, and the precipitated protein pellet was stored and later resuspended in 1 ml of 0.5 M NaOH. Protein concentration was determined by Bradford assay with BSA as a standard. Supernatant was transferred to an Eppendorf tube and neutralized by precipitating ClO4− with K2HPO4. The suspension was vortexed, kept on ice for 10 min and then centrifuged to remove salt. Supernatant was immediately used or stored at −80°C until analysis.
HPLC separation and measurement of adenine nucleotides
Nucleotide analysis was performed by modifying a method previously described in [24, 25]. The HPLC system consisted of a Gold HPLC model equipped with System Gold 168 Detector and System Gold Autosampler 507 from Beckman Coulter Inc. (Fullerton, CA). The analytical column was a Supelcosil LC-18-T, (150 × 4.6 mm ID, particle size 3 μm) from Sigma. Analytical runs were processed by 32 Karat Software (version 8.0) also from Beckman Coulter Inc. The chromatographic separation was performed at ambient temperature with gradient elution. The mobile phase flow rate was set at 0.9 ml/min and consisted of 65 mM potassium phosphate buffer and 3 mM TBAHS adjusted to pH 6.0 with orthophosphoric acid (buffer A), and 30% methanol in 65 mM potassium phosphate buffer with 4 mM TBAHS adjusted to pH 6.0 with orthophosphoric acid (buffer B). The buffers were delivered in a linear gradient as follows: 0–2 min 30% buffer B, 2–16 min to 90% buffer B, 16–20 min to 90% buffer B, 20–21 min returned to 30% buffer B, 21–24 min 30% buffer B; with a 1 min equilibration between injections. The injection volume was 10 μl. Nucleotides were detected using a UV spectrum sweep between 200 and 400 nm, and detection peaks were compared to the UV spectra of ATP and ADP standards at different concentrations. Nucleotides were monitored at A254 and A262 nm. Standard ATP and ADP samples were prepared by dissolving in buffer A. Standards were prepared at a range of 2 to 100 μM to create a standard curve against which to compare the experimental samples. Standards were not filtered prior to injection. Experimental samples were prepared as follows: A volume of 150 μl of nucleotide extract suspension was mixed with 150 μl of buffer A, and filtered prior to injection in HPLC.
Statistical analysis
Data are reported as means ± SEM. Comparisons between two groups were performed with unpaired Student’s t-test. Comparisons between multiple groups were performed by one-way analysis of variance with Bonferroni post-hoc test where applicable. A p value of less than 0.05 was considered statistically significant.
RESULTS
PDGF increases glycolysis and smooth muscle cell proliferation
In the first series of experiments, we examined acute changes in glycolysis caused by PDGF. PDGF was added to VSMC and extracellular acidification rates (ECAR) were recorded for a period of approximately 1 h. As shown in Fig. 1A, PDGF addition to VSMC acutely increased ECAR by 2-fold over cells treated with vehicle control. Addition of oligomycin was then used to examine the glycolytic response to inhibition of mitochondrial ATP production. Interestingly, cells treated acutely with PDGF had an ~8-fold greater response to oligomycin compared with control cells, suggesting a greater ability to increase glycolytic flux under conditions of increased ATP demand.
Figure 1. PDGF increases glycolytic flux and the proliferation of vascular smooth muscle cells (VSMC).

(A) Acute effects of PDGF on extracellular acidification rates (ECAR): VSMC were treated acutely with vehicle (Veh, CTRL) or PDGF (10 ng/ml) at the indicated time (arrow) and ECAR was measured over time. Oligomycin (1 μg/ml) was injected at the indicated time (arrow) to determine the glycolytic response to inhibition of mitochondrial ATP synthesis. (B) Chronic effects of PDGF stimulation on ECAR: VSMC were seeded at densities of 20–60K cells per well followed by treatment with vehicle or PDGF (10 ng/ml) for 24 h. The PDGF or vehicle-containing medium was then removed and the ECAR was measured. The ECAR values from each group were then plotted against final lysate protein that was recovered following the XF assays. The vertical error bars represent the variation in ECAR for each group, and the horizontal error bars represent the variation in cellular protein for each group seeded at a specific cell density. The data were fit using linear regression analysis; CTRL, R2 = 0.97 and PDGF, R2 = 0.99. (C) VSMC were treated with vehicle (CTRL) or PDGF (10 ng/ml) in the absence or presence of 2-deoxy-D-glucose (2-DG, 20 mM). Cell number was measured by cell counting after 48 h. (D) After 24 h of the indicated treatment, acidification rates were recorded without and with oligomycin (1 μg/ml) treatment. Data are presented as means ± SEM; n=3–5 per group, *p<0.05 vs. CTRL, #p<0.05 vs. PDGF.
Next, the chronic effects of PDGF on glycolytic flux were examined. VSMC were seeded at five different cell densities (in the range of 20K–60K cells per well) and then treated with vehicle or PDGF for 48 h. The PDGF-containing medium was then removed, and the glycolytic rates were examined by extracellular flux (XF) analysis. Because PDGF is known to cause rapid proliferation of VSMC, the ECAR values were normalized to total protein in cell lysates recovered after the assay. Consistent with increased cell proliferation, PDGF induced a significant increase in total protein over time compared with control cells (Fig. 1B). Despite removal of the PDGF stimulus prior to the XF assay, cells treated with PDGF demonstrated elevated ECAR values, suggesting that metabolic reprogramming to a more glycolytic phenotype had occurred.
To examine cell proliferation induced by PDGF, cell proliferation was measured by cell counting after 48 h of PDGF treatment. Under these conditions, PDGF increased cell number by ~1.3 fold (Fig. 1C). An inhibitor of glycolysis, 2-deoxy-D-glucose (2-DG), was used to determine whether glycolysis was required for the hyperplastic response. As shown in Fig. 1C, 2-DG inhibited both basal and PDGF-stimulated cell hyperplasia. To confirm that the changes in cell number were due to loss of a proliferative response and not cytotoxicity, cell viability was assessed after 48 h and found to be unchanged (Supplementary Fig. 1). The lower cell proliferation occurring in the cells treated with 2-DG was associated with a lower rate of glycolysis and an inability to mount a glycolytic response upon loss of mitochondrial ATP synthesis (Fig. 1D).
Effect of PI3K/Akt signaling on PDGF-dependent VSMC proliferation
The PI3K/Akt signaling axis has been shown to be activated and required for PDGF-dependent VSMC proliferation [26]. As shown in Fig. 2A, PDGF increased PI3K-dependent Akt phosphorylation significantly compared to vehicle control; this effect was prevented by pre-treatment of VSMC with LY-294002 (LY, 10 μM) without changes in total Akt protein. We also examined the role of PI3K inhibition on VSMC proliferation and found that its activation is required for PDGF-dependent cell growth (Fig. 2B). The lower cell counts in the LY-treated cells were due to loss of a proliferative response and not cytotoxicity, as indicated by LDH viability assay (Fig. 2C).
Figure 2. Effect of inhibition of the PI3K pathway on PDGF-dependent Akt activation and proliferation.
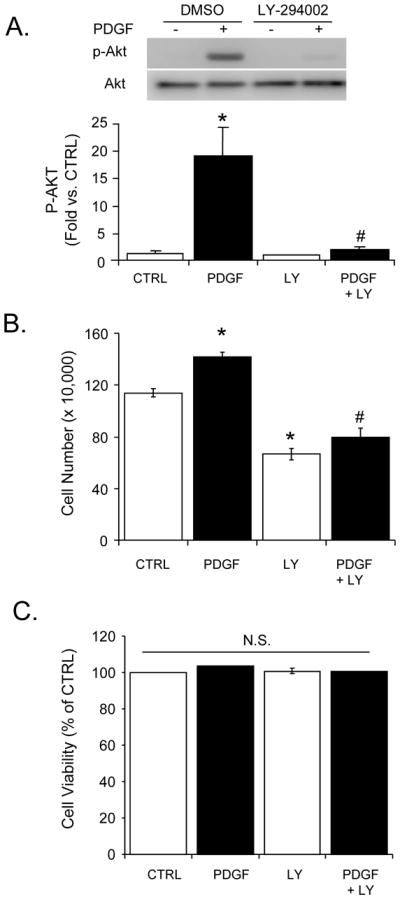
Akt phosphorylation, cell proliferation, and viability measurements of VSMC exposed to PDGF (10 ng/ml) in the absence or presence of LY-294002 (LY; 10 μM). (A) Phosphorylated (p-) and total Akt were examined by Western blotting after PDGF treatment for 10 min in the absence or presence of LY-294002. Top panel: Representative Western blots. Bottom panel: Group data of band intensities acquired by densitometry. The p-Akt values were normalized to their respective total Akt values and expressed as fold change vs. vehicle control (CTRL). (B) VSMC proliferation induced by PDGF after 48 h treatment in the absence or presence of LY was assessed by cell counting. (C) Cell viability of the indicated groups was assessed by LDH assay after 48 h of treatment. Data are presented as means ± SEM; n=3–5 per group, *p<0.05 vs. CTRL, #p<0.05 vs. PDGF.
Effects of PI3K and glycolysis on VSMC cell cycle proteins
PDGF-stimulated PI3K signaling and increased glycolytic activity have been shown to regulate cell cycle proteins that are responsible for growth [26]. We also examined the effect of PDGF on retinoblastoma protein (Rb) phosphorylation and cyclin D1 expression in the absence or presence of LY-294002 and 2-DG. As shown in Supplementary Fig. 2, PDGF induced Rb phosphorylation at Ser807/811 and increased cyclin D1 expression (~2.5 fold compared to control) significantly. In response to PI3K inhibition by LY-294002, cyclin D1 expression, but not PDGF-dependent Rb phosphorylation, was inhibited. Similar results were shown previously [27]. As expected, 2-DG inhibited Rb phosphorylation, but increased cyclin D1 expression as compared to control. The 2-DG-dependent induction of cyclin D1 was not induced further by PDGF treatment. The reasons for increased cyclin D1 expression with 2-DG are still unclear. Nevertheless, these results are in agreement with previously published reports suggesting that both LY-294002 and 2-DG can affect cell cycle proteins and inhibit VSMC proliferation [10, 28].
PDGF increases LDH expression in a PI3K-dependent manner
To determine if the effects of PDGF on glycolysis were accompanied by reported changes in the levels of glycolytic enzymes, VSMC were treated with PDGF for 24 h in the absence or presence of LY-294002 or 2-DG. LDH and GAPDH protein levels were then measured by Western blotting. As shown in Figure 3A, PDGF had no effect on GAPDH protein expression; this finding is contrary to previous reports which showed that PDGF increased GAPDH expression and activity in growth-arrested cells [6]. Under the complete serum culture conditions used throughout this study, PDGF increased LDH protein expression (Fig. 3B) and enzyme activity (Fig. 3C), and pre-treatment with LY-294002 and 2-DG prevented this response. These data suggest that the upregulation of LDH by PDGF is dependent on the PI3K pathway. The lack of upregulation in 2-DG-treated cells is still unclear but may be due to limited levels of ATP (see Supplementary Fig. 3).
Figure 3. Effect of PDGF on glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and lactate dehydrogenase (LDH).

VSMC were treated with PDGF (10 ng/ml) for 24 h in the absence or presence of LY-294002 (LY, 10 μM) or 2-deoxy-D-glucose (2-DG, 20 mM). Lysates from the cells were used to examine GAPDH and LDH expression by immunoblotting. (A and B) Representative Western blots of GAPDH, LDH and β-actin expression. Group data for each data set are shown below the respective immunoblot. (C) Intracellular LDH-specific activity was measured in cell lysates 24 h after the indicated treatment. The rates of NADH oxidation were normalized to protein and expressed as nanomoles of NADH oxidized per min per milligram protein. The data shown are presented as means ± SEM; n=3 per group. N.S. is used to express non-significant differences between groups. *p<0.05 vs. CTRL, #p<0.05 vs. PDGF.
Effect of PDGF on cellular ATP levels
Because actively proliferating cells are expected to have higher energetic requirements than quiescent cells, we examined the levels of ATP and ADP in cells treated without and with PDGF. As shown in Supplementary Fig, 3A, ATP levels were similar between control and PDGF-treated cells. Interestingly, 2-DG caused a significant decrease in ATP, presumably due to the fact that 2-DG becomes phosphorylated by hexokinase upon entering the cells, cannot be metabolized, and may inhibit other energy requiring processes [29]. In the presence of PDGF, 2-DG-treated cells had a similar level of ATP as that of 2-DG alone. Interestingly, LY-294002 treatment had no effect on ATP levels, whether in the presence or absence of PDGF treatment. Shown in Fig. 4B is the ATP to ADP ratio for each treatment. PDGF increased the ATP to ADP ratio, and this was prevented by 2-DG and LY-294002 treatment.
Figure 4. Inhibition of PI3K partially blocks PDGF-induced changes in glycolytic flux.

ECAR values of VSMC treated with vehicle (CTRL) or PDGF (10 ng/ml) in the absence or presence of LY-294002 (LY; 10 μM). (A) Basal and oligomycin-stimulated ECAR values from cells treated for 24 h with vehicle or PDGF in the absence or presence of LY. The rates were normalized to total protein to give mpH units/min/μg protein. (B) Baseline ECAR group data for the treatment groups in panel A. (C) Group data of the oligomycin-dependent ECAR values for treatment groups in panel A. (D) Effects of acute PDGF exposure on glycolytic flux: VSMC were exposed to vehicle (CTRL) or PDGF in the absence or presence of LY for 5 min and ECAR rates were measured. (E) Effects of acute PDGF exposure on glycolytic flux after oligomycin addition. After acute administration of PDGF, cells were exposed to oligomycin (1 μg/ml) and ECAR rates were measured. Data are presented as means ± SEM; n=3–5 per group, *p<0.05 vs. CTRL, #p<0.05 vs. PDGF.
Inhibition of PI3K decreases PDGF-induced glycolytic flux
We next examined how inhibition of PI3K affects glycolysis under basal and PDGF-stimulated conditions. VSMC were pre-treated without and with LY and exposed to PDGF for 24 h. As shown in Fig. 4A–C, LY alone mildly stimulated glycolysis; however, LY prevented the PDGF-dependent stimulation of glycolysis at 24 h as well as the stimulation of glycolysis after oligomycin addition. When cells were treated acutely with LY (1 h), the baseline levels of glycolysis were found to be no different from control cells (Fig. 4D, left panel). After baseline measurement of ECAR, PDGF (10 ng/ml) was added to cells and glycolysis was assessed. As shown in Fig. 4D (right panel), PDGF treatment enhanced glycolysis significantly in VSMC within 5 minutes, and this effect was partially prevented by PI3K inhibition. Similarly, oligomycin-stimulated glycolysis, measured 90 min after injection with PDGF or vehicle, was enhanced in the presence of PDGF and inhibited by LY (Fig. 4E).
PDGF increases mitochondrial respiration
Next, the effect of PDGF on mitochondrial oxygen consumption in the absence or presence of 2-DG was examined. VSMC were treated without or with 2-DG for 1 h, followed by treatment with PDGF for 24 h. Mitochondrial function was assessed using the mitochondrial function assay described in Supplementary Fig. 4. As shown in Fig. 5A and B, treatment with PDGF significantly increased the basal rate of oxygen consumption. The presence of 2-DG inhibited the basal stimulation of OCR by PDGF; however, 2-DG alone did not affect the rate of oxygen consumption compared to control cells. Furthermore, the maximal respiratory capacity induced by FCCP was ~2-fold higher in PDGF-treated cells compared with control cells, and 2-DG decreased the maximal respiratory capacity of cells treated with PDGF to levels comparable to control cells (Fig. 5A and C). The increase in maximal respiratory capacity by PDGF resulted in a substantial increase in the mitochondrial reserve capacity, and 2-DG abolished this mitochondrial bioenergetic reserve in PDGF-treated cells (Fig. 5D).
Figure 5. Effects of PDGF on mitochondrial respiration.

Oxygen consumption rates (OCR) in VSMC: (A) VSMC were pre-treated without or with 2-deoxy-D-glucose (2-DG, 20 mM) followed by exposure to vehicle (CTRL) or PDGF (10 ng/ml) for 24 h. Baseline OCR values were first recorded followed by sequential injection of oligomycin (1 μg/ml), FCCP (1 μM) and antimycin A (10 μM). The measurements were normalized to protein following the assay. (B–D) Group data for basal OCR (panel B), maximal respiratory capacity (panel C), and mitochondrial reserve capacity (panel D), respectively. Data are presented as means ± SEM; n=4–5 per group, *p<0.05 vs. CTRL, #p<0.05 vs. PDGF.
Because the PI3K/Akt pathway regulates glycolysis and therefore substrate supply to mitochondria, i.e., pyruvate [30], we examined the effect of LY-294002 on PDGF-induced changes in mitochondrial respiration (Fig. 6A). Similar to direct inhibition of glycolysis with 2-DG, PI3K blockade with LY-294002 abrogated the effect of PDGF on basal oxygen consumption (Fig. 6B), maximal respiratory capacity (Fig. 6C), and mitochondrial reserve capacity (Fig. 6D). Collectively, these results suggest that PI3K-mediated stimulation of glycolysis is required for PDGF to increase mitochondrial respiration and smooth muscle cell proliferation.
Figure 6. Effect of inhibition of the PI3K pathway on PDGF-dependent changes in mitochondrial respiration.

Oxygen consumption rates (OCR) in VSMC: (A) VSMC pre-treated without or with LY-294002 (LY, 10 μM) were exposed to vehicle (CTRL) or PDGF (10 ng/ml) for 24 h. The baseline OCR was then measured, followed by the sequential addition of oligomycin (1 μg/ml), FCCP (1 μM), and antimycin A (10 μM). The data were normalized to total protein from each well collected after the experiment. (B–D) Group data for basal OCR (panel B), maximal respiratory capacity (panel C), and mitochondrial reserve capacity (panel D), respectively. Data are presented as means ± SEM; n=4–5 per group, *p<0.05 vs. CTRL, #p<0.05 vs. PDGF.
Effect of glucose deprivation on glycolysis, mitochondrial respiration, and smooth muscle cell proliferation
Because 2-DG and PI3K inhibitors could have off-target effects, we further examined the role of glycolysis in this response by substituting L-glucose for D-glucose. Briefly, L-glucose, the non-metabolizable isomer of D-glucose, was added to the cell medium in place of D-glucose, and its effects on oxygen consumption, proliferation, and glycolysis were examined. Interestingly, L-glucose increased basal oxygen consumption in VSMCs (Fig. 7A and B); however, L-glucose did not support PDGF-induced increases in basal (Fig. 7B) or maximal respiratory capacities (Fig. 7C). The VSMC treated with PDGF in L-glucose-containing media showed a remarkably decreased mitochondrial reserve capacity (Fig. 7D). Furthermore, as shown in Fig. 7E, cell proliferation induced by PDGF in D-glucose-containing medium was abolished completely in L-glucose containing medium. In addition, PDGF was unable to increase ECAR in cells cultured in L-glucose-containing medium, and these cells were not responsive to oligomycin (Fig. 7F). It should be noted that in all experiments, pyruvate remained present in the medium at 1 mM concentrations. Taken together, these data suggest that glycolysis is required for enhancing mitochondrial reserve capacity to support PDGF-stimulated VSMC growth.
Figure 7. L-glucose prevents PDGF-induced increases in VSMC reserve capacity and proliferation.

Oxygen consumption rates in VSMC: (A) VSMC were treated with PDGF (10 ng/ml) for 24 h in the presence of D-glucose (1 g/L) or L-glucose (1 g/L). The baseline OCR was then measured, followed by the sequential addition of oligomycin (1 μg/ml), FCCP (1 μM), and antimycin A (10 μM). The data were normalized to total protein from each well collected after the experiment. (B–D) Group data for basal OCR (panel B), maximal respiratory capacity (panel C), and mitochondrial reserve capacity (panel D), respectively. (E) Proliferation of VSMC in D- or L-glucose-containing media after treatment with vehicle (CTRL) or PDGF (10 ng/ml) for 48 h. (F) Glycolytic flux in VSMC cultured in D- or L-glucose-containing media exposed to vehicle (CTRL) or PDGF for 24 h. Data are presented as means ± SEM; n=3–5 per group, *p<0.05 vs. CTRL, #p<0.05 vs. PDGF.
PDGF does not affect mitochondrial membrane potential, mitochondrial mass, or respiratory complex expression/activity
Because the effects of PDGF could be explained by increases in mitochondrial mass or respiratory complex activity, we examined the direct effects of PDGF on mitochondria. JC-1 staining was used to evaluate the membrane potential (ΔΨM) in VSMC treated with PDGF for 24 h. As shown in Supplementary Fig. 5, PDGF did not affect the mitochondrial ΔΨ. Citrate synthase and cytochrome c oxidase activities were also measured after PDGF treatment. As shown in Supplementary Fig. 5B and C, PDGF increased citrate synthase activity by ~10%; however, there was no significant change in cytochrome c oxidase activity, suggesting that the activity of these key mitochondrial proteins were not increased to an appreciable extent due to PDGF treatment. We further examined several mitochondrial complex proteins (Supplementary Fig. 5D) and observed no changes in their expression in response to PDGF.
To determine whether the effects of PDGF on mitochondrial oxygen consumption could derive from changes in pyruvate dehydrogenase (PDH) activation, protein phosphorylation of the E1-α subunit (Ser293) was examined after treatment with PDGF for 24 h in the absence or presence of LY-294002 or 2-DG. No changes in protein phosphorylation occurred (Supplementary Fig. 5E), suggesting similar levels of PDH activation in all treatments. Similarly, total PDH expression and the levels of the pyruvate dehydrogenase kinase-1 (PDK-1) enzyme remained unchanged. These results are interesting in light of published reports that suggest that PDH activity can be inhibited under certain conditions in proliferating cells [31].
Metabolism of L-lactate is sufficient to support PDGF-dependent increases in respiration and cell proliferation
We next examined the effect of L-lactate on PDGF-dependent mitochondrial function and VSMC proliferation. For this, L-lactate (18 mM) was substituted for D-glucose in the culture medium. After a 1 h acclimation to substrate conditions, the cells were exposed to PDGF for 24 h. As shown in Fig. 8A, PDGF increased basal OCR in the presence of both glucose and lactate. There were no differences in the basal respiratory state between lactate and glucose media alone. However, the maximal respiratory and reserve capacities induced by lactate were significantly higher than those observed with glucose (Fig. 8B and C). Interestingly, basal and PDGF-stimulated cell proliferation was lower in cells cultured in lactate compared with those cultured in glucose (Fig. 8D). Nevertheless, the stimulation of cell proliferation by PDGF was preserved in cells cultured in lactate.
Figure 8. L-lactate is sufficient to support increases in mitochondrial reserve capacity and cell proliferation by PDGF.

Oxygen consumption rates and cell proliferation of VSMC treated with PDGF (10 ng/ml) for 24 h in the absence or presence of L-lactate (18 mM; substituted for D-glucose). (A–C) Group data for basal OCR (panel A), maximal respiratory capacity (panel B), and mitochondrial reserve capacity (panel C), respectively, when cells were treated with D-glucose or L-lactate. Data are presented as means ± SEM; n=3–5 per group, *p<0.05 vs. D-gluc CTRL, #p<0.05 vs. D-gluc + PDGF, $p<0.05 vs. L-lac CTRL. (D) Cell proliferation was assessed by cell counting after 48 h of the indicated treatment. (E) Reserve capacity in VSMC treated without or with PDGF (10 ng/ml) for 24 h in the presence of L-lactate (18 mM); oxamate (100 mM) was used to inhibit LDH in the indicated groups. All OCR data (panels A, B, C, and E) were normalized to total protein from each well collected after the experiment. (F) Cell proliferation was assessed by cell counting after 48 h of the indicated treatment. Data are presented as means ± SEM; n=3–5 per group, *p<0.05 vs. L-lac CTRL, #p<0.05 vs. L-lac + PDGF.
The observation that PDGF increased LDH protein and activity (Fig. 3) and that lactate was sufficient to support respiration and PDGF-dependent proliferation suggested that LDH itself may be one of the key factors regulating VSMC proliferation. To examine this, we pre-treated VSMC with sodium oxamate (an inhibitor of LDH) in lactate-containing media 1 h prior to PDGF treatment and examined mitochondrial respiration after 24 h. It should be noted that oxamate, in other studies using the XF24 [16], inhibited extracellular acidification by ~80%, providing further confirmation that our acidification measurements were indeed associated with lactate production. Inhibition of LDH with oxamate resulted in loss of the mitochondrial reserve capacity in PDGF-treated cells (Fig. 8E). LDH inhibition was also associated with loss of basal and PDGF-stimulated proliferation, without affecting cell viability (Fig. 8F and data not shown).
DISCUSSION
Platelet-derived growth factor stimulates cell migration and proliferation and has been studied extensively over the past 20 years [3, 32–35]. Nevertheless, the mechanisms by which PDGF promotes proliferation in vascular disease have not been fully defined. In the present study, we examined the roles of glycolysis and mitochondrial respiration in PDGF-induced VSMC proliferation using extracellular flux analysis. We show for the first time that PDGF modulates LDH activity and mitochondrial oxygen consumption, and that this regulation appears to be due, in part, to activation of the PI3K pathway. Consistent with published studies [10], glycolysis was found to be required for PDGF-induced proliferation. Importantly, data presented here also reveal that mitochondrial respiration is enhanced in response to PDGF and that an enhanced mitochondrial reserve capacity is a feature common to the hyperplastic VSMC phenotype.
Glucose metabolism has been implicated to play an important role in VSMC proliferation. Interestingly, in VSMC, it was shown that 90% of the glucose transported into the cell is converted to lactate [36] and that glucose is the sole source for lactic acid production [8]. We exploited this characteristic of VSMC in this study by using extracellular flux technology to measure glycolytic flux. Under both acute and chronic treatment conditions, PDGF was found to increase extracellular acidification, suggesting that glycolytic flux was increased; this response was inhibited in the presence of 2-DG, a non-metabolizable glucose analog (Fig. 1). In addition, glycolytic flux was also inhibited in cells cultured in L-glucose instead of D-glucose (Fig. 7F), adding validity to our glycolytic flux measurements and the conclusion that glycolytic flux is increased by PDGF. Using oligomycin, an inhibitor of mitochondrial ATP synthase, we also examined the glycolytic response to inhibition of mitochondrial ATP synthesis. Acute treatment of VSMC with PDGF resulted in a several-fold increase in the oligomycin-stimulated rate of extracellular acidification, indicating that the cells had a substantial glycolytic reserve that could be called upon to meet the energy demands of cell division. Chronic treatment of cells with PDGF resulted in higher basal levels of glycolytic flux; yet these cells did not demonstrate as large of a glycolytic reserve as cells treated acutely with PDGF.
Previous studies have also shown that control of glycolysis occurs at different sites, demonstrating multisite metabolic modulation [5]. We found that PDGF stimulation resulted in a significant upregulation of LDH that led to a comparable increase in enzyme activity (Fig. 3). This finding complements gene expression studies showing that the LDH enzyme is increased in rat aortic VSMC with PDGF treatment [39]. Hence, our finding suggests that the lactate dehydrogenase enzyme may be a critical point of modulation utilized by highly proliferating VSMC. However, these results differ from those of Ranganna et al. [6], where glyceraldehyde-3-phosphate dehydrogenase was found to be upregulated after PDGF treatment; we did not observe a comparable increase in GAPDH in the course of our study. The reasons for this disparity are unclear, but may relate to the culture conditions used during PDGF treatment. Diverging from the typical serum deprivation and growth arrest protocol, we incubated cells in PDGF under complete serum conditions. The reason for using this culture condition relates back to the original purpose of this study, which was to measure the changes in metabolism caused by PDGF treatment and to determine their relationship with VSMC proliferation. Under low serum conditions (0.1% FBS), basal mitochondrial function and glycolysis were below the level of detection under these conditions. One interpretation of our findings is that the enhanced expression of LDH with PDGF stimulation results in augmentation of the NAD+-dependent conversion of lactate to pyruvate, an activity generally associated with the H-type (also known as the A-type) isoform of LDH [41], which could increase substrate delivery to mitochondria.
A novel finding of our study is that mitochondrial respiration is increased with PDGF treatment. To the best of our knowledge, this is the first report demonstrating that PDGF modulates mitochondrial oxygen consumption. However, previous reports have shown that mitochondria could be directly affected by PDGF signaling. For example, PDGF signaling was shown to be responsible for the phosphorylation of the ATP synthase delta subunit, but the significance of that finding remains unclear [37, 38]. Interestingly, in our study, the treatments that inhibited PDGF-dependent changes in glycolysis (i.e., 2-DG, PI3K inhibitors, L-glucose, and oxamate) were sufficient to prevent both proliferation and the augmentation of mitochondrial oxygen consumption. In particular, treatment with PDGF resulted in a large increase in the mitochondrial reserve capacity. The possible explanations for this increase in mitochondrial spare respiratory capacity include: (1) that PDGF increased mitochondrial mass, (2) that PDGF increased the activity of enzymes critical to energy transduction, or (3) that PDGF increased respiratory substrates. To test the idea that PDGF could augment mitochondrial mass, MitoTracker Green and confocal microscopy were used to estimate mitochondrial mass (data not shown). No differences were apparent between the treatment groups. Moreover, key mitochondrial proteins showed no change in activity or expression after PDGF treatment (Supplementary Fig. 5). We also examined pyruvate dehydrogenase (PDH), an enzyme through which the bulk of substrates for ATP generation is produced by the oxidation of pyruvate in the tricarboxylic acid cycle. PDH demonstrated no significant changes in its expression or phosphorylation status in response to PDGF (Supplementary Fig. 5E). Similarly, the expression of other PDH subunits (data not shown) as well as PDK-1 was found to remain constant with PDGF treatment. Thus, these findings partially dissociate our results from a Warburg “like” effect, where a combination of increased PDH phosphorylation and PDK protein expression inhibits mitochondrial respiratory function. Therefore, our results suggest an alternate mechanism of action that may couple glycolytic and mitochondrial metabolism in the presence of PDGF.
It appears, then, that metabolic modulation of glycolysis through enhanced LDH may be key to delivering substrate to mitochondria—the hypothesis being that this increases the mitochondrial reserve capacity, which is critical for PDGF-dependent growth. This was examined further by omitting D-glucose from the culture medium, while culturing the cells in excess L-lactate during treatment. Under these conditions, the cells retained their ability to proliferate when stimulated with PDGF and demonstrated an enhanced mitochondrial reserve capacity (Fig. 8). Interestingly, cells cultured in lactate had a higher maximal and spare respiratory capacity even in the absence of PDGF stimulation, yet maintained basal rates of oxygen consumption similar to that of cells cultured in D-glucose. This may be due to the fact that enzymatic conversion from lactate to pyruvate lacks the multifaceted allosteric control compared to glycolysis, which would involve several points of control, including hexokinase, PFK, and pyruvate kinase [5]. Control at LDH would then allow the cell to easily obtain pyruvate in one step rather than having to oxidize glucose through several steps. That oxamate inhibited both the proliferative response to PDGF and the mitochondrial metabolic response further supports the concept that LDH may be key to a hyperplastic VSMC phenotype. Therefore, we propose a role for extracellular lactate as a ‘reservoir’ source for mitochondrial respiration in VSMC in response to PDGF. We speculate that LDH may play an important role in lactate utilization to support oxidative phosphorylation through the conversion of lactate into pyruvate [41–43]. Indeed, evidence in the literature supports a role for lactate uptake and oxidation in muscle cells through the action of a family of monocarboxylate transport proteins (MCTs) which can be coupled to LDH activity [42, 44–46].
What remains to be examined in future studies is how this finely tuned and integrated metabolic response is controlled. It is well-known that Akt signaling stimulated upon PI3K activation is required for proliferation in both cancer and smooth muscle cells [12, 47]. Our findings suggest that PI3K/Akt signaling is required for all steps leading to the proliferative VSMC phenotype, including the upregulation of LDH, (Fig. 3), the increase in glycolytic flux (Fig. 4), and the increase in mitochondrial oxygen consumption and reserve capacity (Fig. 6) occurring upon PDGF stimulation. Moreover, we observed that inhibition of PI3K with LY-294002 prevented cyclin D1 up-regulation by PDGF (Supplementary Fig. 2), which in itself could inhibit the cell cycle and suggests regulation beyond glycolysis. Hence, disentangling the relative contribution of PI3K-mediated changes in energy metabolism from other epiphenomena regulating cell growth will be challenging. Nevertheless, these studies indicate that PDGF signaling coordinates glycolytic flux, cell cycle protein activity, and mitochondrial metabolism to maximize cell proliferation. It appears that PI3K/Akt signaling and LDH are integral to PDGF-induced metabolic changes and the proliferative response. Targeting these bioenergetic responses could be a useful strategy for preventing or ameliorating the abnormal proliferation of smooth muscle cells in vascular disease.
Supplementary Material
Acknowledgments
The authors would like to acknowledge Michelle S. Johnson and Karina C. Ricart for technical support.
GRANTS
This work was supported by National Institutes of Health (NIH) grants ES10167 (to V.D.U.), and NIH training grants T32 HL07457 (B.G.H.) and T32 HL007918 (B.P.D., and J.P.).
Abbreviations
- VSMC
vascular smooth muscle cells
- PDGF
platelet-derived growth factor
- PI3K
phosphoinositide 3-kinase
- NADH
nicotinamide adenine dinucleotide, reduced
- 2-DG
2-deoxy-D-glucose
- DTNB
5,5′-dithiobis-(2-nitrobenzoic) acid
- LY
LY-294002
- GAPDH
glyceraldehyde-phosphate dehydrogenase
- PDH
pyruvate dehydrogenase
- PDK-1
pyruvate dehydrogenase kinase-1
- LDH
lactate dehydrogenase
- FBS
fetal bovine serum
- PBS
phosphate buffered saline
- OCR
oxygen consumption rate
- ECAR
extracellular acidification rate
- FCCP
p-trifluoromethoxy carbonyl cyanide phenyl hydrazone
- EDTA
ethylenediamine tetraacetic acid
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- DTPA
Diethylene triamine pentaacetic acid
- SDS
sodium dodecyl sulfate
- JC-1
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide
- TNB
2-nitro-5-thiobenzoic acid
- TCA
tricarboxylic acid cycle
- TBAHS
tetrabutylammonium hydrogen sulfate
- ATP
adenosine 5-triphosphate
- ADP
adenosine 5-diphosphate
Footnotes
DISCLOSURES
Authors have no additional affiliations to disclose.
References
- 1.Grotendorst GR, Chang T, Seppa HE, Kleinman HK, Martin GR. Platelet-derived growth factor is a chemoattractant for vascular smooth muscle cells. J Cell Physiol. 1982;113:261–266. doi: 10.1002/jcp.1041130213. [DOI] [PubMed] [Google Scholar]
- 2.Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:812–819. doi: 10.1161/ATVBAHA.107.159327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williams LT, Tremble P, Antoniades HN. Platelet-derived growth factor binds specifically to receptors on vascular smooth muscle cells and the binding becomes nondissociable. Proc Natl Acad Sci USA. 1982;79:5867–5870. doi: 10.1073/pnas.79.19.5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marmur JD, Poon M, Rossikhina M, Taubman MB. Induction of PDGF-responsive genes in vascular smooth muscle. Implications for the early response to vessel injury. Circulation. 1992;86:III53–60. [PubMed] [Google Scholar]
- 5.Werle M, Kreuzer J, Hofele J, Elsasser A, Ackermann C, Katus HA, Vogt AM. Metabolic control analysis of the Warburg-effect in proliferating vascular smooth muscle cells. J Biomed Sci. 2005;12:827–834. doi: 10.1007/s11373-005-9010-5. [DOI] [PubMed] [Google Scholar]
- 6.Ranganna K, Yatsu FM. Inhibition of platelet-derived growth factor BB-induced expression of glyceraldehyde-3-phosphate dehydrogenase by sodium butyrate in rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1997;17:3420–3427. doi: 10.1161/01.atv.17.12.3420. [DOI] [PubMed] [Google Scholar]
- 7.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lynch RM, Paul RJ. Compartmentation of glycolytic and glycogenolytic metabolism in vascular smooth muscle. Science. 1983;222:1344–1346. doi: 10.1126/science.6658455. [DOI] [PubMed] [Google Scholar]
- 9.Hall JL, Gibbons GH, Chatham JC. IGF-I promotes a shift in metabolic flux in vascular smooth muscle cells. Am J Physiol Endocrinol Metab. 2002;283:E465–471. doi: 10.1152/ajpendo.00072.2002. [DOI] [PubMed] [Google Scholar]
- 10.Nef HM, Mollmann H, Joseph A, Troidl C, Voss S, Vogt A, Weber M, Hamm CW, Elsasser A. Effects of 2-deoxy-D-glucose on proliferation of vascular smooth muscle cells and endothelial cells. J Int Med Res. 2008;36:986–991. doi: 10.1177/147323000803600515. [DOI] [PubMed] [Google Scholar]
- 11.Zhong D, Xiong L, Liu T, Liu X, Liu X, Chen J, Sun SY, Khuri FR, Zong Y, Zhou Q, Zhou W. The glycolytic inhibitor 2-deoxyglucose activates multiple prosurvival pathways through IGF1R. J Biol Chem. 2009;284:23225–23233. doi: 10.1074/jbc.M109.005280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- 14.Merida I, Avila-Flores A. Tumor metabolism: new opportunities for cancer therapy. Clin Transl Oncol. 2006;8:711–716. doi: 10.1007/s12094-006-0117-6. [DOI] [PubMed] [Google Scholar]
- 15.Pandey NR, Benkirane K, Amiri F, Schiffrin EL. Effects of PPAR-gamma knock-down and hyperglycemia on insulin signaling in vascular smooth muscle cells from hypertensive rats. J Cardiovasc Pharmacol. 2007;49:346–354. doi: 10.1097/FJC.0b013e31804654d7. [DOI] [PubMed] [Google Scholar]
- 16.Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:C125–136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- 17.Hill BG, Dranka BP, Zou L, Chatham JC, Darley-Usmar VM. Importance of the bioenergetic reserve capacity in response to cardiomyocyte stress induced by 4-hydroxynonenal. Biochem J. 2009;424:99–107. doi: 10.1042/BJ20090934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hill BG, Higdon AN, Dranka BP, Darley-Usmar VM. Regulation of vascular smooth muscle cell bioenergetic function by protein glutathiolation. Biochim Biophys Acta. 1797:285–295. doi: 10.1016/j.bbabio.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dranka BP, Hill BG, Darley-Usmar VM. Mitochondrial reserve capacity in endothelial cells: The impact of nitric oxide and reactive oxygen species. Free Radic Biol Med. 48:905–914. doi: 10.1016/j.freeradbiomed.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yadava N, Nicholls DG. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J Neurosci. 2007;27:7310–7317. doi: 10.1523/JNEUROSCI.0212-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 22.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 23.Darley-Usmar V, Capaldi RA, Takamiya S, Millett F, Wilson MT, Malatesta F, Sarti P. Reconstitution and molecular analysis of the respiratory chain. In: Darley-Usmar VM, Rickwood D, Wilson MT, editors. Mitochondria: a practical approach. IRL Press; Washington DC: 1987. pp. 113–152. [Google Scholar]
- 24.Coolen EJ, Arts IC, Swennen EL, Bast A, Stuart MA, Dagnelie PC. Simultaneous determination of adenosine triphosphate and its metabolites in human whole blood by RP-HPLC and UV-detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;864:43–51. doi: 10.1016/j.jchromb.2008.01.033. [DOI] [PubMed] [Google Scholar]
- 25.Zur Nedden S, Eason R, Doney AS, Frenguelli BG. An ion-pair reversed-phase HPLC method for determination of fresh tissue adenine nucleotides avoiding freeze-thaw degradation of ATP. Anal Biochem. 2009;388:108–114. doi: 10.1016/j.ab.2009.02.017. [DOI] [PubMed] [Google Scholar]
- 26.Goncharova EA, Ammit AJ, Irani C, Carroll RG, Eszterhas AJ, Panettieri RA, Krymskaya VP. PI3K is required for proliferation and migration of human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2002;283:L354–363. doi: 10.1152/ajplung.00010.2002. [DOI] [PubMed] [Google Scholar]
- 27.Page K, Li J, Wang Y, Kartha S, Pestell RG, Hershenson MB. Regulation of cyclin D(1) expression and DNA synthesis by phosphatidylinositol 3-kinase in airway smooth muscle cells. Am J Respir Cell Mol Biol. 2000;23:436–443. doi: 10.1165/ajrcmb.23.4.3953. [DOI] [PubMed] [Google Scholar]
- 28.Isenovic ER, Kedees MH, Tepavcevic S, Milosavljevic T, Koricanac G, Trpkovic A, Marche P. Role of PI3K/AKT, cPLA2 and ERK1/2 signaling pathways in insulin regulation of vascular smooth muscle cells proliferation. Cardiovasc Hematol Disord Drug Targets. 2009;9:172–180. doi: 10.2174/187152909789007034. [DOI] [PubMed] [Google Scholar]
- 29.Ralser M, Wamelink MM, Struys EA, Joppich C, Krobitsch S, Jakobs C, Lehrach H. A catabolic block does not sufficiently explain how 2-deoxy-D-glucose inhibits cell growth. Proc Natl Acad Sci USA. 2008;105:17807–17811. doi: 10.1073/pnas.0803090105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stiles BL. PI-3-K and AKT: Onto the mitochondria. Adv Drug Deliv Rev. 2009;61:1276–1282. doi: 10.1016/j.addr.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Q, Lam PY, Han D, Cadenas E. c-Jun N-terminal kinase regulates mitochondrial bioenergetics by modulating pyruvate dehydrogenase activity in primary cortical neurons. J Neurochem. 2008;104:325–335. doi: 10.1111/j.1471-4159.2007.04957.x. [DOI] [PubMed] [Google Scholar]
- 32.Yang XP, Pei ZH, Ren J. Making Up or Breaking Up - The Tortuous Role of Platelet-Derived Growth Factor (PDGF) In Vascular Aging. Clin Exp Pharmacol Physiol. 2009;36:739–47. doi: 10.1111/j.1440-1681.2009.05182.x. [DOI] [PubMed] [Google Scholar]
- 33.Wang Z, Kong D, Li Y, Sarkar FH. PDGF-D signaling: a novel target in cancer therapy. Curr Drug Targets. 2009;10:38–41. doi: 10.2174/138945009787122914. [DOI] [PubMed] [Google Scholar]
- 34.Millette E, Rauch BH, Kenagy RD, Daum G, Clowes AW. Platelet-derived growth factor-BB transactivates the fibroblast growth factor receptor to induce proliferation in human smooth muscle cells. Trends Cardiovasc Med. 2006;16:25–28. doi: 10.1016/j.tcm.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 35.Barst RJ. PDGF signaling in pulmonary arterial hypertension. J Clin Invest. 2005;115:2691–2694. doi: 10.1172/JCI26593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Michelakis ED, Weir EK. The metabolic basis of vascular oxygen sensing: diversity, compartmentalization, and lessons from cancer. Am J Physiol Heart Circ Physiol. 2008;295:H928–H930. doi: 10.1152/ajpheart.00697.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ko YH, Pan W, Inoue C, Pedersen PL. Signal transduction to mitochondrial ATP synthase: evidence that PDGF-dependent phosphorylation of the delta-subunit occurs in several cell lines, involves tyrosine, and is modulated by lysophosphatidic acid. Mitochondrion. 2002;1:339–348. doi: 10.1016/s1567-7249(01)00036-8. [DOI] [PubMed] [Google Scholar]
- 38.Zhang FX, Pan W, Hutchins JB. Phosphorylation of F1F0 ATPase delta-subunit is regulated by platelet-derived growth factor in mouse cortical neurons in vitro. J Neurochem. 1995;65:2812–2815. doi: 10.1046/j.1471-4159.1995.65062812.x. [DOI] [PubMed] [Google Scholar]
- 39.Green RS, Lieb ME, Weintraub AS, Gacheru SN, Rosenfield CL, Shah S, Kagan HM, Taubman MB. Identification of lysyl oxidase and other platelet-derived growth factor-inducible genes in vascular smooth muscle cells by differential screening. Lab Invest. 1995;73:476–482. [PubMed] [Google Scholar]
- 40.Janero DR, Hreniuk D, Sharif HM. Hydroperoxide-induced oxidative stress impairs heart muscle cell carbohydrate metabolism. Am J Physiol. 1994;266:C179–188. doi: 10.1152/ajpcell.1994.266.1.C179. [DOI] [PubMed] [Google Scholar]
- 41.Brooks GA, Dubouchaud H, Brown M, Sicurello JP, Butz CE. Role of mitochondrial lactate dehydrogenase and lactate oxidation in the intracellular lactate shuttle. Proc Natl Acad Sci USA. 1999;96:1129–1134. doi: 10.1073/pnas.96.3.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hashimoto T, Brooks GA. Mitochondrial lactate oxidation complex and an adaptive role for lactate production. Med Sci Sports Exerc. 2008;40:486–494. doi: 10.1249/MSS.0b013e31815fcb04. [DOI] [PubMed] [Google Scholar]
- 43.Hashimoto T, Hussien R, Cho HS, Kaufer D, Brooks GA. Evidence for the mitochondrial lactate oxidation complex in rat neurons: demonstration of an essential component of brain lactate shuttles. PLoS One. 2008;3:e2915. doi: 10.1371/journal.pone.0002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hashimoto T, Hussien R, Brooks GA. Colocalization of MCT1, CD147, and LDH in mitochondrial inner membrane of L6 muscle cells: evidence of a mitochondrial lactate oxidation complex. Am J Physiol Endocrinol Metab. 2006;290:E1237–1244. doi: 10.1152/ajpendo.00594.2005. [DOI] [PubMed] [Google Scholar]
- 45.Brooks GA. Lactate shuttle -- between but not within cells? J Physiol. 2002;541:333–334. doi: 10.1113/jphysiol.2002.023705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brooks GA. Lactate shuttles in nature. Biochem Soc Trans. 2002;30:258–264. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 47.Stabile E, Zhou YF, Saji M, Castagna M, Shou M, Kinnaird TD, Baffour R, Ringel MD, Epstein SE, Fuchs S. Akt controls vascular smooth muscle cell proliferation in vitro and in vivo by delaying G1/S exit. Circ Res. 2003;93:1059–1065. doi: 10.1161/01.RES.0000105086.31909.1B. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.