

Abstract
This review highlights a symposium on stress risk factors and stress susceptibility, presented at the Neurobiology of Stress workshop in Boulder, Colorado, June 2010. This symposium addressed factors linking stress plasticity and reactivity to stress pathology in animal models and in humans. Dr. Jason Radley discussed studies demonstrating prefrontal cortical neuroplasticity and prefrontal control of hypothalamo-pituitary-adrenocortical axis function in rat, highlighting emerging evidence for a critical role of this region in normal and pathological stress integration. Dr. Mohamed Kabbaj summarized his studies of possible epigenetic mechanisms underlying behavioral differences in rat populations bred for differential stress reactivity. Dr. Lauren Jacobson described studies using a mouse model to explore the diverse actions of antidepressant action in brain, suggesting mechanisms whereby antidepressants may be differentially effective in treating specific depression endophenotypes. Dr. Rachel Yehuda discussed the role of glucocorticoids in post-traumatic stress disorder (PTSD), indicating that low cortisol may be a trait that predisposes the individual to development of the disorder. Furthermore, she presented evidence indicating that traumatic events can have transgenerational impact on cortisol reactivity and development of PTSD symptoms. Together, the symposium highlighted emerging themes regarding the role of brain reorganization, individual differences and epigenetics in determining stress plasticity and pathology.
Keywords: antidepressants, chromatin modification, depression, glucocorticoids, hippocampus, individual differences, prefrontal cortex, PTSD
INTRODUCTION
Adaptation in the face of physical or psychological stress is a major priority for all biological systems. As a result, natural selection has favored an efficient and highly conserved set of interlocking systems that maintain physiologic integrity even in the most demanding of circumstances. The adaptive strategy recruits systems that act as rapid and delayed effectors, and strategies to limit or avoid stressor exposure.
Physiological stress responses are initiated rapidly and are designed to optimize mobilization of resources and restoration of homeostasis (Munck et al., 1984). Activation of the sympathoadrenomedullary system occurs within seconds of perceived stress. Sympathetic nervous system (SNS) excitation promotes norepinephrine-induced changes in numerous bodily systems, including increases in heart rate and blood pressure, and causes adrenomedullary epinephrine release, promoting hepatic glycogenolysis. The hypothalamo-pituitary-adrenocortical (HPA) axis response is initiated on a slightly longer time scale (due to its neuroendocrine components). The HPA axis introduces glucocorticoid hormones into the circulation to provide further redistribution of energy resources (e.g., hepatic gluconeogenesis), while also serving to limit the duration and impact of the initial stress response. Both of these ‘emergency’ responses are of immediate benefit, but are potentially damaging if either prolonged or shortened in time, necessitating feedback mechanisms aimed at limiting duration of neural responses and hormone secretion.
Stressors elicit cognitive responses that are important in the overall interpretation of stimuli. Cognitive interpretations are likely based on innate response predispositions, prior experience with stimulus arrays that constitute the ‘stressor’, and the intensity of the stressor itself, and will affect the nature and intensity of the physiological response. Cognitive and physiological reactions to stressors are highly influenced by genetics, early-life environment and trauma, and contribute to individual differences in stress reactivity and in the case of PTSD, stress pathology.
The majority of the stress responses involve mobilization of resources to adapt to the adverse conditions. Generally, response patterns are appropriately regulated to ‘fit’ the prevailing threat. For example, responses to acute stress generally ebb soon after the stressor is stopped, generally via effective neural or hormonal feedback mechanisms (e.g., glucocorticoid negative feedback)(c.f., (Keller-Wood and Dallman, 1984)). An exception to this pattern can be seen in the case of PTSD, in which an acute event that elicits the need for an intense reaction (e.g., rape, combat, life-threating accident) leaves lingering physiological response patterns though the immediate threat associated with the event has passed. However, prolonged stress exposure recruits long-term adaptive mechanisms to cope with increased life adversity, many of which involve neuroplastic adaptations (e.g., changes in gene regulation, altered neuronal structure) c.f., (Cook and Wellman, 2004, McEwen, 2001). While many plastic mechanisms are adaptive, and promote processes such as HPA axis habituation and reduced susceptibility to glutamate neurotoxicity (McEwen, 2001), increased pressure on the relevant neuronal systems (i.e., ‘allostatic load’ (McEwen, 1998, McEwen and Stellar, 1993)) can render the organism more sensitive or susceptible to new challenges. Thus, neuroplastic events triggered by stress may render stress systems hyperresponsive or indeed, hyporesponsive, placing the organism at risk for stress-related diseases such as depression and PTSD.
Responses to stress are ultimately based on the predispositions of the organism. The magnitude, duration and pathological consequences of physiological ‘stress responses’ differ markedly between individuals, based on stress history, genetic background, and early-life programming. Thus, resilience and susceptibility to stress are dictated by a variety of factors that ultimately determine whether neuroplastic adaptations can effectively promote coping or lead to loss of appropriate stress control and perhaps pathology.
This symposium was intended to provide understanding of factors that link stress plasticity and reactivity to stress pathology in animal models and in humans. Dr. Radley’s work explores neuroplastic pathways in the forebrain that are targeted by stressors and play a critical role in limiting the net impact of stress on physiological responses (as well as behavior). Dr. Kabbaj describes studies of the neural mechanisms that predispose individuals to stress-reactive or non stress-reactive phenotypes, focusing on differences in epigenetic programming of stress-related gene expression in brain. Dr. Jacobson addresses the key problem of HPA glucocorticoid hyper- vs. hyposecretion within the context of depressive disorders, and explains how drugs with very different mechanisms of action may be selectively beneficial for the glucocorticoid ‘endophenotypes’. Finally, Dr. Yehuda provides evidence demonstrating that PTSD pathology may be linked to an inadequate glucocorticoid response to an initial traumatic stressor which then does not enable the person to engage mechanisms associated with physiological homeostasis. Interestingly, recent evidence suggests that this inadequate glucocorticoid response may have epigenetic origins.
NEUROPLASTICITY AND LIMBIC CIRCUITRY: ROLE IN STRESS INTEGRATION JASON RADLEY, UNIVERSITY OF IOWA
Neural regulation of adaptive responses to stress requires complex integrative processes across activational and modulatory networks in the brain. Cell groups in the limbic forebrain, notably aspects of the amygdala, hippocampal formation (HF), and medial prefrontal cortex (mPFC), are implicated not only in cognitive/affective responses to stress, but also in endocrine adjustments, by modulating activity of the HPA axis (Cullinan et al., 1995, Dayas et al., 2001, Herman and Cullinan, 1997, Li and Sawchenko, 1998). Limbic forebrain cell groups are also targets of glucocorticoids (GCs), and exhibit the capacity to limit the HPA response following stress exposure via GC receptor-mediated negative feedback (Diorio et al., 1993, Kovacs and Makara, 1988, Sapolsky et al., 1984). Thus, alterations in HPA function that have been documented to result from exposure to chronic stress (Herman et al., 1995, Ottenweller et al., 1989, Willner, 1997,) and are likely to involve the failure of the restraining influences exerted by this modulatory network. The prevalence of HPA axis hyperactivity in depressive illness (see Jacobson, this article) raises the prospect that clarifying the mechanisms of stress neuroendocrine systems in animal studies may help to understand the pathophysiology of this disorder (Carroll et al., 1976, Sapolsky, 1996, Sheline et al., 1996). To this end, our work has focused on the relationship between structural and neuroendocrine plasticity triggered by chronic stress. We have attacked this problem on two fronts: the first is understanding the nature of chronic stress’ effects on structural plasticity in limbic cortical regions (most notably in mPFC), and the second is the elucidation of the neural pathways that provide for their HPA-inhibitory influences.
Stress Plasticity in Limbic Cortical Circuitry
A substantial body of research over the past several decades has implicated the hippocampus as a central player in chronic stress-induced plasticity and HPA axis dysregulation (Conrad, 2008, Jacobson and Sapolsky, 1991, McEwen, 2001). These studies have generally found that the hippocampus plays an important role in limiting the HPA axis response following acute stress, at least in part via GC negative feedback, whereas chronic stress and GCs impair hippocampal structure and function. The histopathological features in animal models of chronic stress parallel neuroimaging studies showing gray matter volume and functional impairments in the hippocampus of depressed patients (Sheline et al., 2003, Sheline et al., 1996, Watanabe et al., 1992)(although not observed in all studies: see (Fink, 2011) for discussion of this controversy). Sustained elevations in GCs have even been implicated in neurodegenerative mechanisms by rendering neurons more susceptible to excitotoxic insults (Conrad, 2008, Conrad et al., 2007). Thus, while stress-induced neuroplasticity in the hippocampus has remained as the prime suspect in the disruption and/or maintenance of restraining influences on the HPA axis, direct support for this idea has not been forthcoming. Furthermore, evidence in recent years has emerged that complicates the model placing the hippocampus at the epicenter of stress pathology. Multiple limbic forebrain regions, e.g., septum, mPFC, posterior paraventricular nucleus of the thalamus (PVTp)(Diorio et al., 1993, Feldman and Conforti, 1980, Jaferi and Bhatnagar, 2006, Kovacs and Makara, 1988), are also target sites for GC negative feedback influences over the HPA axis, suggesting that restraining influences over the HPA axis may be exerted more broadly via multiple cell groups, bringing these regions into play as targets for chronic stress-induced plasticity. Moreover, neuroimaging studies in depression have also shown comparable gray matter volume deficits in mPFC as previously shown in the hippocampus (Drevets et al., 1997), which led us to examine the relationship between chronic stress and structural plasticity in mPFC.
These studies entailed performing intracellular injections of fluorescent dye (Lucifer Yellow) in pyramidal neurons in mPFC (notably within the anterior cingulate and prelimbic areas; ACd and PL, respectively) after 3 weeks of daily restraint stress, followed by analysis of digitally reconstructed neurons using high-resolution confocal laser-scanning microscopy. Chronic stress reduced total apical dendritic length by 20%, and apical dendritic spine densities by 16% (Radley et al., 2005, Radley et al., 2006b, Radley et al., 2004). These effects were most pronounced in the outermost aspect of layer I, in the distal portion of the dendritic tree (Fig. 1). Given that spines comprise the vast majority of sites of postsynaptic contacts for excitatory synapses in the mammalian prefrontal cortex, if the effects of stress on dendritic length and spine density are taken together, as much as one-third of the entire population of excitatory synaptic inputs into mPFC may be compromised following chronic stress (Radley et al., 2006b). Next, we utilized a Rayburst-based automated approach (NeuronStudio; (Rodriguez et al., 2003, Wearne et al., 2005)) to analyze the effects of chronic stress on spine morphometric features, which revealed chronic stress-induced decreases in mean apical dendritic spine volume and surface area throughout the dendritic tree in mPFC pyramidal neurons (Radley et al., 2008) (Fig. 1). This analysis revealed an overall shift in the population of spines, manifested by a reduction in large spines and an increase in small spines. Collectively, these studies illustrate how chronic stress may disrupt the net excitatory synaptic input into mPFC, leaving the available population of axospinous synapses with a diminished functional capacity, in terms of biochemical compartmentalization, receptor expression, and synaptic efficacy.
Figure 1.

Diagram illustrating the effects of chronic stress (3 weeks of restraint) on structural plasticity in mPFC pyramidal neurons. Fluorescent dye-injections of pyramidal neurons were made in the dorsal anterior cingulate (ACd) and prelimbic (PL) areas of the rat. An atlas plate (lower left) depicts the approximate region within mPFC that neurons were filled for morphologic analyses. Distance in millimeters relative to bregma is indicated; adapted from Swanson (1992). Schematic neurons are shown for control (left) and chronic restraint stress (right), with arrows highlighting the fact that dendritic atrophy and spine/excitatory synapse loss is most prominent on distal apical dendrites (right). Also shown in each panel are examples of confocal laser-scanning microscopy images of dendritic segments. fa, forceps anterior, corpus callosum.
Some evidence to date supports the possibility that stress-induced morphological changes also have functional consequences. One report has linked chronic stress-induced dendritic atrophy in mPFC pyramidal neurons to reduced serotonin- and hypocretin-evoked EPSCs (Liu and Aghajanian, 2008). Another study showed that long-term potentiation in the hippocampal-PFC pathway is disrupted following chronic stress (Cerqueira et al., 2007). Several reports have correlated chronic stress-induced dendritic remodeling in mPFC with impaired performance in several prefrontal-dependent tasks (Dias-Ferreira et al., 2009, Liston et al., 2006). These studies help to provide a more cellular-based perspective for understanding how stress may trigger a sequelae of changes in mPFC, that produces the disordered thought and affect that is characteristic of these illnesses.
Limbic Cortex and Stress Regulation: Functional Connectivity and Heterogeneity
Despite the advances in understanding the role of the hippocampus and mPFC in stress pathology and depression, less progress has been made in clarifying the relationship of stress-induced plasticity and the withdrawal of HPA-restraining influences. One major impediment has been the inability to unravel the neural circuitry providing for limbic cortical modulation of the HPA axis. Although both of these regions issue projections throughout numerous basal forebrain and hypothalamic structures, they lack any direct innervation of HPA effector neurons within the paraventricular hypothalamic nucleus (PVH)(Cullinan et al., 1993, Herman et al., 2003, Sesack et al., 1989). Moreover, mPFC and hippocampal outputs are predominantly excitatory, utilizing the neurotransmitter glutamate (Swanson and Cowan, 1977, Walaas and Fonnum, 1980), implicating GABAergic relays (Cullinan et al., 1993, Herman et al., 2003). Whereas attempts at localizing the source of stress-inhibitory influences from the hippocampal formation have been successful (Herman et al., 1995), the cortical subfield(s) within mPFC that provide the source of inhibition have been more elusive. This may be due to the fact that the role of the mPFC in stress regulation was thought to be unitary (Akana et al., 2001, Diorio et al., 1993, Figueiredo et al., 2003); however, several conflicting reports have implicated the mPFC as exerting an excitatory influence on HPA output (Spencer et al., 2005, Sullivan and Gratton, 1999).
Through a series of carefully directed studies employing discrete excitotoxin lesions in cortical subfields of mPFC, we found that mPFC influences over acute emotional (restraint) stress-induced HPA output are differentiated in a dorsal-ventral manner (Radley et al., 2006a). Lesions to PL enhanced, whereas infralimbic (IL) lesions inhibited, HPA activation in response to acute emotional (restraint) stress. Furthermore, PL lesions resulted in a prolonged elevation of plasma corticosterone after the cessation of restraint, consistent with its role as a target site for GC negative feedback under normal conditions (Diorio et al, 2003). IL lesions also enhanced activation of a distinct subpopulation of neurons in PVH that issue projections to brainstem and spinal cord regions implicated in autonomic output. Subsequent experiments performed in rats bearing retrograde tracer deposits to label PVH-autonomic projections, confirmed that IL lesions selectively increased stress-induced activation in this cell group (Radley et al., 2006a). These data suggest that IL normally provides an excitatory influence, whereas PL has the capacity to restrain, HPA axis activation in response to acutely stressful experiences. Moreover, these studies implicate differentiated regions of mPFC in the inhibitory control of autonomic and neuroendocrine aspects of stress-induced PVH outflow (Fig. 2).
Figure 2.

Schematic drawing of a sagittal section through the rat basal forebrain highlighting possible circuitry by which the medial prefrontal cortex (mPFC) may influence stress-related hypothalamic paraventricular nucleus (PVH) effector mechanisms. These studies have also established a contingent of GABAergic neurons in anterior bed nucleus of the stria terminalis (aBST) as the likely source of relays for prelimbic cortex (PL) inhibitory influences over acute stress-induced hypothalamo-pituitary-adrenocortical (HPA) axis output. The dashed line from the infralimbic cortex (IL) indicates that the projection has not been formally established. ac, anterior commissure; Ant. Pit., anterior pituitary; ACd, anterior cingulate cortex, dorsal subdivision; aBST, bed nucleus of the stria terminalis, anterior subdivision; cc, corpus callosum; CRH, corticotropin-releasing hormone; mPFC, medial prefrontal cortex; IL, infralimbic area; ot, optic tract; Pre-Auto, preautonomic subdivisions; PVH, paraventricular nucleus of the hypothalamus; PL, prelimbic area.
Follow-up work was focused on defining the possible neural mechanisms providing for HPA-inhibitory influences from PL. A series of ablation and anatomical tract tracing experiments, and assay of central and hormonal indices of HPA axis responses to acute emotional stress, revealed that a discrete population of GABAergic neurons in the anterior subdivision of the bed nucleus of the stria terminalis (aBST; corresponding to the dorsomedial and fusiform subdivisions of (Dong et al., 2001) is likely to subserve PL influences over acute stress-induced HPA output (Radley et al., 2009). This region of aBST contains a cluster of stress-sensitive, PVH-projecting, GABAergic neurons, that show a diminished activation following PL lesions (Radley et al., 2009). Moreover, selective ablation of these neurons produces an enhancement of stress-induced HPA activation similar to that observed following PL lesions (Radley et al., 2009). To date, one other study also suggests that IL modulation of stress responses is relayed via contiguous regions within aBST (Spencer et al., 2005), presumably via a contiguous or interposed group of PVH-projecting excitatory neurons (Choi et al., 2007).
Another issue concerns the broader organization of cortical limbic modulation of the HPA axis, since stress-induced structural plasticity at multiple regions may contribute to its dysregulation and stress pathology. Evidence gathered from a series of experiments employing the aforementioned technical approaches, suggests that stress-inhibitory influences of mPFC and HF are exerted principally via convergence onto the same population of GABAergic neurons in aBST (Radley and Sawchenko, 2011). This conclusion is based upon three lines of evidence: (1) anatomical tracing experiments indicate that extrinsic projections from HF and mPFC converge onto stress-sensitive, PVH-projecting neurons in aBST; (2) GABAergic PVH-projecting cell groups in aBST show a diminished functional activation following acute stress in animals bearing excitotoxin lesions of either vSUB or PL; (3) aBST plays a more prominent inhibitory role than the ventral subiculum (vSUB) over stress-induced increases in plasma corticosterone.
There are a number of hypotheses that derive from this “aBST convergence network” organization that should help to inform future studies (Fig. 3). One is that aBST serves as a neural hub for receiving and integrating stress-modulatory influences from additional limbic forebrain regions (i.e., PVTp, septum, amygdala). Another prediction is that this network, notably via GABAergic relays in aBST, may serve to integrate GC receptor-mediated negative feedback signals from these limbic forebrain regions. In this regard, none of these regions provide any appreciable innervation of PVH, although each projects to the aBST (Shin et al., 2008). The fact that the circuits and mechanisms highlighted by our work are largely inhibitory, this may help to set these apart from HPA-activating networks yet to be clarified (e.g., one that conveys HPA-excitatory influences of IL). Finally, chronic stress-induced neuroplasticity in key nodal points (e.g., dendritic atrophy, synapse loss in mPFC/HF) may diminish their weighting in the modulatory network, via altering integration in aBST GABA neurons, and subsequent disinhibition of the HPA activity (e.g., sensitization, facilitation). In summary, this work should help in deciphering how these systems are organized, and to provide a framework for understanding how stress-induced neuroplasticity may alter the integration of modulatory and activational systems for adaptation and pathology.
Figure 3.

Proposed role of the anterior bed nucleus of the stria terminalis (aBST) as an integrator of limbic cortical influences during emotional stress-induced hypothalmo-pituitary-adrenocortical (HPA) axis output in the rat. Anatomical and lesion data support the pathways highlighted in red with aBST providing an important source of GABAergic innervation of PVH, and relaying limbic cortical influences from the hippocampal formation (HF) and medial prefrontal cortex (mPFC) (i.e., prelimbic cortex (PL)). The paraventricular thalamic nucleus (PVT) is shown (highlighted in black), as it is known to influence HPA output, notably via glucocorrticoid receptor-mediated negative feedback (Jaferi and Bhatnagar, 2006). Like the ventral subliculum (vSUB) and PL, PVT does not provide any direct innervation of the hypothalamic paraventricular nucleus (PVH), but does issue projections to the aBST. Chronic stress may compromise these inhibitory influences over PVH/HPA output via aBST, manifesting in corticosterone hypersecretion, as evident in depression.
INDIVIDUAL DIFFERENCES IN STRESS SENSITIVITY: EPIGENETIC MECHANISMS MOHAMMED KABBAJ, FLORIDA STATE UNIVERSITY
Depression is a growing problem worldwide that possesses variation in symptoms and response to treatment. This problem is particularly acute not only because so many individuals go undiagnosed and therefore untreated, but also because the underlying mechanisms of antidepressant resistance are still unclear. Clinical evidence indicates that personality traits can be used to predict vulnerability to mood disorders such as depression (Cloninger, 2006). As a depressive episode is often preceded by stress (Kessler et al., 2005), it is possible that individual differences in response to stress may contribute to such observed individual variation in the behavior and pathology of depression.
Social Stress and Individual Differences in Anxiety- and Depression-like Behaviors
In humans, depression-inducing stressors tend towards a psychological nature (Bjorkqvist, 2001; Kessler, 1997; Kessler et al., 1985). As such, we utilize an animal model of intraspecies social conflict termed social defeat. This stressor is a modified version of the resident-intruder paradigm, where a male intruder replaces the female cohabitant in the home cage of an aggressive, dominant male. As repeated social defeat does not result in habituation upon repeated presentation (Tidey and Miczek, 1997), this model has the advantage of providing ethologically- and ecologically-relevant forms of persistent emotional stress. When flight is barred, the intruder will assume a supine position and emit loud, and frequent ultrasonic distress calls (Blanchard and Blanchard, 1977). Chronic exposure to social defeat has been found to induce both short- and long-term behavioral and physiological changes. These include decreased locomotor and exploratory activity (Koolhaas et al., 1997, Meerlo et al., 1996); reduced aggression and sexual behavior (Meerlo et al., 1996), increased submissive behavior and anxiety (Ruis et al., 1999). Chronically defeated rats show reduced mobility in a forced swim test, and reduced preference for sweet sucrose solution (anhedonia) as well (Rygula et al., 2005). Reduced locomotor and exploratory activity implies a deficit in motivation while reduced mobility in the forced swim test is interpreted to reflect depressive-like behavior. Decreased sucrose preference may indicate desensitization of the brain reward circuitry. Taken together, these findings suggest that chronic social defeat in rats is an appropriate model for depressive disorders.
As individuals vary in their response to depression and subsequent treatment, so too may they show differences in the response to stress. A multitude of animal paradigms that model human mood disorders have been developed, based on persistent inter-individual differences in response to stress, in order to study the neurobiology of human affect disorders (for review, see (Harro, 2010)). One of these models relies on the response to the mild stress of a novel environment, where some rats, known as high responders (HR), exhibit high rates of exploratory locomotion while others, known as low responders (LR), exhibit low rates of locomotor activity (Hooks et al., 1992b, Kabbaj and Akil, 2001, Piazza et al., 1989, Pierre and Vezina, 1997). The locomotor response to a novel environment not only predicts subsequent behavioral responses to drugs such as amphetamine and cocaine (Hooks et al., 1992a, Hooks et al., 1991a, Hooks et al., 1991b, Kabbaj and Akil, 2001, Piazza et al., 1989, Pierre and Vezina, 1997), but also predicts anxiety-related behavior in these animals (Dellu et al., 1996, Kabbaj et al., 2000). Indeed, LR rats display higher levels of anxiety in an elevated T-maze, a response that is more strongly enhanced in LR than HR animals following repeated exposure to the test(Verheij et al., 2009). HR and LR rats also appear to exhibit different behaviors in the forced swim test at basal conditions and following antidepressant treatment, although these points are still unclear. Indeed, Taghzouti and colleagues reported individual differences in the test phase of the FST together with differential effects of subchronic fluoxetine injections (Taghzouti et al., 1999), whereas a recent study using the same injection protocol revealed no individual differences in the test phase of the FST procedure, an equal effectiveness of fluoxetine in reducing behavioral despair, but an antidepressant effect of desipramine in LR only (Jama et al., 2008). Taken together, this evidence strongly suggests that HR and LR animals possess differential vulnerabilities to the social defeat paradigm that could model the individual differences in vulnerabilities to stress-related mood disorders.
In our behavioral investigations, we focused on basal and defeat-induced differences between HR and LR animals on anhedonia, anxiety, social approach and avoidance, and contextual fear memory. We therefore analyzed the vulnerability of HR and LR animals to several aspects of social defeat-induced depressive-like behaviors. First, analysis of global locomotor and exploratory behavior in an open-field confirmed previous reports that non-defeated HR animals exhibit reduced anxiety levels compared to their LR counterparts (Dellu et al., 1996, Kabbaj et al., 2000). Exposure to repeated social defeat, however, strongly reduces the time spent by HR animals in the center of the arena to that of the level of LR rats. Moreover, HR and LR animals display similar responses to repeated social defeat in terms of general mobility, exploratory and stereotyped behaviors, demonstrating that the observed reduction of time spent in the center exhibited by HR animals is related to anxiety. Although this suspected higher vulnerability to stress-induced anxiety remains to be confirmed and analyzed in detail using more anxiety-specific behavioral procedures, this point is of particular interest when considering other observations in LR and HR animals. Indeed, while HR animals self-administer higher levels of cocaine than LR individuals under basal conditions, no individual differences can be observed following social defeat exposure (Kabbaj and Akil, 2001). Moreover, while HR animals display a higher preference than LR individuals for a 0.25% sucrose solution during the first presentation, both HR and LR rats exhibit similar sucrose preferences following exposure to social defeat. The stereotypic decrease in immobility duration displayed by HR animals during the first exposure to FST is lost upon repeated FST presentations (Taghzouti et al., 1999). While repeated defeat exposure did not affect sucrose preference, or social avoidance in LR animals, HR animals consistently demonstrated behavioral susceptibilities to this stressor. Finally, when re-exposed to the context of social defeat, repeatedly defeated LR and HR animals both demonstrated similar freezing responses to this context. Following acute social defeat exposure, however, only HR animals exhibited freezing behavior when tested four weeks later. Together, these observations suggest an increased sensitivity to stress in HR animals related to a heightened emotional reactivity (see Table 1).
Table 1.
Behavioral Characteristics of High Responder (HR) vs. Low Responder (LR) Rat Strains
| HR control | LR control | Defeat effects in HR rats | Defeat effects in LR rats | |
|---|---|---|---|---|
| Locomotion in Novel environment | High | Low | NS | NS |
| Cocaine self-administration | High | Low | Decreased | Increased |
| Anxiety in open field | Low | High | Increased | No effects |
| Anhedonia-sucrose preference | High | Low | Decreased | No effects |
| Contextual freezing after chronic defeat | -- | -- | Increased | Increased |
| Contextual freezing after acute defeat | -- | -- | Increased | No effects |
Summary of the behavioral characteristics of rat strains showing high (HR) or low (LR) locomotor responses to novelty stress. Control=behavioral responses in naïve HR and LR subjects; Defeat effects= response patterns seen after exposure to social defeat. NS=no significant difference from control pattern.
Epigenetic Factors and Individual Differences
Individual behavioral responses to social defeat underscore the potential for underlying neurobiological alterations. Such behavioral changes may be the products of chromatin modification in response to the experience of social defeat. Chromatin modification is a dynamic process that regulates gene expression without alteration of the DNA sequences (Crosio et al., 2003, Guan et al., 2002). This modification is primarily accomplished through modifications of histone N-terminal tails at the promoter regions of specific genes (Cheung et al., 2000). Recent studies have found changes in modifications at specific gene promoter regions in association with social defeat (Covington et al., 2009, Tsankova et al., 2006, Tsankova et al., 2004, Wilkinson et al., 2009). One such modification is histone acetylation. Acetylation has been widely studied and it is generally accepted that hyperacetylation leads to an increase in gene expression, while hypoacetylation leads to gene silencing (Forsberg and Bresnick, 2001, Ito and Adcock, 2002). Histone acetylation is a stress-regulated process, as acute psychological stressors cause acetylation of lysine 14 of histone H3 (H3K14) in the dentate gyrus (Reul and Chandramohan, 2007). Furthermore, increased acetylation at specific promoter regions of genes such as brain derived neurotrophic factor (BDNF) is associated with a reversal of depressive-like behavior following electroconvulsive shock therapy (ECS), while overall increased acetylation in the nucleus accumbens has been associated with depressive-like symptoms in mice (Covington et al., 2009, Tsankova et al., 2006).
We examined potential epigenetic correlates of LR-HR behavioral differences in three brain areas that are relevant to human depression: the hippocampus, the amygdala, and the medial prefrontal cortex. As a first step to explore a possible role for histone modification in epigenetic modulation of stress reactivity, we examined basal differences in histone acetylation between HR and LR animals where we found higher levels of H3 and H2B acetylation in HR hippocampi. Such increased acetylation may account for the observed increases in hippocampal gene expression in HR rats shown previously (Kabbaj, 2004). We also looked at differences in histone acetylation in HR and LR rats following exposure to acute and repeated social defeat. Following acute social defeat, we found an interesting difference in the timing of this acetylation between HR and LR animals. In the hippocampus, while histone H3 hyperacetylation (acetylation of H3K9 and K14) is significantly increased in both HR and LR animals, this increase is seen 30 minutes following defeat exposure in HR’s but 2h and 30 minutes after exposure in LR animals. In the amygdala, there is a similar pattern in the timing of acetylation, but in the opposite direction with HR animals exhibiting a transient decrease in H3 hyperacetylation 30 minutes after defeat and LR animals exhibiting a sustained decrease 2h and 30 minutes later. Our findings suggest that there is differential regulation of gene expression between the hippocampus and the amygdala, with HR animals appearing to respond to the stressor faster at the molecular level. These changes appear to be specific to the amygdala and hippocampus, as we found no changes in acetylation in the medial prefrontal cortex. Additionally, acute defeat appears to have no effect on histone H4 acetylation, as we found no significant changes in acetylation on this terminal tail. Following repeated social defeat exposure, we found differential changes in acetylation in the hippocampus, with histone H3 acetylation decreasing in HR rats, but increasing in LR rats and significant increases in histone H4 acetylation in both HR and LR animals. While the timing of these acetylation changes did not differ between HR and LR rats as seen following acute defeat, both groups had increased histone H3 acetylation 24 hours after the last exposure to defeat in the hippocampus. Interestingly, we did not see any changes in the amygdala following repeated defeat in either HR or LR rats. This could mean that the amygdala is involved only in the initial fear response to defeat, but not in repeated presentations, although a thorough investigation of specific nuclei of the amygdala is still needed. We also did not see any changes in the medial prefrontal cortex following repeated defeat, again suggesting that acetylation in this area is unaffected by defeat.
We sought to uncover the mechanism behind this differential change in acetylation by investigating levels of expression of one histone acetyltransferase (HATs) and several histone deacetylases (HDACs). We chose these two classes of enzymes as they are both directly responsible for changes in acetylation on histones (Wade, 2001). HAT enzymes use an acetyltransferase catalytic domain to add acetyl groups to lysine residues on histone N-terminal tails (Roth et al., 2001), thereby weakening their interaction with the DNA and facilitating transcriptional activation ((Bannister and Kouzarides, 1996, Ogryzko et al., 1996); for review see (Roth et al., 2001)). One such HAT is the CREB-binding protein (CBP), a transcriptional coactivator that interacts with numerous transcriptional regulators and facilitates the assembly of the transcriptional machinery (Chrivia et al., 1993, Janknecht, 2002). Importantly, CBP preferentially acetylates H3K14 (Cheung et al., 2000, Lo et al., 2000, McManus and Hendzel, 2001). HDAC enzymes exert their effect by removing the negatively charged acetyl groups from acetylated histones, thereby increasing the net positive charge and the affinity of histones for the negatively charged DNA. This strengthening of histone-DNA contacts upon histone deacetylation interferes with transcriptional activation. Thus HDACs are considered active transcriptional repressors. Accordingly, we investigated HDACs recently linked to regulation of CBP. We observed a significant increase in CBP and decrease in HDAC3 in HR animals, as compared to LR rats. Although this regulation remains to be confirmed at the protein level, HR animals appear to exhibit an increase in CBP and a decrease in HDAC3, both acting in a coherent manner to explain the higher acetylation level of histones H3 and H2B. In contrast, we didn’t find any regulation of the other investigated targets following social defeat. While this result may appear surprising in light of the observed modifications in histone acetylation, it is in line with similar analyses of several class I and II HDAC mRNA levels, including HDACs 4 and 5, in hippocampus after chronic social defeat in mice (Tsankova et al., 2006). Indeed, the authors reported no significant influence of social defeat alone on the levels of HDAC expression (Tsankova et al., 2006). It may be the case that post-translational modification of HDACs may underlie the observed changes in histone acetylation as class II HDACs have the ability to shuttle between the nucleus and cytoplasm depending on the received signal (de Ruijter et al., 2003). Also, we can not rule out the possibility that other HDACs, HATs, or other brain structures are not implicated. Recent work has implicated HDAC2 in the regulation of acetylation in the nucleus accumbens following repeated exposure to social defeat (Covington et al., 2009). Also, GCN5 is a HAT that preferentially acetylates lysine 14 on H3 (Lo et al., 2000). Both of these are potential targets for future investigation. Clearly, follow-up studies will be necessary to link changes in histone acetylation/deacetylation in regulation of specific stress-regulatory genes.
STRESS, ANTIDEPRESSANTS AND GLUCOCORTICOID ‘ENDOPHENOTYPES’ LAUREN JACOBSON, ALBANY MEDICAL COLLEGE
HPA activity is of interest as a biomarker for treatment response in depression, with up to 80% of depressed patients exhibiting evidence of elevated HPA activity. Moreover, the failure of treatment to normalize HPA activity is a significant predictor of depression recurrence (Ising et al., 2007). The “corticosteroid hypothesis of depression” attributes HPA hyperactivity in depression to impaired brain glucocorticoid receptor (GR) expression and function that can be reversed or compensated by antidepressants (Holsboer, 2000). However, the contrasts between melancholic and atypical depression raise questions as to whether impaired GR function is universal, and enhanced GR function necessarily beneficial, to all depressed individuals. Melancholic and atypical depression have largely opposing psychiatric features (insomnia and appetite loss in melancholia; hypersomnia and weight gain in atypical depression) that tend to be associated with differences in HPA activity. Melancholia is more likely to correlate with HPA hyperactivity and glucocorticoid feedback resistance, whereas atypical depressed patients often exhibit HPA hypoactivity and greater sensitivity to dexamethasone suppression (American Psychiatric Association. and American Psychiatric Association. Task Force on DSM-IV., 2000, Gold et al., 2002, Pariante and Miller, 2001, Stewart et al., 2009). Intriguingly, the conflicting psychiatric and endocrine characteristics of these two subtypes also correlate with differential responsiveness to older-generation antidepressants, with melancholics benefiting from both tricyclic antidepressants (TCA) and monoamine oxidase inhibitors (MAOI), but atypical depressed patients, particularly those with early-onset, chronic depression, responding significantly better to MAOIs than to TCAs (Stewart et al., 2009). These differences in antidepressant efficacy suggest that atypical and other forms of depression lacking HPA hyperactivity are not just troubling exceptions to the corticosteroid hypothesis of depression, but potentially instructive models to make this hypothesis more inclusive and useful for predicting antidepressant response.
In light of the evidence for opposing psychiatric and endocrine features in depression, we have hypothesized that antidepressants effective for these different types of depression should have drug- and brain region-specific actions, rather than uniform effects, on HPA activity and GR expression. To test this hypothesis, we measured HPA hormones and performed in situ hybridization analysis of GR and GR target genes after chronic antidepressant treatment in male C57BL/6 mice. Mice were adrenalectomized and replaced with fixed corticosterone levels to control for indirect, autoregulatory changes in GR due to antidepressant effects on glucocorticoid secretion. Mice were also subjected to the forced-swim test of depression-like immobility behavior to verify that drug doses were sufficient for clinically-relevant effects. Under these conditions, we observed little change in hippocampal mineralocorticoid receptor (MR) gene expression or in hippocampal GR gene expression (Heydendael and Jacobson, 2008, Heydendael and Jacobson, 2009, Heydendael and Jacobson, 2010), suggesting that previously-reported changes in these receptor populations (Pariante and Miller, 2001) might have indeed reflected autoregulation (Herman, 1993) secondary to antidepressant-induced changes in glucocorticoid secretion.
However, supporting our predictions, we found that imipramine (a tricyclic antidepressant) and phenelzine (a monoamine oxidase inhibitor) had opposing effects on GR gene expression and HPA activity. Imipramine facilitated inhibition of HPA activity at low glucocorticoid levels (Mukherjee et al., 2004), whereas phenelzine impaired corticosterone inhibition of ACTH and corticotropin-releasing hormone (CRH; (Kier et al., 2005)). Imipramine significantly increased GR gene expression in the paraventricular hypothalamus and prefrontal cortex (Heydendael and Jacobson, 2008), two regions shown to be capable of mediating HPA inhibition by localized glucocorticoid implants (Diorio et al., 1993, Sawchenko, 1987). In contrast to the effects of imipramine (as well as effects predicted by the corticosteroid hypothesis of depression), phenelzine significantly reduced GR expression in these same regions (Heydendael and Jacobson, 2008). The selective serotonin reuptake inhibitor (SSRI) fluoxetine had no significant effects on basal HPA activity, although it exhibited the potential to influence HPA feedback regulation in also inhibiting prefrontal cortex GR gene expression (Heydendael and Jacobson, 2010). The actions of imipramine and phenelzine could respectively improve feedback sensitivity of the HPA axis to the modest cortisol elevations typically observed in melancholic depression (Young et al., 1994), and normalize the HPA hypoactivity and enhanced sensitivity to glucocorticoid feedback reported in atypical depression (Levitan et al., 2002, Stewart et al., 2009). The lack of fluoxetine effects on HPA activity may indicate, as occasional studies have suggested (Perry, 1996), that this class of drugs is less effective for HPA-dysregulated depression.
We have further examined antidepressant effects on GR gene expression in brainstem monoaminergic nuclei connected with depression pathology or antidepressant action (Heydendael and Jacobson, 2009). In both the locus coeruleus and dorsal raphé nucleus, we found consistent but drug-specific effects to inhibit GR gene expression and increase expression of genes such as tyrosine hydroxylase and tryptophan hydroxylase-2 (Brady et al., 1992, Makino et al., 2002), the rate-limiting enzymes for synthesis of norepinephrine and serotonin, respectively. Phenelzine significantly inhibited GR and increased tyrosine hydroxylase gene expression in the locus coeruleus, while imipramine had no effects on GR but inhibited tyrosine hydroxylase gene expression in this region. In contrast, imipramine significantly inhibited GR and increased tryptophan hydroxylase-2 gene expression in the dorsal raphé nucleus, whereas phenelzine had no effects on either gene product in this region (Heydendael and Jacobson, 2009). Fluoxetine shared the actions of both imipramine and phenelzine in having significant effects to inhibit GR gene expression and stimulate monoamine-synthesizing enzyme gene expression in the locus coeruleus as well as the dorsal raphé nucleus (Heydendael and Jacobson, 2010). Supporting the likelihood that antidepressant effects on enzyme expression required GR activity, gene expression of tyrosine hydroxylase and tryptophan hydroxylase-2 was unaffected by antidepressant treatment in adrenalectomized mice without glucocorticoid replacement (Heydendael and Jacobson, 2009, Heydendael and Jacobson, 2010). Drug effects were also brain region-specific, as we did not observe significant antidepressant effects in the dopaminergic ventral tegmental nucleus or the serotonergic median raphé nucleus (Heydendael and Jacobson, 2009).
Alterations in noradrenergic or serotonergic tone potentially resulting from changes in tyrosine hydroxylase or tryptophan hydroxylase expression could affect both HPA activity and mood. Effects on HPA activity may be complex due to the context-specific and multi-synaptic influence of the locus coeruleus and dorsal raphé nucleus on this axis (Lowry, 2002, Ziegler et al., 1999). However, since defects in norepinephrine and serotonin, at least in vulnerable individuals, are thought to be involved in depression (Heninger et al., 1996), regulation of tyrosine hydroxylase and tryptophan hydroxylase expression suggests a novel, GR-related mechanism for antidepressant effects on mood. Since atypical depression has been specifically linked with norepinephrine deficiency (Brady et al., 1992, Gold et al., 2002), the potential to increase norepinephrine transmission could account for the efficacy of phenelzine in atypical depression (Stewart et al., 2009). The apparent overlap of fluoxetine effects with those of both imipramine and phenelzine, particularly on brainstem GR and monoamine-synthesizing enzyme expression, could contribute to the widespread efficacy reported for SSRIs (Pande et al., 1996, Vaswani et al., 2003). Notably, since glucocorticoid excess can itself cause depression symptoms (Warrington and Bostwick, 2006), the ability to attenuate HPA activity could augment the effects on serotonergic tone of drugs such as imipramine.
Our results therefore support an expansion of the corticosteroid hypothesis of depression to include drug- and brain region-specific effects of antidepressants on GR signaling in the brainstem as well as in forebrain regions besides the hippocampus (Table 2). Although their underlying pharmacology remains to be defined, these effects suggest that HPA tests have renewed value for predicting and monitoring antidepressant response. For example, the choice between antidepressants with TCA- or MAOI-like mechanisms of action might be dictated by whether the diagnosis of depression is accompanied by evidence of greater or lesser sensitivity to dexamethasone suppression. Because glucocorticoids can affect mood (Stewart et al., 2009), our findings further imply that effects on HPA activity could contribute to the therapeutic actions of antidepressants (Figure 4).
Table 2.
Effects of Antidepressants on Expression of Glucocorticoid Receptor (GR) and GR Target Genes
| Paraventricular hypothalamus | Prefrontal Cortex | Locus Coeruleus | Dorsal Raphé Nucleus | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TCA | MAOI | SSRI | TCA | MAOI | SSRI | TCA | MAOI | SSRI | TCA | MAOI | SSRI | |
| GR | ↑ | ↓ | --- | ↑ | ↓ | ↓ | --- | ↓ | ↓ | ↓ | --- | ↓ |
| GR target gene | ---/↓ | ↑ | --- | N/A | N/A | N/A | ↓ | ↑ | ↑ | ↑ | --- | ↑ |
Summary of the effects of representative antidepressants on GR gene expression and GR target gene expression in selected forebrain and brainstem regions of the mouse brain. GR target genes examined were corticotropin-releasing hormone (CRH) for the paraventricular hypothalamus, tyrosine hydroxylase for the locus coeruleus, and tryptophan hydroxylase-2 for the dorsal raphé nucleus.
TCA=tricyclic antidepressant administration (imipramine); MAOI= monoamine oxidase inhibitor administration (phenylzine); SSRI= selective serotonin reuptake inhibitor administration (fluoxetine).
↑, significant increase; ↓ significant decrease; ---, no significant effect; ---/↓ nonsignificant trend towards a decrease (0.05 < P < 0.1); N/A, not applicable.
Figure 4.
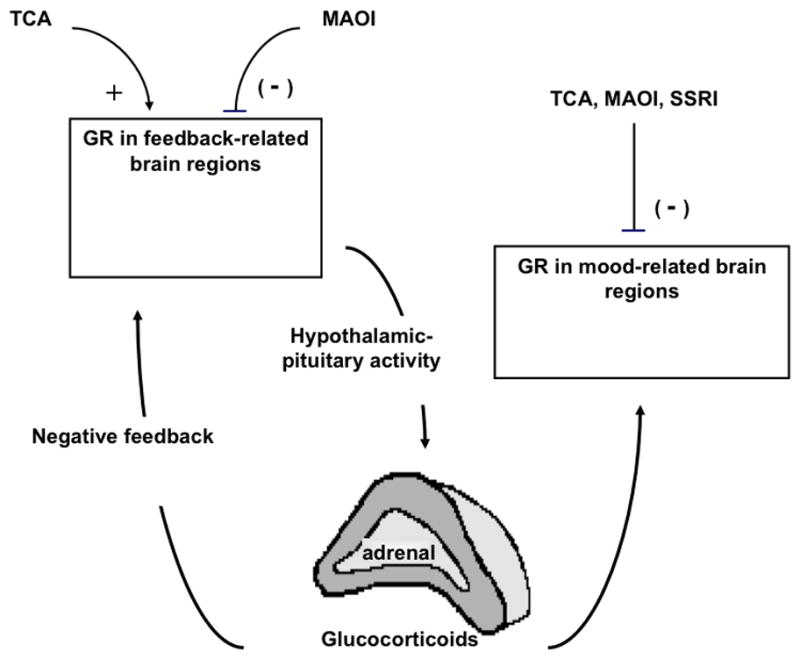
Proposed model for glucocorticoid receptor (GR) -related effects of antidepressant effects on hypothalamo-pituitary-adrenocortical (HPA) axis activity and mood. Representative tricyclic (TCA) (imipramine) and monamine oxidase inhibitor (MAOI)(phenylzine) antidepressants have opposing effects on HPA activity, potentially via opposing effects on GR in feedback sites within the forebrain. The ability of TCA to facilitate and MAOI to impair glucocorticoid feedback could contribute to normalizing HPA activity in melancholic or atypical depression, respectively. The selective serotonin reuptake inhibitor (SSRI) fluoxetine did not exhibit marked effects on HPA activity and is not included in this part of the model. Antidepressants could also regulate mood through direct and indirect effects on GR. TCA, MAOI, and SSRI antidepressants had similar effects to inhibit GR expression and relieve GR-mediated inhibition of monoamine-synthesizing enzymes in the locus coeruleus and dorsal raphé nucleus, which could contribute to the increases in norepinephrine and serotonin thought to be important to antidepressant response. Alternatively or in addition, changes in GR in HPA feedback-related regions (observed primarily for TCA and MAOI) could indirectly affect glucocorticoid-sensitive mood centers by raising or lowering glucocorticoid levels.
GLUCOCORTICOID ALTERATIONS IN PTSD RACHEL YEHUDA, JAMES J. PETERS VA MEDICAL CENTER AND MT. SINAI SCHOOL OF MEDICINE
The initial conception of PTSD (as first described in the Diagnostic and Statistical Manual of Mental Disorders (DSM)-III) indicated that it represented a universal human response to exposure to extremely traumatic experiences, defined as both markedly distressing, and unusual or rare in a person’s experience (American Psychiatric Association., 1980). Three distinct, but co-occurring, symptom groups are present in PTSD. Re-experiencing symptoms are intrusions of the traumatic memory in the form of distressing images, nightmares, or dissociative experiences such as flashbacks. Avoidance symptoms include actively avoiding reminders of the traumatic event including persons, places, or things associated with the trauma and more passive behaviors reflecting emotional numbing and constriction. Hyperarousal symptoms such as insomnia, irritability, impaired concentration, hypervigilance, and increased startle responses concern more “physiologic” manifestations of trauma exposure. In DSM-IV, these symptoms must impair social, occupational or interpersonal function, and persist in tandem for at least a month following the trauma (American Psychiatric Association., 1994).
It was implicit in both the original and revised definitions of PTSD that the most important predictor of PTSD would be the type and severity of trauma exposure. Reciprocally, the presence of PTSD provided post hoc confirmation of trauma exposure. The strong theoretical link between exposure to events at the extreme end of the stress response spectrum and PTSD provided the rationale for hypothesizing that biologic alterations in this disorder, particularly those associated with glucocorticoids, would be similar to those observed in basic science models of stress (Yehuda et al., 1998a).
The normal fear response is characterized by a series of biological reactions that help the body cope with stress. Activation of the sympathetic nervous system (SNS) and the release of adrenaline allow the organism to increase its physiological capacity for “fight-or-flight” in response to threat (Cannon, 1914) and facilitate consolidation of the threat memory (McGaugh and Roozendaal, 2002). The simultaneous activation of the HPA axis, culminating in the release of cortisol, helps contain the stress response when the threat is removed (Munck et al., 1984), as described above. As stress-activated biological reactions are suppressed as a result of cortisol inhibition, elevated cortisol levels also exert a negative feedback inhibition at the pituitary, hypothalamus, and amygdala—sites initially responsible for the stimulation of cortisol release (McEwen, 1992). Negative feedback inhibition occurs due to the presence of glucocorticoid receptors (GR), and leads to the restoration of basal hormone levels (de Kloet et al., 1986).
Early biological studies in PTSD hypothesized increases in both cortisol and catecholamines. However, only the latter were found to be elevated (Kosten et al., 1987, Southwick et al., 1997, Yehuda et al., 1998b). The first neuroendocrine study of cortisol in PTSD demonstrated lower 24-hr urinary cortisol excretion in PTSD compared to persons with other psychiatric diagnoses (Mason et al., 1986). Although these results were initially difficult to interpret, it soon became clear that 1) only a minority of persons exposed to trauma failed to recover from initial fear reactions and move on to develop PTSD, and that 2) traumatic exposures that had been presumed to be rare were common occurrences (Kessler et al., 1995). These epidemiologic observations resulted in a paradigm shift with respect to the conceptual link between trauma exposure and PTSD (Yehuda, 2002). That is, rather than representing the normative response to trauma, it seemed more appropriate to describe PTSD as a failure to engage the biological mechanisms associated with recovery and physiologic homeostasis (Yehuda and LeDoux, 2007, Yehuda and McFarlane, 1995). This conceptual change provided an important context for understanding glucocorticoid alterations in PTSD. Indeed, if PTSD represented a specific biological phenotype representing a failure of recovery, lower cortisol levels could represent an important biological correlate of this phenomenon.
Glucocorticoid Alterations in chronic PTSD: Overview
Alterations of the HPA axis are a predominant feature of PTSD pathophysiology. While there are exceptions in the literature, it appears that PTSD is associated with a unique profile in that corticotropin releasing hormone (CRH) levels appear to be increased (Baker et al., 1999) while urinary and plasma levels of cortisol are lower or at least not elevated, compared to non-exposed persons without PTSD (for review, see (Yehuda, 2002)). Increased cortisol suppression in response to dexamethasone (DEX) has also been observed in most studies, reflecting an enhanced negative feedback inhibition. An important feature of this profile is that it differs from that observed in acute and chronic stress and depression, which has been classically associated with increased CRH and cortisol levels, reduced cortisol suppression to DEX, and reduced GR responsiveness.
Indeed it is presumed that the enhanced negative feedback inhibition reflects a greater responsiveness of GR, as suggested by demonstrations of changes in GR number and responsiveness in response to DEX administration and other challenges (Yehuda et al., 1995). Although it is easy to reach the erroneous conclusion that the lower cortisol levels reflect a less active HPA axis, this does not appear to be the case (Yehuda et al., 1996). Circadian rhythm of cortisol reflects a more dynamic regulatory pattern, as confirmed by mathematical modeling of cortisol oscillations over the diurnal cycle (Elzinga et al., 2003). Furthermore, hormonal responses to naturalistic and laboratory provocations suggest an exaggerated hormonal (and subjective) response to stress. In some cases, but more rarely, elevated cortisol levels have been reported in PTSD. Together, these findings imply that although cortisol levels may be generally lower, the adrenal gland is certainly capable of producing adequate amounts of cortisol in response to challenge (Yehuda, 2005).
Origin of Glucocorticoid Related Alterations in PTSD
Though initial observations related low cortisol levels and related findings of HPA axis alterations to PTSD pathophysiology, several converging findings suggest that these cortisol-related alterations may reflect a pre-existing vulnerability trait that increase the probability of developing PTSD following trauma exposure. In some longitudinal studies evaluating trauma survivors in the immediate aftermath of exposure and thereafter, there was an attenuated rise in cortisol immediately after the trauma in those at greater risk for developing PTSD or who actually did develop PTSD (Yehuda et al., 1998a). This finding has tended to be confined to samples evaluating cortisol in response to a similar event exposure. Thus, lower cortisol levels were associated with prior assault in a study of consecutively appearing rape victims to an emergency room hours after rape (Resnick et al., 1995). Lower cortisol levels were also observed in association with PTSD symptoms in a group of motor vehicle accidents (Delahanty et al., 2003), and in response to the natural disaster of an ice storm (Anisman et al., 2001). It was not replicated in a large cohort of civilians appearing to the emergency department in response to a wide range of events ranging from terrorism to accidents (Shalev et al., 2008). Possibly such a differentiation requires a relatively homogenous sample with similar risk factors for exposure to a particular traumatic event or PTSD following that event.
Interestingly, in studies finding evidence of low cortisol in association with PTSD or PTSD risk, there was also evidence of greater SNS arousal as reflected by catecholamine levels. It may be that increased SNS activation reflects an inadequately contained acute stress response. In the above study of civilians, neither lower cortisol levels, nor elevated catecholamines were observed in the acute aftermath of trauma (Shalev et al., 2008).
That lower cortisol levels reflect PTSD risk has also been supported by studies of individuals at risk for PTSD based not on trauma exposure, but rather, on risk factor of parental PTSD. Parental PTSD has been demonstrated to increase the prevalence of PTSD following trauma exposure by as much as three-fold (Yehuda and Bierer, 2008). In a sample of Holocaust offspring, lower cortisol levels were also observed in association with parental PTSD (Yehuda and LeDoux, 2007). Offspring of Holocaust survivors with PTSD also showed significantly enhanced cortisol responses to 0.50 mg DEX compared to offspring of Holocaust survivors without PTSD and demographically comparable controls (Yehuda et al., 2007). In addition, urinary cortisol levels were negatively correlated with severity of parental PTSD symptoms, even after controlling for PTSD symptoms in offspring. The finding that low cortisol is associated with parental PTSD was recently replicated in a cohort of infants of mothers exposed while pregnant to the World Trade Center attacks on September 11, 2001, in whom significantly lower salivary cortisol levels were observed in the subgroup of offspring born to mothers who developed PTSD from this event compared to infants whose mothers did not (Yehuda et al., 2005).
An attenuated cortisol response in the acute aftermath of trauma in those at greater risk for PTSD may result in a cascade in which there is increased SNS activation, leading to an exaggerated catecholamine response to the trauma. This, in turn, could initiate a process in which traumatic memories become “over-consolidated” or inappropriately remembered due to an exaggerated level of distress. The primary mechanism through which catecholamines facilitate memory formation is by maintaining organisms at a high level of arousal. This response is modulated by adrenal steroids (Roozendaal et al., 2008). Thus, if there is insufficient cortisol signaling, catecholamine-induced arousal might be prolonged and distress increased. Both circumstances might impair memory consolidation or lead to increased fear conditioning to more generalized stimuli. If low cortisol levels represent a pre-existing characteristic, reinforced by “over-consolidation” at the time of the trauma, then failing to properly contain the SNS response to traumatic reminders could perpetuate the intrusive and hyperarousal symptoms of chronic PTSD, leading to the elaboration of avoidance symptoms that commonly occur in the disorder even over years or decades.
The above discussion is important because it suggests that cortisol levels (and related glucocorticoid alterations) in PTSD reflect pre-trauma characteristics. Thus, the role of glucocorticoid alterations in PTSD pathophysiology may be more as precipitants or facilitators of disorder following trauma exposure than as consequences of exposure per se. This formulation is appealing because a central question in trying to understand the biological basis of PTSD as a response to extreme stress involves resolving why only a minority of those exposed develop the disorder. That there is a biological vulnerability explains why some persons experience exaggerated arousal and distress and move on to develop a condition characterized by sustained arousal, while others are able to achieve homeostasis in response to exposures of similar magnitude. Because there are clearly risk factors for PTSD, some of which may be constitutional, it is important to consider a wide range of biological contributors to the HPA axis alterations that have been observed. These include genetic polymorphisms, permanent changes in gene expression resulting from epigenetic modifications, and transient and reversible changes reflecting day-to-day alterations in biological processes, as well as the role of ongoing environmental perturbations on these latter processes.
SUMMARY
The work presented in this symposium highlights several import considerations for understanding the genesis of stress pathology or induction of stress resistance in animal models: neuroplastic responses in cortical and hippocampal nodes controlling stress responses; epigenetic factors that influence stress coping; and the contribution of glucocorticoid hypo-, as well as hypersecretion to stress phenotypes and circuit organization. Taken together, these lines of research highlight a considerable variance in organismic ‘strategies’ for stress coping. Dr. Kabbaj’s work on individual differences in the HR-LR model highlights the heterogeneity of stress response patterning across acute vs. chronic exposure regimens, with the LR showing enhanced anxiety to acute stress exposure, and HRs showing more pronounced sensitivity to chronic stress in the form of social defeat. Dr. Jacobson’s studies suggest that depression may be composed of both hypo-active and hyper-active HPA axis endophenotypes, which involve different underlying neural circuit activity and may be selectively accessed by different classes of antidepressant drugs. Dr. Radley’s work suggests that stress responses are controlled by convergent inputs from prelimbic and hippocampal inputs to subcortical relays. There are known individual differences in brain region reactivity to stress, illustrated in Dr. Kabbaj’s work highlighting stress-related HR-LR histone acetylation/deacetylation in hippocampus but not prefrontal cortex. Thus, it stands that genetic, or in this case, epigenetic factors may alter the relatively role of the hippocampus and prefrontal cortex in stress integration, thereby influencing the circuitry that underlies an individual’s stress response strategy. Finally, Dr. Yehuda’s work presents evidence for altered HPA axis control as a trait, rather than state variable for development of PTSD, wherein low cortisol represents a significant risk factor for ‘overconsolidation’ of stressful experiences and hence, disease development.
Overall, these sets of studies are illustrative of the importance of individual differences in determination of stress responsiveness, particularly with regard to determining how prolonged or chronic stress will influence the physiology and behavior of the organism. The importance of prior experience and genetics on individual stress reactivity is an emergent theme in the stress field, and work in this area promises to illuminate factors determining stress resilience vs. susceptibility in heterogeneous populations.
Acknowledgments
The 2010 Neurobiology of Stress Workshop was supported by NIH grant R13MH090623 (JPH). Research reported in this review was supported by NARSAD and Anxiety Disorders Association of America Young Investigator Awards (JJR); NIH grants R01MH087583, R21MH081046, and R21MH083128 (MK); NIH grant R01MH080394 (LJ); and VISN3 MIRECC, DOD award #W81XWH-06-2-0032, NIH ARRA grant RC1MH088101, and the Lightfighter Trust (RY).
Footnotes
DECLARATION OF INTEREST
The authors have no conflict of interest.
References
- Akana SF, Chu A, Soriano L, Dallman MF. Corticosterone exerts site-specific and state-dependent effects in prefrontal cortex and amygdala on regulation of adrenocorticotropic hormone, insulin and fat depots. J Neuroendocrinol. 2001;13:625–637. doi: 10.1046/j.1365-2826.2001.00676.x. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 3. Washington, DC: 1980. (DSM-III) [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 3. Washington, DC: 1994. text revision (DMS-III-TR) [Google Scholar]
- American Psychiatric Association. and American Psychiatric Association. Task Force on DSM-IV. Diagnostic and statistical manual of mental disorders : DSM-IV-TR. xxxvii. Washington, DC: 2000. p. 943. [Google Scholar]
- Anisman H, Griffiths J, Matheson K, Ravindran AV, Merali Z. Posttraumatic stress symptoms and salivary cortisol levels. Am J Psychiatry. 2001;158:1509–2511. doi: 10.1176/appi.ajp.158.9.1509. [DOI] [PubMed] [Google Scholar]
- Baker DG, West SA, Nicholson WE, Ekhator NN, Kasckow JW, Hill KK, Bruce AB, Orth DN, Geracioti TD., Jr Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatry. 1999;156:585–588. doi: 10.1176/ajp.156.4.585. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- Blanchard RJ, Blanchard DC. Aggressive behavior in the rat. Behav Biol. 1977;21:197–224. doi: 10.1016/s0091-6773(77)90308-x. [DOI] [PubMed] [Google Scholar]
- Brady LS, Gold PW, Herkenham M, Lynn AB, Whitfield HJ., Jr The antidepressants fluoxetine, idazoxan and phenelzine alter corticotropin-releasing hormone and tyrosine hydroxylase mRNA levels in rat brain: therapeutic implications. Brain Res. 1992;572:117–125. doi: 10.1016/0006-8993(92)90459-m. [DOI] [PubMed] [Google Scholar]
- Cannon WB. The emergency function of the adrenal medulla in pain and the major emotions. Am J Physiol. 1914;33:356–372. [Google Scholar]
- Carroll BJ, Curtis GC, Mendels J. Neuroendocrine regulation in depression, I: limbic system-adrenocortical dysfunction. Arch Gen Psychiatry. 1976;33:1039–1044. doi: 10.1001/archpsyc.1976.01770090029002. [DOI] [PubMed] [Google Scholar]
- Cerqueira JJ, Mailliet F, Almeida OF, Jay TM, Sousa N. The prefrontal cortex as a key target of the maladaptive response to stress. J Neurosci. 2007;27:2781–2787. doi: 10.1523/JNEUROSCI.4372-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung WL, Briggs SD, Allis CD. Acetylation and chromosomal functions. Curr Opin Cell Biol. 2000;12:326–333. doi: 10.1016/s0955-0674(00)00096-x. [DOI] [PubMed] [Google Scholar]
- Choi DC, Furay AR, Evanson NK, Ostrander MM, Ulrich-Lai YM, Herman JP. Bed nucleus of the stria terminalis subregions differentially regulate hypothalamic-pituitary-adrenal axis activity: implications for the integration of limbic inputs. J Neurosci. 2007;27:2025–2034. doi: 10.1523/JNEUROSCI.4301-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- Cloninger CR. The science of well being: An integrated approach to mental health and its disorders. Psychiatr Danub. 2006;18:218–224. [PubMed] [Google Scholar]
- Conrad CD. Chronic stress-induced hippocampal vulnerability: the glucocorticoid vulnerability hypothesis. Rev Neurosci. 2008;19:395–411. doi: 10.1515/revneuro.2008.19.6.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad CD, McLaughlin KJ, Harman JS, Foltz C, Wieczorek L, Lightner E, Wright RL. Chronic glucocorticoids increase hippocampal vulnerability to neurotoxicity under conditions that produce CA3 dendritic retraction but fail to impair spatial recognition memory. J Neurosci. 2007;27:8278–8285. doi: 10.1523/JNEUROSCI.2121-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook SC, Wellman CL. Chronic stress alters dendritic morphology in rat medial prefrontal cortex. J Neurobiol. 2004;60:236–248. doi: 10.1002/neu.20025. [DOI] [PubMed] [Google Scholar]
- Covington HE, 3rd, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, Fass DM, Renthal W, Rush AJ, 3rd, Wu EY, Ghose S, Krishnan V, Russo SJ, Tamminga C, Haggarty SJ, Nestler EJ. Antidepressant actions of histone deacetylase inhibitors. J Neurosci. 2009;29:11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosio C, Heitz E, Allis CD, Borrelli E, Sassone-Corsi P. Chromatin remodeling and neuronal response: multiple signaling pathways induce specific histone H3 modifications and early gene expression in hippocampal neurons. J Cell Sci. 2003;116:4905–4914. doi: 10.1242/jcs.00804. [DOI] [PubMed] [Google Scholar]
- Cullinan WE, Herman JP, Battaglia DF, Akil H, Watson SJ. Pattern and time course of immediate early gene expression in rat brain following acute stress. Neuroscience. 1995;64:477–505. doi: 10.1016/0306-4522(94)00355-9. [DOI] [PubMed] [Google Scholar]
- Cullinan WE, Herman JP, Watson SJ. Ventral subicular interaction with the hypothalamic paraventricular nucleus: evidence for a relay in the bed nucleus of the stria terminalis. J Comp Neurol. 1993;332:1–20. doi: 10.1002/cne.903320102. [DOI] [PubMed] [Google Scholar]
- Dayas CV, Buller KM, Crane JW, Xu Y, Day TA. Stressor categorization: acute physical and psychological stressors elicit distinctive recruitment patterns in the amygdala and in medullary noradrenergic cell groups. Eur J Neurosci. 2001;14:1143–1152. doi: 10.1046/j.0953-816x.2001.01733.x. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Reul JM, de Ronde FS, Bloemers M, Ratka A. Function and plasticity of brain corticosteroid receptor systems: action of neuropeptides. J Steroid Biochem. 1986;25:723–731. doi: 10.1016/0022-4731(86)90301-8. [DOI] [PubMed] [Google Scholar]
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delahanty DL, Raimonde AJ, Spoonster E, Cullado M. Injury severity, prior trauma history, urinary cortisol levels, and acute PTSD in motor vehicle accident victims. J Anxiety Disord. 2003;17:149–164. doi: 10.1016/s0887-6185(02)00185-8. [DOI] [PubMed] [Google Scholar]
- Dellu F, Piazza PV, Mayo W, Le Moal M, Simon H. Novelty-seeking in rats--biobehavioral characteristics and possible relationship with the sensation-seeking trait in man. Neuropsychobiology. 1996;34:136–145. doi: 10.1159/000119305. [DOI] [PubMed] [Google Scholar]
- Dias-Ferreira E, Sousa JC, Melo I, Morgado P, Mesquita AR, Cerqueira JJ, Costa RM, Sousa N. Chronic stress causes frontostriatal reorganization and affects decision-making. Science. 2009;325:621–625. doi: 10.1126/science.1171203. [DOI] [PubMed] [Google Scholar]
- Diorio D, Viau V, Meaney MJ. The role of the medial prefrontal cortex (cingulate gyrus) in the regulation of hypothalamic-pituitary-adrenal responses to stress. J Neurosci. 1993;13:3839–3847. doi: 10.1523/JNEUROSCI.13-09-03839.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong HW, Petrovich GD, Watts AG, Swanson LW. Basic organization of projections from the oval and fusiform nuclei of the bed nuclei of the stria terminalis in adult rat brain. J Comp Neurol. 2001;436:430–455. doi: 10.1002/cne.1079. [DOI] [PubMed] [Google Scholar]
- Drevets WC, Price JL, Simpson JR, Jr, Todd RD, Reich T, Vannier M, Raichle ME. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997;386:824–827. doi: 10.1038/386824a0. [DOI] [PubMed] [Google Scholar]
- Elzinga BM, Schmahl CG, Vermetten E, van Dyck R, Bremner JD. Higher cortisol levels following exposure to traumatic reminders in abuse-related PTSD. Neuropsychopharmacology. 2003;28:1656–1665. doi: 10.1038/sj.npp.1300226. [DOI] [PubMed] [Google Scholar]
- Feldman S, Conforti N. The role of the medial septal nucleus in mediating adrenocortical responses to somatosensory stimulation. J Neurosci Res. 1980;5:19–23. doi: 10.1002/jnr.490050104. [DOI] [PubMed] [Google Scholar]
- Figueiredo HF, Bruestle A, Bodie B, Dolgas CM, Herman JP. The medial prefrontal cortex differentially regulates stress-induced c-fos expression in the forebrain depending on type of stressor. Eur J Neurosci. 2003;18:2357–2364. doi: 10.1046/j.1460-9568.2003.02932.x. [DOI] [PubMed] [Google Scholar]
- Fink G. Stress controversies: Post-traumatic stress disorder, hippocampal volume, gastroduodenal ulceration. J Neuroendo. 2011;23:107–117. doi: 10.1111/j.1365-2826.2010.02089.x. [DOI] [PubMed] [Google Scholar]
- Forsberg EC, Bresnick EH. Histone acetylation beyond promoters: long-range acetylation patterns in the chromatin world. Bioessays. 2001;23:820–830. doi: 10.1002/bies.1117. [DOI] [PubMed] [Google Scholar]
- Gold PW, Gabry KE, Yasuda MR, Chrousos GP. Divergent endocrine abnormalities in melancholic and atypical depression: clinical and pathophysiologic implications. Endocrinol Metab Clin North Am. 2002;31:37–62. vi. doi: 10.1016/s0889-8529(01)00022-6. [DOI] [PubMed] [Google Scholar]
- Guan Z, Giustetto M, Lomvardas S, Kim JH, Miniaci MC, Schwartz JH, Thanos D, Kandel ER. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;111:483–493. doi: 10.1016/s0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- Harro J. Inter-individual differences in neurobiology as vulnerability factors for affective disorders: implications for psychopharmacology. Pharmacol Ther. 2010;125:402–422. doi: 10.1016/j.pharmthera.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Heninger GR, Delgado PL, Charney DS. The revised monoamine theory of depression: a modulatory role for monoamines, based on new findings from monoamine depletion experiments in humans. Pharmacopsychiatry. 1996;29:2–11. doi: 10.1055/s-2007-979535. [DOI] [PubMed] [Google Scholar]
- Herman JP. Regulation of adrenocorticosteroid receptor mRNA expression in the central nervous system. Cell Mol Neurobiol. 1993;13:349–372. doi: 10.1007/BF00711577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP, Adams D, Prewitt C. Regulatory changes in neuroendocrine stress-integrative circuitry produced by a variable stress paradigm. Neuroendocrinology. 1995;61:180–190. doi: 10.1159/000126839. [DOI] [PubMed] [Google Scholar]
- Herman JP, Cullinan WE. Neurocircuitry of stress: Central control of the hypothalamo-pituitary-adrenocortical axis. TINS. 1997;20:78–83. doi: 10.1016/s0166-2236(96)10069-2. [DOI] [PubMed] [Google Scholar]
- Herman JP, Cullinan WE, Morano MI, Akil H, Watson SJ. Contribution of the ventral subiculum to inhibitory regulation of the hypothalamo-pituitary-adrenocortical axis. J Neuroendocrinol. 1995;7:475–482. doi: 10.1111/j.1365-2826.1995.tb00784.x. [DOI] [PubMed] [Google Scholar]
- Herman JP, Figueiredo H, Mueller NK, Ulrich-Lai Y, Ostrander MM, Choi DC, Cullinan WE. Central mechanisms of stress integration: hierarchical circuitry controlling hypothalamo-pituitary-adrenocortical responsiveness. Front Neuroendocrinol. 2003;24:151–180. doi: 10.1016/j.yfrne.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Heydendael W, Jacobson L. Differential effects of imipramine and phenelzine on corticosteroid receptor gene expression in mouse brain: potential relevance to antidepressant response. Brain Res. 2008;1238:93–107. doi: 10.1016/j.brainres.2008.08.018. [DOI] [PubMed] [Google Scholar]
- Heydendael W, Jacobson L. Glucocorticoid status affects antidepressant regulation of locus coeruleus tyrosine hydroxylase and dorsal raphe tryptophan hydroxylase gene expression. Brain Res. 2009;1288:69–78. doi: 10.1016/j.brainres.2009.06.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heydendael W, Jacobson L. Widespread hypothalamic-pituitary-adrenocortical axis-relevant and mood-relevant effects of chronic fluoxetine treatment on glucocorticoid receptor gene expression in mice. Eur J Neurosci. 2010;31:892–902. doi: 10.1111/j.1460-9568.2010.07131.x. [DOI] [PubMed] [Google Scholar]
- Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharm. 23:477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- Hooks MS, Jones GH, Liem BJ, Justice JB., Jr Sensitization and individual differences to IP amphetamine, cocaine, or caffeine following repeated intra-cranial amphetamine infusions. Ann N Y Acad Sci. 1992a;654:444–447. doi: 10.1111/j.1749-6632.1992.tb25993.x. [DOI] [PubMed] [Google Scholar]
- Hooks MS, Jones GH, Liem BJ, Justice JB., Jr Sensitization and individual differences to IP amphetamine, cocaine, or caffeine following repeated intracranial amphetamine infusions. Pharmacol Biochem Behav. 1992b;43:815–823. doi: 10.1016/0091-3057(92)90413-a. [DOI] [PubMed] [Google Scholar]
- Hooks MS, Jones GH, Smith AD, Neill DB, Justice JB., Jr Individual differences in locomotor activity and sensitization. Pharmacol Biochem Behav. 1991a;38:467–470. doi: 10.1016/0091-3057(91)90308-o. [DOI] [PubMed] [Google Scholar]
- Hooks MS, Jones GH, Smith AD, Neill DB, Justice JB., Jr Response to novelty predicts the locomotor and nucleus accumbens dopamine response to cocaine. Synapse. 1991b;9:121–128. doi: 10.1002/syn.890090206. [DOI] [PubMed] [Google Scholar]
- Ising M, Horstmann S, Kloiber S, Lucae S, Binder EB, Kern N, Kunzel HE, Pfennig A, Uhr M, Holsboer F. Combined dexamethasone/corticotropin releasing hormone test predicts treatment response in major depression - a potential biomarker? Biol Psychiatry. 2007;62:47–54. doi: 10.1016/j.biopsych.2006.07.039. [DOI] [PubMed] [Google Scholar]
- Ito K, Adcock IM. Histone acetylation and histone deacetylation. Mol Biotechnol. 2002;20:99–106. doi: 10.1385/MB:20:1:099. [DOI] [PubMed] [Google Scholar]
- Jacobson L, Sapolsky RM. The role of the hippocampus in feedback regulation of the hypothalamo-pituitary-adrenocortical axis. Endocrine Rev. 1991;12:118–134. doi: 10.1210/edrv-12-2-118. [DOI] [PubMed] [Google Scholar]
- Jaferi A, Bhatnagar S. Corticosterone can act at the posterior paraventricular thalamus to inhibit hypothalamic-pituitary-adrenal activity in animals that habituate to repeated stress. Endocrinology. 2006;147:4917–4930. doi: 10.1210/en.2005-1393. [DOI] [PubMed] [Google Scholar]
- Jama A, Cecchi M, Calvo N, Watson SJ, Akil H. Inter-individual differences in novelty-seeking behavior in rats predict differential responses to desipramine in the forced swim test. Psychopharmacology (Berl) 2008;198:333–340. doi: 10.1007/s00213-008-1126-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janknecht R. The versatile functions of the transcriptional coactivators p300 and CBP and their roles in disease. Histol Histopathol. 2002;17:657–668. doi: 10.14670/HH-17.657. [DOI] [PubMed] [Google Scholar]
- Kabbaj M. Neurobiological bases of individual differences in emotional and stress responsiveness: high responders-low responders model. Arch Neurol. 2004;61:1009–1012. doi: 10.1001/archneur.61.7.1009. [DOI] [PubMed] [Google Scholar]
- Kabbaj M, Akil H. Individual differences in novelty-seeking behavior in rats: a c-fos study. Neuroscience. 2001;106:535–545. doi: 10.1016/s0306-4522(01)00291-3. [DOI] [PubMed] [Google Scholar]
- Kabbaj M, Devine DP, Savage VR, Akil H. Neurobiological correlates of individual differences in novelty-seeking behavior in the rat: differential expression of stress-related molecules. J Neurosci. 2000;20:6983–6988. doi: 10.1523/JNEUROSCI.20-18-06983.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller-Wood M, Dallman MF. Corticosteroid inhibition of ACTH secretion. Endocrine Rev. 1984;5:1–24. doi: 10.1210/edrv-5-1-1. [DOI] [PubMed] [Google Scholar]
- Kessler D, Sharp D, Lewis G. Screening for depression in primary care. Br J Gen Pract. 2005;55:659–660. [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB. Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry. 1995;52:1048–1060. doi: 10.1001/archpsyc.1995.03950240066012. [DOI] [PubMed] [Google Scholar]
- Kier A, Han J, Jacobson L. Chronic treatment with the monoamine oxidase inhibitor phenelzine increases hypothalamic-pituitary-adrenocortical activity in male C57BL/6 mice: relevance to atypical depression. Endocrinology. 2005;146:1338–1347. doi: 10.1210/en.2004-0650. [DOI] [PubMed] [Google Scholar]
- Koolhaas JM, Meerlo P, De Boer SF, Strubbe JH, Bohus B. The temporal dynamics of the stress response. Neurosci Biobehav Rev. 1997;21:775–782. doi: 10.1016/s0149-7634(96)00057-7. [DOI] [PubMed] [Google Scholar]
- Kosten TR, Mason JW, Giller EL, Ostroff RB, Harkness L. Sustained urinary norepinephrine and epinephrine elevation in post-traumatic stress disorder. Psychoneuroendocrinology. 1987;12:13–20. doi: 10.1016/0306-4530(87)90017-5. [DOI] [PubMed] [Google Scholar]
- Kovacs KJ, Makara GB. Corticosterone and dexamethasone act at different brain sites to inhibit adrenalectomy-induced adrenocorticotropin hypersecretion. Brain Res. 1988;474:205–210. doi: 10.1016/0006-8993(88)90435-0. [DOI] [PubMed] [Google Scholar]
- Levitan RD, Vaccarino FJ, Brown GM, Kennedy SH. Low-dose dexamethasone challenge in women with atypical major depression: pilot study. J Psychiatry Neurosci. 2002;27:47–51. [PMC free article] [PubMed] [Google Scholar]
- Li HY, Sawchenko PE. Hypothalamic effector neurons and extended circuitries activated in “neurogenic” stress: a comparison of footshock effects exerted acutely, chronically, and in animals with controlled glucocorticoid levels. J Comp Neurol. 1998;393:244–266. [PubMed] [Google Scholar]
- Liston C, Miller MM, Goldwater DS, Radley JJ, Rocher AB, Hof PR, Morrison JH, McEwen BS. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J Neurosci. 2006;26:7870–7874. doi: 10.1523/JNEUROSCI.1184-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RJ, Aghajanian GK. Stress blunts serotonin- and hypocretin-evoked EPSCs in prefrontal cortex: role of corticosterone-mediated apical dendritic atrophy. Proc Natl Acad Sci U S A. 2008;105:359–364. doi: 10.1073/pnas.0706679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo WS, Trievel RC, Rojas JR, Duggan L, Hsu JY, Allis CD, Marmorstein R, Berger SL. Phosphorylation of serine 10 in histone H3 is functionally linked in vitro and in vivo to Gcn5-mediated acetylation at lysine 14. Mol Cell. 2000;5:917–926. doi: 10.1016/s1097-2765(00)80257-9. [DOI] [PubMed] [Google Scholar]
- Lowry CA. Functional subsets of serotonergic neurones: implications for control of the hypothalamic-pituitary-adrenal axis. J Neuroendocrinol. 2002;14:911–923. doi: 10.1046/j.1365-2826.2002.00861.x. [DOI] [PubMed] [Google Scholar]
- Makino S, Smith MA, Gold PW. Regulatory role of glucocorticoids and glucocorticoid receptor mRNA levels on tyrosine hydroxylase gene expression in the locus coeruleus during repeated immobilization stress. Brain Res. 2002;943:216–223. doi: 10.1016/s0006-8993(02)02647-1. [DOI] [PubMed] [Google Scholar]
- Mason JW, Giller EL, Kosten TR, Ostroff RB, Podd L. Urinary free-cortisol levels in posttraumatic stress disorder patients. J Nerv Ment Dis. 1986;174:145–149. doi: 10.1097/00005053-198603000-00003. [DOI] [PubMed] [Google Scholar]
- McEwen B. Re-examination of the glucocorticoid hypothesis of stress and aging. Prog Brain Res. 1992;93:365–381. doi: 10.1016/s0079-6123(08)64585-9. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Stress, adaptation, and disease. Allostasis and allostatic load. Ann N Y Acad Sci. 1998;840:33–44. doi: 10.1111/j.1749-6632.1998.tb09546.x. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Plasticity of the hippocampus: adaptation to chronic stress and allostatic load. Ann N Y Acad Sci. 2001;933:265–277. doi: 10.1111/j.1749-6632.2001.tb05830.x. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Stellar E. Stress and the individual. Mechanisms leading to disease. Arch Intern Med. 1993;153:2093–2101. [PubMed] [Google Scholar]
- McGaugh JL, Roozendaal B. Role of adrenal stress hormones in forming lasting memories in the brain. Curr Opin Neurobiol. 2002;12:205–210. doi: 10.1016/s0959-4388(02)00306-9. [DOI] [PubMed] [Google Scholar]
- McManus KJ, Hendzel MJ. CBP, a transcriptional coactivator and acetyltransferase. Biochem Cell Biol. 2001;79:253–266. [PubMed] [Google Scholar]
- Meerlo P, Overkamp GJ, Daan S, Van Den Hoofdakker RH, Koolhaas JM. Changes in behaviour and body weight following a single or double social defeat in rats. Stress. 1996;1:21–32. doi: 10.3109/10253899609001093. [DOI] [PubMed] [Google Scholar]
- Mukherjee K, Knisely A, Jacobson L. Partial glucocorticoid agonist-like effects of imipramine on hypothalamic-pituitary-adrenocortical activity, thymus weight, and hippocampal glucocorticoid receptors in male C57BL/6 mice. Endocrinology. 2004;145:4185–4191. doi: 10.1210/en.2004-0147. [DOI] [PubMed] [Google Scholar]
- Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relations to pharmacological actions. Endocrine Rev. 1984;5:25–44. doi: 10.1210/edrv-5-1-25. [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- Ottenweller JE, Natelson BH, Pitman DL, Drastal SD. Adrenocortical and behavioral responses to repeated stressors: toward an animal model of chronic stress and stress-related mental illness. Biol Psychiatry. 1989;26:829–841. doi: 10.1016/0006-3223(89)90123-6. [DOI] [PubMed] [Google Scholar]
- Pande AC, Birkett M, Fechner-Bates S, Haskett RF, Greden JF. Fluoxetine versus phenelzine in atypical depression. Biol Psychiatry. 1996;40:1017–1020. doi: 10.1016/0006-3223(95)00628-1. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry. 2001;49:391–404. doi: 10.1016/s0006-3223(00)01088-x. [DOI] [PubMed] [Google Scholar]
- Pego JM, Morgado P, Pinto LG, Cerqueira JJ, Almeida OF, Sousa N. Dissociation of the morphological correlates of stress-induced anxiety and fear. Eur J Neurosci. 2008;27:1503–1516. doi: 10.1111/j.1460-9568.2008.06112.x. [DOI] [PubMed] [Google Scholar]
- Perry PJ. Pharmacotherapy for major depression with melancholic features: relative efficacy of tricyclic versus selective serotonin reuptake inhibitor antidepressants. J Affect Disord. 1996;39:1–6. doi: 10.1016/0165-0327(96)00014-6. [DOI] [PubMed] [Google Scholar]
- Piazza PV, Deminiere JM, Le Moal M, Simon H. Factors that predict individual vulnerability to amphetamine self-administration. Science. 1989;245:1511–1513. doi: 10.1126/science.2781295. [DOI] [PubMed] [Google Scholar]
- Pierre PJ, Vezina P. Predisposition to self-administer amphetamine: the contribution of response to novelty and prior exposure to the drug. Psychopharmacology (Berl) 1997;129:277–284. doi: 10.1007/s002130050191. [DOI] [PubMed] [Google Scholar]
- Radley JJ, Arias CM, Sawchenko PE. Regional differentiation of the medial prefrontal cortex in regulating adaptive responses to acute emotional stress. J Neurosci. 2006a;26:12967–12976. doi: 10.1523/JNEUROSCI.4297-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radley JJ, Gosselink KL, Sawchenko PE. A discrete GABAergic relay mediates medial prefrontal cortical inhibition of the neuroendocrine stress response. J Neurosci. 2009;29:7330–7340. doi: 10.1523/JNEUROSCI.5924-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radley JJ, Rocher AB, Janssen WG, Hof PR, McEwen BS, Morrison JH. Reversibility of apical dendritic retraction in the rat medial prefrontal cortex following repeated stress. Exp Neurol. 2005;196:199–203. doi: 10.1016/j.expneurol.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Radley JJ, Rocher AB, Miller M, Janssen WG, Liston C, Hof PR, McEwen BS, Morrison JH. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb Cortex. 2006b;16:313–320. doi: 10.1093/cercor/bhi104. [DOI] [PubMed] [Google Scholar]
- Radley JJ, Rocher AB, Rodriguez A, Ehlenberger DB, Dammann M, McEwen BS, Morrison JH, Wearne SL, Hof PR. Repeated stress alters dendritic spine morphology in the rat medial prefrontal cortex. J Comp Neurol. 2008;507:1141–1150. doi: 10.1002/cne.21588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radley JJ, Sawchenko PE. A common substrate for prefrontal and hippocampal inhibition of the neuroendocrine stress response. J Neurosci. 31:9683–9695. doi: 10.1523/JNEUROSCI.6040-10.2011. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radley JJ, Sisti HM, Hao J, Rocher AB, McCall T, Hof PR, McEwen BS, Morrison JH. Chronic behavioral stress induces apical dendritic reorganization in pyramidal neurons of the medial prefrontal cortex. Neuroscience. 2004;125:1–6. doi: 10.1016/j.neuroscience.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Resnick HS, Yehuda R, Pitman RK, Foy DW. Effect of previous trauma on acute plasma cortisol level following rape. Am J Psychiatry. 1995;152:1675–1677. doi: 10.1176/ajp.152.11.1675. [DOI] [PubMed] [Google Scholar]
- Reul JMHM, Chandramohan Y. Epigentic mechansims in stress-related-memory formation. Psychoneuroendocrinology. 2007;32:521–525. doi: 10.1016/j.psyneuen.2007.03.016. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger D, Kelliher K, Einstein M, Henderson SC, Morrison JH, Hof PR, Wearne SL. Automated reconstruction of three-dimensional neuronal morphology from laser scanning microscopy images. Methods. 2003;30:94–105. doi: 10.1016/s1046-2023(03)00011-2. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Barsegyan A, Lee S. Adrenal stress hormones, amygdala activation, and memory for emotionally arousing experiences. Prog Brain Res. 2008;167:79–97. doi: 10.1016/S0079-6123(07)67006-X. [DOI] [PubMed] [Google Scholar]
- Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- Ruis MA, te Brake JH, Buwalda B, De Boer SF, Meerlo P, Korte SM, Blokhuis HJ, Koolhaas JM. Housing familiar male wildtype rats together reduces the long-term adverse behavioural and physiological effects of social defeat. Psychoneuroendocrinology. 1999;24:285–300. doi: 10.1016/s0306-4530(98)00050-x. [DOI] [PubMed] [Google Scholar]
- Rygula R, Abumaria N, Flugge G, Fuchs E, Ruther E, Havemann-Reinecke U. Anhedonia and motivational deficits in rats: impact of chronic social stress. Behav Brain Res. 2005;162:127–134. doi: 10.1016/j.bbr.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM. Why stress is bad for your brain. Science. 1996;273:749–750. doi: 10.1126/science.273.5276.749. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Krey LC, McEwen BS. Glucocorticoid-sensitive hippocampal neurons are involved in terminating the adrenocortical stress response. Proc Natl Acad Sci U S A. 1984;81:6174–6177. doi: 10.1073/pnas.81.19.6174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM, Krey LC, McEwen BS. Prolonged glucocorticoid exposure reduces hippocampal neuron number: implications for aging. J Neurosci. 1985;5:1222–1227. doi: 10.1523/JNEUROSCI.05-05-01222.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawchenko PE. Evidence for a local site of action for glucocorticoids in inhibiting CRF and vasopressin expression in the paraventricular nucleus. Brain Res. 1987;403:213–223. doi: 10.1016/0006-8993(87)90058-8. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Deutch AY, Roth RH, Bunney BS. Topographical organization of the efferent projections of the medial prefrontal cortex in the rat: an anterograde tract-tracing study with Phaseolus vulgaris leucoagglutinin. J Comp Neurol. 1989;290:213–242. doi: 10.1002/cne.902900205. [DOI] [PubMed] [Google Scholar]
- Shalev AY, Videlock EJ, Peleg T, Segman R, Pitman RK, Yehuda R. Stress hormones and post-traumatic stress disorder in civilian trauma victims: a longitudinal study. Part I: HPA axis responses. Int J Neuropsychopharmacol. 2008;11:365–372. doi: 10.1017/S1461145707008127. [DOI] [PubMed] [Google Scholar]
- Sheline YI, Gado MH, Kraemer HC. Untreated depression and hippocampal volume loss. Am J Psychiatry. 2003;160:1516–1518. doi: 10.1176/appi.ajp.160.8.1516. [DOI] [PubMed] [Google Scholar]
- Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW. Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci U S A. 1996;93:3908–3913. doi: 10.1073/pnas.93.9.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JW, Geerling JC, Loewy AD. Inputs to the ventrolateral bed nucleus of the stria terminalis. J Comp Neurol. 2008;511:628–657. doi: 10.1002/cne.21870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwick SM, Krystal JH, Bremner JD, Morgan CA, 3rd, Nicolaou AL, Nagy LM, Johnson DR, Heninger GR, Charney DS. Noradrenergic and serotonergic function in posttraumatic stress disorder. Arch Gen Psychiatry. 1997;54:749–758. doi: 10.1001/archpsyc.1997.01830200083012. [DOI] [PubMed] [Google Scholar]
- Spencer SJ, Buller KM, Day TA. Medial prefrontal cortex control of the paraventricular hypothalamic nucleus response to psychological stress: possible role of the bed nucleus of the stria terminalis. J Comp Neurol. 2005;481:363–376. doi: 10.1002/cne.20376. [DOI] [PubMed] [Google Scholar]
- Stewart JW, McGrath PJ, Quitkin FM, Klein DF. DSM-IV depression with atypical features: is it valid? Neuropsychopharmacology. 2009;34:2625–2632. doi: 10.1038/npp.2009.99. [DOI] [PubMed] [Google Scholar]
- Sullivan RM, Gratton A. Lateralized effects of medial prefrontal cortex lesions on neuroendocrine and autonomic stress responses in rats. J Neurosci. 1999;19:2834–2840. doi: 10.1523/JNEUROSCI.19-07-02834.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson LW, Cowan WM. An autoradiographic study of the organization of the efferent connections of the hippocampal formation in the rat. J Comp Neurol. 1977;172:49–84. doi: 10.1002/cne.901720104. [DOI] [PubMed] [Google Scholar]
- Taghzouti K, Lamarque S, Kharouby M, Simon H. Interindividual differences in active and passive behaviors in the forced-swimming test: implications for animal models of psychopathology. Biol Psychiatry. 1999;45:750–758. doi: 10.1016/s0006-3223(98)00156-5. [DOI] [PubMed] [Google Scholar]
- Tidey JW, Miczek KA. Acquisition of cocaine self-administration after social stress: role of accumbens dopamine. Psychopharmacology (Berl) 1997;130:203–212. doi: 10.1007/s002130050230. [DOI] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Tsankova NM, Kumar A, Nestler EJ. Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J Neurosci. 2004;24:5603–5610. doi: 10.1523/JNEUROSCI.0589-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaswani M, Linda FK, Ramesh S. Role of selective serotonin reuptake inhibitors in psychiatric disorders: a comprehensive review. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:85–102. doi: 10.1016/s0278-5846(02)00338-x. [DOI] [PubMed] [Google Scholar]
- Verheij MM, Veenvliet JV, Groot Kormelink T, Steenhof M, Cools AR. Individual differences in the sensitivity to serotonergic drugs: a pharmacobehavioural approach using rats selected on the basis of their response to novelty. Psychopharmacology (Berl) 2009;205:441–455. doi: 10.1007/s00213-009-1552-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade PA. Transcriptional control at regulatory checkpoints by histone deacetylases: molecular connections between cancer and chromatin. Hum Mol Genet. 2001;10:693–698. doi: 10.1093/hmg/10.7.693. [DOI] [PubMed] [Google Scholar]
- Walaas I, Fonnum F. Biochemical evidence for glutamate as a transmitter in hippocampal efferents to the basal forebrain and hypothalamus in the rat brain. Neuroscience. 1980;5:1691–1698. doi: 10.1016/0306-4522(80)90088-3. [DOI] [PubMed] [Google Scholar]
- Warrington TP, Bostwick JM. Psychiatric adverse effects of corticosteroids. Mayo Clin Proc. 2006;81:1361–1367. doi: 10.4065/81.10.1361. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Gould E, McEwen BS. Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res. 1992;588:341–345. doi: 10.1016/0006-8993(92)91597-8. [DOI] [PubMed] [Google Scholar]
- Wearne SL, Rodriguez A, Ehlenberger DB, Rocher AB, Henderson SC, Hof PR. New techniques for imaging, digitization and analysis of three-dimensional neural morphology on multiple scales. Neuroscience. 2005;136:661–680. doi: 10.1016/j.neuroscience.2005.05.053. [DOI] [PubMed] [Google Scholar]
- Wilkinson MB, Xiao G, Kumar A, LaPlant Q, Renthal W, Sikder D, Kodadek TJ, Nestler EJ. Imipramine treatment and resiliency exhibit similar chromatin regulation in the mouse nucleus accumbens in depression models. J Neurosci. 2009;29:7820–7832. doi: 10.1523/JNEUROSCI.0932-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willner P. Validity, reliability and utility of the chronic mild stress model of depression: a 10-year review and evaluation. Psychopharmacology (Berl) 1997;134:319–329. doi: 10.1007/s002130050456. [DOI] [PubMed] [Google Scholar]
- Yehuda R. Post-traumatic stress disorder. N Engl J Med. 2002;346:108–114. doi: 10.1056/NEJMra012941. [DOI] [PubMed] [Google Scholar]
- Yehuda R. Neuroendocrine aspects of PTSD. In: Holsboer F, Ströhle A, editors. Anxiety and AnxiolyticDrugs. Berlin: Springer; 2005. pp. 371–403. [Google Scholar]
- Yehuda R, Bierer LM. Transgenerational transmission of cortisol and PTSD risk. Prog Brain Res. 2008;167:121–135. doi: 10.1016/S0079-6123(07)67009-5. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Blair W, Labinsky E, Bierer LM. Effects of parental PTSD on the cortisol response to dexamethasone administration in their adult offspring. Am J Psychiatry. 2007;164:163–166. doi: 10.1176/ajp.2007.164.1.163. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Boisoneau D, Lowy MT, Giller EL., Jr Dose-response changes in plasma cortisol and lymphocyte glucocorticoid receptors following dexamethasone administration in combat veterans with and without posttraumatic stress disorder. Arch Gen Psychiatry. 1995;52:583–593. doi: 10.1001/archpsyc.1995.03950190065010. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Engel SM, Brand SR, Seckl J, Marcus SM, Berkowitz GS. Transgenerational effects of posttraumatic stress disorder in babies of mothers exposed to the World Trade Center attacks during pregnancy. J Clin Endocrinol Metab. 2005;90:4115–4118. doi: 10.1210/jc.2005-0550. [DOI] [PubMed] [Google Scholar]
- Yehuda R, LeDoux J. Response variation following trauma: a translational neuroscience approach to understanding PTSD. Neuron. 2007;56:19–32. doi: 10.1016/j.neuron.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Yehuda R, McFarlane AC. Conflict between current knowledge about posttraumatic stress disorder and its original conceptual basis. Am J Psychiatry. 1995;152:1705–1713. doi: 10.1176/ajp.152.12.1705. [DOI] [PubMed] [Google Scholar]
- Yehuda R, McFarlane AC, Shalev AY. Predicting the development of posttraumatic stress disorder from the acute response to a traumatic event. Biol Psychiatry. 1998a;44:1305–1313. doi: 10.1016/s0006-3223(98)00276-5. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Siever LJ, Teicher MH, Levengood RA, Gerber DK, Schmeidler J, Yang RK. Plasma norepinephrine and 3-methoxy-4-hydroxyphenylglycol concentrations and severity of depression in combat posttraumatic stress disorder and major depressive disorder. Biol Psychiatry. 1998b;44:56–63. doi: 10.1016/s0006-3223(98)80007-3. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Teicher MH, Trestman RL, Levengood RA, Siever LJ. Cortisol regulation in posttraumatic stress disorder and major depression: a chronobiological analysis. Biol Psychiatry. 1996;40:79–88. doi: 10.1016/0006-3223(95)00451-3. [DOI] [PubMed] [Google Scholar]
- Young EA, Haskett RF, Grunhaus L, Pande A, Weinberg VM, Watson SJ, Akil H. Increased evening activation of the hypothalamic-pituitary-adrenal axis in depressed patients. Arch Gen Psychiatry. 1994;51:701–707. doi: 10.1001/archpsyc.1994.03950090033005. [DOI] [PubMed] [Google Scholar]
- Ziegler DR, Cass WA, Herman JP. Excitatory influence of the locus coeruleus in hypothalamic-pituitary-adrenocortical axis responses to stress. J Neuroendocrinol. 1999;11:361–369. doi: 10.1046/j.1365-2826.1999.00337.x. [DOI] [PubMed] [Google Scholar]