

Abstract
Carbohydrates serve many structural and functional roles in biology. While the majority of monosaccharides are characterized by the chemical composition: (CH2O)n, modifications including deoxygenation, C-alkylation, amination, O- and N-methylation, which are characteristic of many sugar appendages of secondary metabolites, are not uncommon. Interestingly, some sugar molecules are formed via modifications including amine oxidation, sulfur incorporation, and “high-carbon” chain attachment. Most of these unusual sugars have been identified over the past several decades as components of microbially produced natural products, although a few high-carbon sugars are also found in the lipooligosaccharides of the outer cell walls of Gram-negative bacteria. Despite their broad distribution in nature, these sugars are considered “rare” due to their relative scarcity. The biosynthetic steps that underlie their formation continue to perplex researchers to this day and many questions regarding key transformations remain unanswered. This review will focus on our current understanding of the biosynthesis of unusual sugars bearing oxidized amine substituents, thio-functional groups, and high-carbon chains.
1. Introduction
Carbohydrates are essential biomolecules for all living organisms and exhibit great diversity in their chemical structures. The name, carbohydrate, originally stems from their chemical composition, which is normally (CH2O)n. However, a wide variety of deoxysugars are found in lipopolysaccharides, glycoproteins, glycolipids, and numerous secondary metabolites. In many cases, these unusual sugars have been shown to be indispensable for the activity of the parent molecules. A great number of gene clusters dedicated to the production of deoxysugars have been identified and the pathways leading to their formation have been extensively studied over the past few decades.1,2 Generally, the monosaccharides are first activated at C-1 by the substitution of nucleoside diphosphate (NDP), which functions as a handle for recognition by subsequent biosynthetic enzymes. Except for 2-deoxyribose, which forms the backbone of DNA, most naturally occurring deoxysugars are 6-deoxyhexoses. They are derived from a common precursor, NDP-4-keto-6-deoxy-α-D-hexose, which is further modified by a varied combination of reduction, isomerization, methylation or transamination reactions to give a diverse array of unusual sugars.
The complexity seen in unusual sugar structures is greatly enhanced by the incorporation of heteroatoms, such as nitrogen and sulfur. The intrinsic chemical properties of these unusual sugars can be significantly altered by the installed heteroatom-containing substituents. Moreover, interconversion between the oxidation states of these heteroatom-containing functional groups attenuates the electronegativity, hydrophilicity and steric hindrance of the overall structures. In addition to the substituents they carry, some unusual sugars are distinguished by a longer than usual carbon chain length. Such “high-carbon” sugars have been identified among microbially-derived antibiotic compounds and in the outer cell walls of some Gram-negative bacteria.
Since many natural products are glycosylated, modifying the structure and/or composition of the sugar components holds promise for varying and/or enhancing the biological activities of the parent systems. Thus, it is critical to have a full understanding of the biosynthesis of these unusual sugars so that suitable strategies to control, mimic, or alter their formation can be developed. Monosaccharides that contain oxidized amines, C-S linkages, or high carbon numbers are rare. In many cases, the key biosynthetic transformations leading to these unusual sugars remain obscure or only partially understood to date. This review will cover what is known regarding their biosynthesis and what work remains to be done to achieve a complete understanding.
2. Sugars Containing Oxidized Amines
Deoxyaminosugars comprise an important class of deoxysugars, among which 2-N-acetylamino sugars are widespread. However, many deoxyaminosugars having a primary amino group at either C-3, C-4, or C-6 are also known. The amino groups in these sugars are universally introduced into the corresponding ketosugar substrates via 5′-phosphate (PLP)-dependent transamination reactions. The biosynthesis of deoxyaminosugars and the catalytic mechanism utilized by sugar aminotransferases (SATs) have been recently reviewed.3,4 Despite the abundance of deoxyaminosugars, monosaccharides carrying nitrogen-functional groups with higher oxidation states are rarely observed in nature. In a review highlighting aminosugar diversity and biosynthesis Timmons and Thorson noted that the existence of oxidized aminosugars extends the diversity of glycosylated natural products.5 Coincidentally, after this review was published, several groups demonstrated the in vitro characterization of aminosugar N-oxygenase, which was considered a major breakthrough in the study of deoxyaminosugar biosynthesis. In this section of the review, we summarize current understanding regarding the biosynthesis of sugars containing hydroxylamino-, nitroso- and nitro groups, with a focus on recent progress in the enzymatic oxidation of aminosugars.
2.1. Occurrence and biosynthesis of hydroxyaminosugars
A hydroxyaminosugar is present in the structures of both calicheamicin (1) from Micromonospora echinospora ssp. Calichensis6 and esperamicin (2) from Actinomadura verrucosospora.7,8 These antibiotics belong to the 10-membered enediyne ring family and display potent antitumor activity. Calicheamicin and esperamicin share closely related chemical structures that include a rare 4-thiosugar (3) in addition to the hydroxyaminosugar moiety (4). The elucidation of the gene clusters for the biosynthesis of calicheamicin9 and esperamicin10 have led to proposals regarding the formation of hydroxyaminosugars. Based on well-established studies of deoxyaminosugar biosynthesis3 and an example of P450 N-oxygenase in nocardicin A biosynthesis (NocL),11 Johnson and Thorson proposed two P450-dependent enzymes (CalE10 and CalO2) as candidates for the N-oxygenases in calicheamicin biosynthesis12. Subsequent in vitro studies of recombinant CalE10 and CalO2 using a series of putative thymidine diphosphate (TDP)-sugar substrates demonstrated that CalE10 is indeed an aminosugar N-oxygenase (no substrate binding or oxidation was observed with CalO2). As shown in Scheme 1, CalE10 catalyzes the oxidation of TDP-4-amino-4,6-dideoxy-α-D-glucose (6) to afford two products: the major hydroxyaminosugar (7) and the minor nitrosugar (8).12 This enzymatic reaction shows regiospecificity toward C-4 amino groups and the oxidation occurs at the NDP-sugar stage prior to glycosyltransfer to the enediyne scaffold. Although the nitrosugar is the minor product in this case, CalE10 remains the only known sugar N-oxygenase with demonstrated capability to catalyze an overall 6-e− transfer to fully oxidize an amino group to a nitro group.
Scheme 1.

Proposed pathway for the biosynthesis of hydroxyaminosugar
2.2. Occurrence of nitrososugars and nitrosugars
Only four naturally occurring nitrosugars have been identified.13 These include D-kijanose (9), D-rubranitrose (14), L-evernitrose (16) and L-decilonitrose (18). D-Kijanose (9) is a highly functionalized nitrosugar that was first found in kijanimicin (10) from Actinomadura kijaniata.14 The occurrence of D-kijanose is thus far limited to a family of spirotetronate natural products including kijanimicin (10),14 tetrocarcin (11),15,16 lobophorin (12)17 and arisostatin (13).18 D-Rubranitrose (14) is the sugar component of rubradirin (15), an antibiotic belonging to the ansamycin family produced by Streptomyces achromogenes var. rubradiris.19 The first structural report of rubradirin identified rubranitrose as an L-sugar.20 It was later revised to be a D-sugar when compared to chemically synthesized L- and D-rubranitrose.21,22 The L-isomer of rubranitrose is also called L-evernitrose (16) as it was originally identified in the structure of everninomycins (17) from Micromonospora carbonaceae.23–25 The last example, L-decilonitrose (18) was found in decilorubicin (19), an anthracycline antibiotic from Streptomyces virginiae.26,27 It also exists in several structurally related natural products including arugomycin (20),28,29 cororubicin (21),30 and viriplanin A (22).31,32 Interestingly, these anthracycline-type antibiotics all contain a bicyclo-aminosugar moiety that is attached to the aglycone via a C-glycosidic bond in addition to the typical O-glycosidic linkage.
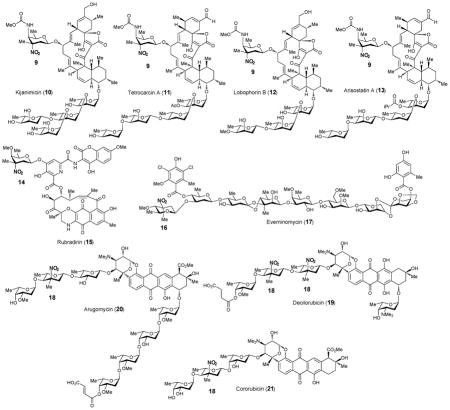
2.3. Biosynthesis of nitrosugars
Two basic routes for the biosynthesis of nitro-containing natural products, including nitrosugars, have been identified: sequential N-oxidation of the respective amines and direct nitration of appropriate precursors.33 In the former case, generation of the nitrated natural product is usually accompanied by the co-production of minor metabolites containing an unoxidized or a partially oxidized amino group. For example, derivatives of kijanimicin (10) bearing a 3-amino-D-kijanose rather than kijanose (9) have been identified.34 A rubradirin analogue, protorubradirin (24), containing a 3-nitroso-D-rubranitrose (23) also exists in nature35.
These findings strongly suggest that the nitro moiety in these sugars is biosynthesized via consecutive N-oxidation of the corresponding aminosugars. In the case of 14, the nitrososugar (23) generated as an intermediate may be prematurely released and then incorporated into 24.35 The isolation and sequencing of the gene clusters for the nitrosugar-containing natural products, kijanimicin (10),34 tetrocarcin (11),36 rubradirin (15)37 and everninomycin (17)38 allowed the postulation of the biosynthetic pathways for their corresponding nitrosugars (see Scheme 2). While minor variations exist for the proposed pathways, the key N-oxidation step is believed to be catalyzed by a flavin-dependent enzyme, encoded by a gene that is conserved among the above gene clusters for nitrosugar-containing secondary metabolites. These include KijD3 of the kijanimicin cluster of A. kijaniata,34 TcaB10 of the tetrocarcin cluster of Micromonospora chalcea,36 RubN8 of the ruradirin cluster of S. achromogenes var. rubradiris,37 and ORF36 (or EvdC) of the everninomycin cluster of M. carbonaceae var. Africana.38
Scheme 2.

Proposed pathways for the biosynthesis of nitrosugars
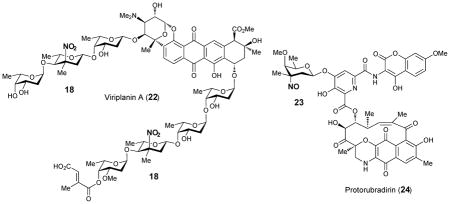
2.4. Characterization of enzymes involved in the biosynthesis of nitrosugars
The biological roles of RubN8 and ORF36 as sugar N-oxygenases were first studied by Bachman and co-workers using chemically synthesized TDP-L-epi-vancosamine (27) as the substrate surrogate.39 The incubation was carried out in the presence of FAD, NADPH, and flavin reductase, with either RubN8 or ORF36. Time course experiments revealed that the amino group of 27 was oxidized to hydroxylamine (29) and then nitroso (30) in sequence (Scheme 3). In a separate experiment, the corresponding 4-ketosugar (26) was shown to be oxidized only to the 3-hydroxylamino-4-ketosugar (31).40 An analogous result was also reported for KijD3, in which partial N-oxidation of a 4-ketosugar substrate (25) to yield a hydroxyamino sugar product (32) was noted (Scheme 3). Crystal structures of both ORF36 and KijD3 reveal a similar protein fold shared with class D flavin-containing monooxygenases.40,41
Scheme 3.
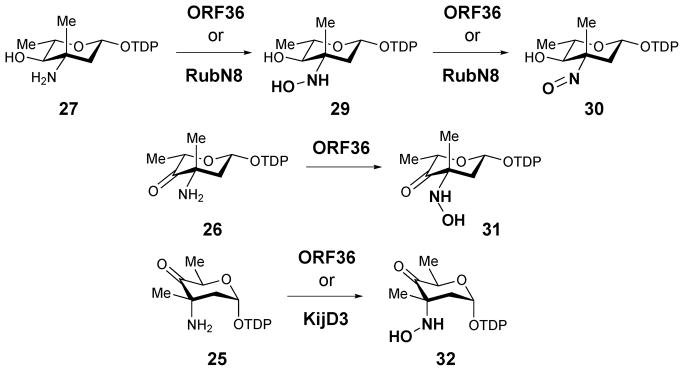
Reactions catalyzed by N-oxygenases
Based on these biochemical and structural data, a mechanism for nitrososugar (30) formation has been proposed (Scheme 4).40 The reaction is initiated by the reduction of the flavin coenzyme in the active site of N-oxygenase by a flavin reductase. In the kij gene cluster, the kijD6 gene, which is located near kijD3 (the N-oxygenase) in the kij gene cluster, encodes a putative NAD(P)H-dependent flavin reductase.34 The gene product of kijD6 was assigned to play such a role in the biosynthesis of kijanose (9). However, the kijD6 homologue is absent in the rubradirin (15) and everninomycin (17) clusters and exogenous flavin reductases may be required. The reduced flavin coenzyme then reacts with molecular oxygen to generate a flavin-C4a-hydroperoxy species (33), which is well known for its ability to hydroxylate a nucleophilic functional group. After transferring the electrophilic hydroxyl group to the amino group of 27, the hydroxylamine product (29) is released and the flavin coenzyme is regenerated. A second round of oxidation of the resultant hydroxylaminosugar (29) along with dehydration of water yields the nitrososugar (30).
Scheme 4.

Proposed mechanism for ORF36-catalyzed N-oxidation
It should be noted that ORF36 and RubN8 convert their respective aminosugar substrate (27) to the corresponding nitrososugar (30) instead of the anticipated nitrosugars (28) during in vitro assays,39 and prolonged incubation under aerobic conditions did not make a difference. Similarly, protorubradirin (24) is produced at a significant level during fermentation, and the nitroso group of its sugar component 23 is readily oxidized to the nitro group (as in 14) upon exposure to ambient light and air.35 It is therefore possible that nitrososugars are the immediate products generated by enzymatic N-oxidation during D-rubranitrose and L-evernitrose biosynthesis and that they are further oxidized to nitrosugars via a latent photochemical process or by reacting with endogeneous reactive oxygen species.
3. Thiosugars
Sulfur is the fifth most abundant element in living organisms, after oxygen, carbon, hydrogen and nitrogen. Sulfur-containing compounds are widely distributed in nature and include amino acids, enzyme cofactors, antioxidants, nucleotides, metal clusters, and numerous secondary metabolites.42 These natural products exhibit a remarkable degree of structural diversity and many of them have potent biological activities. The term, thiosugar, generally describes a carbohydrate moiety that carries a sulfur-containing functional group or is connected to other molecule via a sulfide linkage. Only a few naturally occurring thiosugars have been identified and their relative scarcity has hindered investigation aimed at elucidating their biosynthetic pathways and studying their biological activities.43,44 In this review, the known thiosugars are categorized into four groups based on the chemical nature of their functional groups: sugars containing thiols, sulfides, sulfonium ions, and sulfonic acids. The biosynthetic pathways leading to their formation and the enzymes catalyzing the key sulfur incorporation step will be discussed based on the data that is currently available.
3.1. Group I: Thiosugars carrying a free thiol group
The only members of group I are the angucycline-type antibiotics rhodonocardin A (34) and B isolated from Nocardia sp. No. 53.45 In rhodonocardin A, the benz[a]anthraquinone skeleton is decorated with three sugar units, rhodinose (35), 2-thioglucose (36), and glucose, which are joined via O-glycosidic bonds to C-12b, C-5, and C-4a, respectively. Rhodonocardin B contains only the first two sugars. The biosynthesis of rhodonocardins has not been studied.
3.2. Group II: Thiosugars containing sulfur as part of a sulfide linkage
Sulfides, or thioethers, are compounds containing C-S-C connectivity. The sulfur atom of a thioether can be alkylated to form a sulfonium ion or oxidized to a sulfoxide and/or sulfone under amiable conditions. However, compared to the free thiols, which display a much higher tendency toward oxidation and stronger nucleophilicity, sulfides are more resistant to chemical reactions and are, therefore, more stable. For this reason, most of the known naturally occurring thiosugars belong to group II. In this class of compounds, the sulfur atom either serves as a linkage between the sugar molecule and the aglycone, or replaces the endocyclic ring oxygen of the parent sugar structure. Compounds within group II are further divided into five subgroups according to their overall chemical structure.
3.2.1. Thiosugars containing a thiocycle ring
The first example of this class of compounds, 5-thio-D-glucose, is a synthetic product reported in 1962.46 Since then, a number of 5-thiopyranoses and 4-thiofuranoses have been prepared as inhibitors against enzymes that utilize the parent natural saccharides as substrates.43 Isolated from the marine sponge Clathria pyramida in 1987, 5-thio-D-mannose (37) is the only known free thiocycle-containing monosaccharide identified in nature so far.47 All other thiosugars of this class are found as part of unusual nucleosides or glycopeptides. It has been speculated that 5-thio-D-mannose may be produced by a sulfate reducing bacterial symbiont of C. pyramida, rather than by C. pyramida, itself.
The albomycin antibiotics δ1, δ2 and ε (38a, 38b, and 38c), isolated from Streptomyces subtropicus and several other Streptomyces species, contain a 6-amino-6-deoxy-4-thio-heptofuranose uronic acid nucleoside (39) and a ferrichrome siderophore moiety (40).48,49 These two subunits are linked to one another via a serine residue. The iron-chelating siderophore can be recognized by the bacterial siderophore-dependent iron acquisition system. This allows albomycins to be actively transported across the membranes leading to their designation as “Trojan horse” antibiotics.50 Next, the albomycin is hydrolyzed by cellar peptidases, such as PepN in Escherichia coli, releasing the bioactive nucleoside moiety.51 The seryl-linked 6-amino-6-deoxy-4′-thio-heptofuranose-5′-methyl-4′-imino-cytosyluronic acid fragment, named SB-217452 (41), has been demonstrated to be a potent inhibitor against bacterial seryl-tRNA synthetase (SerRS).52 It was proposed that this peptide-thioribose-nucleoside conjugate (41) mimics an intermediate in the SerRS-catalyzed reaction, thereby inhibiting the aminoacylation activity and disrupting protein biosynthesis. The presence of sulfur in 38 is essential for the observed antimicrobial activity, because the oxygen ring analogue of albomycin is totally inactive.53
Chen and co-workers recently reported the identification of the albomycin biosynthetic gene cluster in Streptomyces sp. ATCC 700974 (abmA-R).54 Gene deletion and complementation experiments indicated that AbmE catalyzes the N4-carbamoylation of cytidine during albomycin biosynthesis. 5 Moreover, AbmI, annotated as a SAM-dependent methyltransferase, was heterologously expressed in E. coli and was shown to be a novel cytidine N3-methyltransferase.
While the cytidine modification steps have been verified and the biosynthesis of the siderophore moiety is also expected to occur via known chemistry, how the 4-thio-heptofuranose uronic acid moiety is biosynthesized is not obvious. Ideas regarding the biosynthesis of the uronic acid moiety itself will be discussed in section 4.8. It is possible that the key sulfur incorporation step proceeds via a mechanism analogous to that employed by biotin (45) synthase (BioB) during biotin biosynthesis (see Scheme 5).55,56 BioB is a member of the radical SAM enzyme superfamily.57 Radical SAM enzymes harbor a [4Fe-4S] cluster that is typically bound by a conserved CX3CX2C motif. In the active +1 redox state, the [4Fe-4S] cluster affects the reductive cleavage of SAM to give methionine and 5′-deoxyadenosyl radical (42). The latter is a powerful oxidant that can abstract a hydrogen atom from substrates to yield substrate radicals, which then undergo transformation via radical-mediated mechanisms. In BioB, 5′-deoxyadenosyl radical (42) abstracts hydrogen from dethiobiotin (43) and the resulting substrate radical (44) abstracts sulfur from a second [2Fe-2S] cluster that is bound to a conserved CX2CX5C motif. Since abmM appears to encode a radical SAM enzyme with both the [4Fe-4S] and [2Fe-2S] cluster binding motifs, the same sulfur insertion mechanism may be operative in AbmM (46 → 47, pathway a). In an alternative hypothetical mechanism, the sulfur is provided by AbmD (a homologue of cysteine desulfhydrase) and incorporated into place by AbmJ, which is also predicted to be a radical SAM enzyme (see Scheme 5, pathway b).
Scheme 5.

Possible mechanism of sulfur incorporation into albomycin catalyzed by radical SAM dependent enzymes
3.2.2. BE-7585A
BE-7585A (48) from Amycolatopsis orientalis subsp. Vinearia is a constitutional isomer of the aforementioned type I thiosugar-containing rhodonocardin A (34).58 Both compounds contain the same benz[a]anthraquinone core and carry the same three sugar subunits. However, in BE-7585A, 2-thioglucose (36) and glucose are joined together as a 1,1′-O-linked disaccharide that is attached to the C-5 of aglycone via a thioether linkage. Since the strains that produce rhodonocardins and BE-7585A are both Actinobacteria, it is possible that the same mechanism of sulfur incorporation is operative during the production of both.
The BE-7585A biosynthetic gene cluster (designated as bex) was recently cloned and sequenced.59 Among all genes in the bex cluster, only the bexX gene appears to be involved in thiosugar biosynthesis since its encoded protein exhibits sequence similarity to thiazole synthase ThiG (38% identity to ThiG from Bacillus subtilis subsp. Subtilis str 168). Because ThiG catalyzes the key sulfur incorporation reaction to form the thiazole moiety (49) during thiamine biosynthesis (see Scheme 6),60,61 BexX is expected to perform an analogous function in the construction of 2-thioglucose in the BE-7585A pathway (Scheme 7). This proposal is supported by trapping experiments using recombinant BexX expressed heterologously in E. coli.62 Upon reduction with NaBH4, the as-isolated BexX was found to form a reduced dehydration adduct with a hexose monophosphate substrate (derived from 51a–51c), later determined to be D-glucose-6-phosphate (50, D-G6P), via an active site lysine residue (K110). When the BexX–D-G6P complex (51c) was further derivatized by the carbonyl-specific labeling reagent, hydroxylamine, the appearance of the corresponding imine BexX–D-G6P–NH2OH was detected by ESI-MS. These results clearly indicate that BexX is the enzyme responsible for priming the sugar to accept a nucleophilic sulfur donor (51c →52, Scheme 7). However, no potential sulfur delivery protein, such as ThiS, which works along with ThiG in thiazole biosynthesis (see Scheme 6), is encoded in the bex gene cluster. It is possible that one of the sulfur carrier proteins utilized in the biosynthesis of primary metabolites may be recruited as the partner protein of BexX to make 2-thioglucose in the biosynthesis of BE-7585A (48).
Scheme 6.
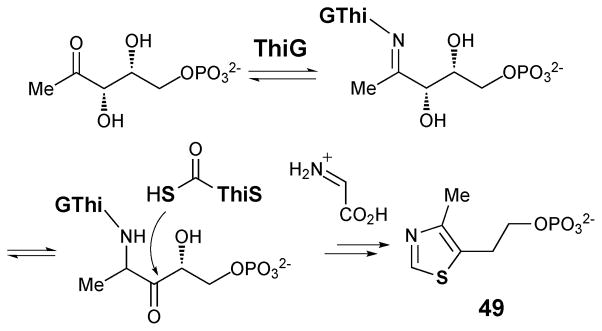
ThiG-catalyzed thiazol formation
Scheme 7.
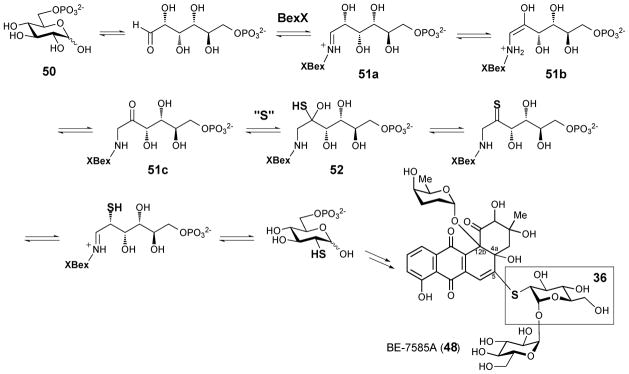
Biosynthesis of 2-thioglucose in BE-7585A
Since the C-5 position of the angucycline core (53) of BE-7585A is highly electrophilic, formation of the thioether linkage may result from a simple Michael addition of the 2-thio group to C-5 of the aglycone followed by a re-aromatization and autoxidation to give 48 (Scheme 8). Such C-S bond formation has been demonstrated in vitro during the conversion of urdamycin A to urdamycin E.63 Interestingly, the 2-thioglucose in rhodonocardin A (34) is attached to C-5 of the aglycone via an O-glycosidic bond. Since a thiol group is a better nucleophile than a hydroxyl group, to avoid the competing thio-addition to the electrophilic C-5 site we propose that sulfur incorporation into rhodonocardin A may occur after the O-glycosylation at C-5. As depicted in Scheme 9, starting from 54, the sulfur incorporation step may follow a similar pathway as seen in the BexX-catalyzed reaction.59,62
Scheme 8.

Possible mechanism for the formation of the thioether linkage in BE-7585 A
Scheme 9.

Proposed biosynthesis of 2-thioglucose in rhodonocardin
3.2.3. Lincosamide-containing antibiotics: lincomycin, celesticetin, and Bu-2545
Lincomycin A (55), isolated from Streptomyces lincolnensis culture media,64,65 displays antibiotic activity against Gram-positive bacteria.66,67 It has been suggested that lincomycin A blocks protein synthesis by binding to the peptidyltransferase domain of the 50S ribosomal subunit due to its structural resemblance to the 3′ end of L-Pro-Met-tRNA and deacylated-tRNA.68 Lincomycin A contains a 1-thiooctose core known as lincosamine (56),69 which is also found in Bu-2545 (57) from Streptomyces strain H230-570 and celesticetin (58) from Streptomyces caelestis.71,72 For many years, lincomycin A (55) and its semi-synthetic chlorinated derivative, clindamycin, were heavily used to treat Gram-positive bacterial infections.66 Although they are rarely prescribed today due to adverse effects, including diarrhea and colitis, they are still used for patients who are allergic to penicillin and when the infection is caused by bacteria that have developed resistance.73
The genes required for lincomycin A biosynthesis have been identified and sequenced in S. lincolnensis strains 78–1174 and ATCC 2546675 (designated as lmb). When S. coelicolor was transformed with a cosmid containing the lmb cluster from S. lincolnensis ATCC 25466, heterologous lincomycin A production was observed.75 Hybridization analysis using probes designed according to the lincomycin A biosynthetic gene cluster allowed the identification of the celesticetin biosynthetic gene cluster in S. caelestis ATCC 1584 (designated as ccb).76
The two structural components of lincomycin A (55), methylthiolincosamide (59, MTL) and propylproline (60), have been proposed to be biosynthesized separately. Thus far, three enzymes involved in the construction of propylproline unit, LmbB1,77 LmbB277 and LmbJ,78 have been characterized in vitro. Progress has also been made in learning the origin of the lincosamine octose core.79 The available data regarding the biosynthesis of the octose core will be discussed in section 4.7. Despite these efforts, how the sulfur atom is incorporated into MTL (59) is still poorly understood. It has been proposed that the thiomethyl substituent of MTL might be transferred from 5′-thiomethyladenosine (MTH), a side product of polyamine biosynthesis derived from SAM.80 However, no obvious gene encoding an enzyme capable of catalyzing the transfer of the thiomethyl group can be found in the lincomycin A biosynthetic gene cluster. Thus, the mechanism of C-1 thiol incorporation remains a challenging problem waiting to be resolved.
3.2.4. Thiosugars found in endiyne-type natural products
Several unusual 4-thiosugars (3, 61, 62) have been found as structural components of four endiyne-containing natural products: calicheamicin (1) from M. echinospora ssp. Calichensis,6 esperamicin (3) from A. verrucosospora,7,8 namenamicin (63) from Polysyncraton lithostrotum,81 and shishijimicin (64) from Didemnum proliferum.82 While the first two are bacterial secondary metabolites, the latter two are isolated from marine invertebrates. On the basis of the analysis of the calicheamicin biosynthetic gene cluster, the translated product of the calS4 (or calK) gene, which encodes a putative C-S lyase, has been proposed to catalyze the conversion of a TDP-2,6-dideoxy-4-ketosugar (65) to the corresponding 4-thiosugar (66, see Scheme 10).9,83 However, this hypothesis has not been verified and it is unclear whether other proteins are also required for the sulfur incorporation step.
Scheme 10.

Proposed pathway for the biosynthesis of 4-thioglucose in calicheamicin
3.2.5. Glucosinolates
Glucosinolates (67) are 1-thiosugar-containing natural products produced by plants and are utilized for defense against insects and pathogens.84 Generally, damage to the plant tissues will trigger the hydrolysis of the C-S glycosidic bond of glucosinolates by myrosinases to release unstable thiohydroximate-O-sulfates.85,86 The sulfate moiety is then lost via a spontaneous Lossen rearrangement resulting in the formation of various sulfur-containing compounds including isothiocyanates, which are toxic to the invaders.
The production of glucosinolates is limited to species of the order Brassicales, which include economically important Brassica crops, such as cabbage, and the model organism Arabidopsis.84 Structurally, glucosinolates are β-thioglucoside N-hydroxysulfates containing a side chain that is derived from an amino acid. The biosynthesis of glucosinolaets has been extensively investigated and reviewed.85–88 As depicted in Scheme 11, the initial step is the oxidation of the amino acid precursor (68) by a cytochrome P450 monooxygenase of the CYP79 family. A second oxidation of the resulting aldoxime (69) catalyzed by an enzyme of the CYP83 family generates activated nitrile oxide (70) or aci-nitro (71) intermediates. The critical sulfur incorporation step has been proposed to occur non-enzymatically with cysteine as the sulfur donor. Subsequent cleavage of the thio-conjugate (72) by PLP-dependent C-S lyase SUR1 yields thiohydroximate (73), which is then glycosylated by UDP-S-glucosyltransferase to give 75. A final sulfonation reaction catalyzed by a sulfotransferase completes the biosynthesis of the glucosinolate core (67).
Scheme 11.

Proposed pathway for the biosynthesis of glucosinolates
Although cysteine has long been considered to be the most probable sulfur donor in glucosinolate biosynthesis, recent reports have indicated that glutathione (GSH) might be a more plausible sulfur donor.89,90 One piece of evidence supporting this possibility was provided by experiments using mutants impaired with respect to GSH biosynthesis. These mutants accumulated a reduced level of glucosinolates upon herbivore challenging and fungal attack, which was restored upon feeding with GSH.89 Moreover, assuming cysteine serves as the sulfur donor, the resulting thio-conjugate (72) would contain a free amino group, which is required for the subsequent PLP-dependent cleavage by C-S lyase SUR1 (72 → 73). On the other hand, if GSH serves as the sulfur donor, the γ-glutamyl moiety of the conjugate (76) would have to be hydrolyzed to yield a free amino group prior to cleavage by C-S lyase SUR1. In fact, engineering experiments of Nicotiana benthamiana have shown that co-transformation of γ-glutamyl peptidase 1 (GGP1) together with the genes for glucosinolate biosynthesis reduces GSH-conjugate accumulation and increases the production of glucosinolates.91 The hypothesis that GSH is the sulfur donor has prompted the search for a specific GSH-S-transferase (GST) that might catalyze the sulfur incorporation step.92
3.3. Group III: Thiosugars containing a sulfonium ion
Several well-known sulfonium-containing compounds including SAM, S-methylmethionine, and dimethylsulfoniopropionate (DMSP) play crucial roles in living systems.93 For example, DMSP from marine algae serves not only as the major source of reduced sulfur for marine microorganisms, but also as the precursor of dimethylsulfide, the primary sulfur source in the global atmosphere.94
Salacinol (77)95 and kotalanol (78)96 isolated from the medicinal herb Salacia reticulate are both naturally occurring thiosugars that contain a sulfonium ion moiety. Several analogues, including ponkolanol (79) and salaprinol (80),97 were later found from other Salacia species. The unique zwitterionic structure of salacinol (77) and its analogues includes a 1-deoxy-4-thio-D-arabinofuranosyl cationic core and 1-deoxy-aldosyl-3-sulfate as the anionic counter ion. These compounds exhibit high α-glucosidase inhibitory activity and have consequently emerged as anti-diabetic drug candidates.95–97 While extensive structure-activity relationship studies have been carried out,43,98 the steps involved in the biosynthetic formation of these sulfonium-containing sugars remain unknown. Since methionine serves as a precursor for the biosynthesis of SAM, S-methylmethionine, and DMSP, it is possible that methionine is also a precursor in the biosynthesis of salacinol, kotalanol, ponkolanol and salaprinol.93
3.4. Group IV: Thiosugars containing a sulfonic acid group
In 1959, Benson and coworkers carried out a series of radioisotope labeling experiments in higher plants leading to the discovery of sulfoquinovosyl diacylglycerol (81, SQDG) as a minor component in chloroplast membranes.99,101 SQDG contains an unusual sulfosugar moiety and comprises about 4–7% of total lipids in most photosynthetic organisms including algae, plants and cyanobacteria.101,102 Subsequent studies using [35S]-sulfate traced the sulfolipid biosynthetic pathway to a nucleotide-activated sulfoquinovose intermediate (82, see Scheme 12).103 Through genetic analysis of the sulfolipid-producing purple bacteria, Rhodobacter sphaeroides,104,105 a small gene cluster with only four genes, sqdA, sqdB, sqdC and sqdD, was recently identified as the likely, the long-sought after sulfolipid biosynthetic gene cluster. Orthologues of sqdB are universally found in other SQDG-producing organisms. It was proposed that SqdB catalyzes the synthesis of NDP-sulfoquinovose (82) based on its sequence similarity to other NDP-sugar modification enzymes.106 The gene sequence of sqdB was utilized to isolate cDNA of the orthologous SQD1 gene in Arabidopsis thaliana.107 SQD1 that had been recombinantly expressed in E. coli could catalyze the insertion of inorganic sulfite into UDP-glucose (74). In addition, an attempt to purify the native SQD1 protein from spinach led to the finding that SQD1 forms a tight complex with ferredoxin-dependent glutamate synthase (FdGOGAT) in vivo.108 The exact function of this large SQD1-FdGOGAT complex and the detailed mechanism of sulfonyl incorporation awaits further investigation.
Scheme 12.

Proposed pathway for the biosynthesis of sulfoquinovosyl diacylglycerol
4. Compounds Containing High-carbon Sugars
Unusual sugars with high carbon numbers are found in an array of microbially produced antibiotics109 and in the outer walls of some Gram-negative bacteria. They are formed via condensation reactions between the C-5 and C-6 sugars of primary metabolism, or derivatives thereof, and an exogenous carbon source. The known mechanisms employed for condensation during the biosynthesis of high-carbon sugars are as varied as the mechanisms for biological C–C bond formation in general. In many cases, the mechanism of sugar chain elongation remains poorly understood.
4.1. Tunicamine containing nucleoside antibiotics
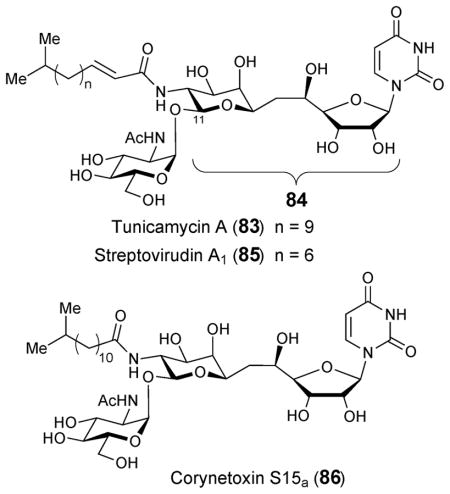
Tunicamycin (83) is a nucleoside antibiotic that was first isolated from culture media of Streptomyces lysosuperificus and subsequently Streptomyces chartreusis NRRL 3882.110,111 Tunicamycin is extracted as a complex composed of closely related components having a general structure that includes an unusual 11-carbon aminodialdolose core, known as tunicamine (84), joined to N-acetylglucosamine (GlcNAc), uracil, and an amide-linked fatty acid.112 Streptovirudins (85) and corynetoxins (86), isolated from Streptomyces griseoflavus113 and Corynebacterium rahayi,114 respectively, are structurally homologous to tunicamycin as are antibiotics MM19290,115 mycospocidin,116 and antibiotic 24010.117 The latter three, however, were isolated as complexes composed of multiple components for which detailed structures remain uncharacterized.
Tunicamycin (83) and related antibiotics disrupt assembly of the bacterial cell wall via inhibition of phospho-N-acetylmuramoyl-pentapeptide transferase (MraY), an enzyme involved in the biosynthesis of the peptidoglycan precursor lipid I.118 The fact that tunicamine-containing compounds can also block N-linked glycoprotein synthesis by inhibiting UDP-N-acetylglucosamine: dolichol phosphate GlcNAc-1-P transferase (also known as GlcNAc-1-P translocase or GPT) in eukaryotes has precluded their use in clinical settings.119 In this case, The α,β-1,11-glycosidically-linked GlcNAc moiety of tunicamycin is thought to resemble the transferred GlcNAc-1-phosphate during the transition state of the GPT reaction.120 Interestingly, tunicamycin has also been shown to block palmitoylation of acyl proteins by inhibition of palmitoyl transferase.121
The biosynthesis of the tunicamycin-like antibiotics is only partially understood. The steps leading to the unique tunicamine core structure (84) are of particular interest as they are predicted to include an unusual C–C bond coupling reaction. Labeling studies with [2-14C]-uridine, [6,6-2H2]-D-glucose, and D-[1–13C]-glucose showed that the tunicamine core is derived from uridine and UDP-GlcNAc (87).122 This led to the proposal of a tentative biosynthetic pathway for tunicamine in which uridine 5′-aldehyde (90) undergoes condensation with UDP-4-keto-6-deoxy-GlcNAc-5,6-ene (88, see Scheme 13A).
Scheme 13.

Proposed pathways for the biosynthesis of the tunicamine core
The S. chartreusis genome was recently sequenced and a gene cluster for tuncamycin biosynthesis was identified by comparative bioinformatics.123 Subsequent introduction of the gene cluster into Streptomyces coelicolor confirmed heterologous production. The clustered genes include 14 open reading frames, designated tunA-N, which the authors claimed to comprise the minimal set of genes required for tunicamycin biosynthesis. A similar gene cluster is present in the MM19290 producer S. clavuligerus and in Actinosynnema mirum, although the latter strain has not been identified as a producer of tunicamine-containing compounds. Within the gene cluster, TunB is identified as a putative member of the radical SAM enzyme superfamily. It was proposed to catalyze the oxidation of uridine (89) to uridine-5′-aldehyde (90, see Scheme 13A).123 TunA and F, which are homologous to NAD-dependent epimerase/dehydratase and UDP-GlcNAc 4-epimerase, respectively, are likely responsible for the conversion of UDP-GlcNAc (87) to UDP-4-keto-6-deoxy-GlcNAc-5,6-ene (88). TunM, a SAM-dependent methyltransferase homologue, was postulated to catalyze the subsequent condensation of this product (88) with uridine-5′-aldehyde (90) to yield UDP-N-acetyltunicamine-uracil adduct (91, Scheme 13A).
The S. chartreusis tunicamycin biosynthetic gene cluster was independently identified using a “high-throughput heterologous expression” approach in which culture media from many strains of Streptomyces lividans transformed with plasmids from a S. chartreusis genomic library were screened for antibiotic activity.124 In this study the authors identified tunA-L (excluding tunM and N) as the minimal set of genes required for tunicamycin biosynthesis. This led to a more reasonable proposal that TunB catalyzes the condensation of uridine (89) with UDP-6-deoxy-GalNAc-5,6-ene (93) to yield UDP-N-acetyltunicamine-uracil (91, Scheme 13B). This hypothesis is supported by the finding that a TunB knockout strain accumulates UDP-6-deoxy-GlcNAc-5,6-ene in culture media.
The in vitro activities of purified recombinant proteins, TunA and TunF, from S. chartreusis were recently reported.125 TunA is homologous to TDP-glucose 4,6-dehydratase, which carries out the conversion of TDP-glucose to TDP-4-keto-6-deoxy-glucose (5) during the biosynthesis of 6-deoxyhexoses.1,2 However, unlike a typical 4,6-dehydratase-catalyzed reaction involving an internal hydrogen transfer from C-4 to C-6, the TunA catalytic cycle is concluded by a 1,2-hydride addition to the 4-keto group of the 4-keto-6-deoxy-GlcNAc-5,6-ene intermediate (88), rather than 1,4-hydride addition to the adjacent C5-C6 double bond, to give UDP-6-deoxy-GlcNAc-5,6-ene (92) as the product (Scheme 13B).
A 1.9 Å crystal structure of TunA in complex with UDP-GlcNAc (87) and NAD+ was also determined.125 Comparisons were drawn between the crystal structures of TunA and other 4,6-dehydratases, which all appear highly similar. It appears that the orientation of the nicotinamide ring in TunA, which places C-4 of NAD+ toward C-4 of UDP-GlcNAc and away from C-6, serves as a “regioselective switch” that determines the identity of the product. On the basis of these results, a biosynthetic pathway was put forward in which UDP-6-deoxy-GalNAc-5,6-ene (93), produced from UDP-GlcNAc (87) by the successive oxidation and epimerization reactions of TunA and TunF, is joined with 5′-uridine radical (94) by TunB (with possible involvement of TunM) to give UDP-N-acetyltunicamine-uracil (91, Scheme 13B).125 It was also proposed that epimerization of UDP-6-deoxy-GlcNAc-5,6-ene (92) to UDP-6-deoxy-GalNAc-5,6-ene (93) by TunF is needed to place σ*(C8′-O8′) LUMO to be parallel to C7′ SOMO in 7′-UDP-N-acetyltunicamine-uracil, thereby stabilizing this transient radical intermediate (95).
4.2. Liposidomycin, caprazamycin, muraymycin, A-90289A and B, and FR900493
Liposidomycin (96),126 caprazamycin (97),127,128 muraymycin (98),129 A-90289A (99) and B,130 and FR900493 (100)131 belong to a class of structurally and biosynthetically related antibiotics that contain a seven-carbon 5′-C-glycyluridine core (101). Like tunicamycins (83), they are potent inhibitors of MraY. Gene clusters have recently been identified for the biosynthesis of liposidomycin in Streptomyces sp. SN1061M (lpmA-Y),132 caprazamycin in Streptomyces sp. MK730-62F2 (cpz9-31),133 muraymycin in Streptomyces sp. NRRL 30471 (mur11-36),134 and A-90289 in Streptomyces sp. SANK 60405 (lipA-B1).130 A gene likely encoding serine hydroxymethyltransferase (SHMT) is found in all four clusters. The well established function of SHMT is to catalyze the reversible formation of β-hydroxy-α-amino acids from an aldehyde and glycine.135 Thus, these SHMT homologues (lpmL, cpz14, mur17, and lipK) are thought to catalyze condensation between uridine 5′-aldehyde (90) and the PLP-bound glycine to give 101 (Scheme 14).
Scheme 14.

Proposed biosynthesis of 5′-C-glycyluridine core
It was previously proposed that uridine is oxidized to uridine 5′-aldehyde by the alcohol dehydrogenase homologues encoded by lpmW, cpz25, lipV (the corresponding gene is absent in the muraymycin gene cluster). Surprisingly, however, LipL, an iron(II)-dependent α-ketoglutarate dioxygenase, was found to be the actual enzyme catalyzing the oxidation of UMP to uridine 5′-aldehyde (90).136 Iron, α-ketoglutarate, and oxygen are necessary for the activity of this enzyme. Thus, LpmM, Cpz15, Mur16, which are LipL homologues, likely serve an analogous role in each respective pathway (Scheme 14). Interestingly, the gene clusters for the 5′-C-carbamoyluridine-containing MraY inhibitors, A-500359 from Streptomyces griseus SANK 60196 (orf1-38)130 and A-503083 from Streptomyces sp. SANK 62799 (capA-W),130 also harbor genes encoding LipL homologues (Orf7 and CapA). This observation suggests that 5′-C-glycyluridine (101) could be an intermediate in the biosynthesis of these antibiotics as well. Additional high-carbon nucleoside antibiotics including A-94964 from Streptomyces sp. SANK 60404137 and A-102395138 from Amycolatopsis sp. SANK 60206, for which genetic data are currently unavailable, are likely produced by similar pathways.
4.3. Octosyl acids and ezomycins
Octosyl acids A (102), B (103), and C (104) are bicyclic, anhydrooctose uronic acid nucleosides isolated from media of Streptomyces cacaoi var. asoensis.139 This strain also produces polyoxins (105, 106), a class of nucleoside antibiotics that are active against phytogenic fungi via inhibition of chitin synthase.140 Isotopic labeling studies suggested that octosyl acids and polyoxins may be derived from a common octofuranulose uronic acid nucleoside intermediate (107). Experiments in which S. cacaoi was fed [U-14C]-uridine or [1-13C]-glucose revealed that uridine is incorporated intact during the biosynthesis of polyoxin C (105) and that glucose is not a direct precursor.141 In addition, C-3 of pyruvate and glycerate were shown to be the source of C-6′. When [1-13C]- or [6-13C]-glucose was fed to an S. cacaoi mutant that accumulates polyoxin C and octosyl acid, 13C enrichment patterns for both compounds were similar.142 Taken together, these observations led to a hypothesis that the biosynthesis of both polyoxins and octosyl acids involves a condensation reaction between phosphoenolpyruvate (PEP, 108) and a uridine derivative (possibly uridine 5′-aldehyde (90), see Scheme 15) to yield the proposed octofuranulose uronic acid nucleoside (107). In this case, cyclization followed by reduction at C-7′ would yield the bicyclic anhydrooctose uronic acid of octosyl acids. Carbons derived from C-1 and C-2 of PEP are lost during polyoxin biosynthesis.
Scheme 15.

Proposed pathways for the biosynthesis of octosyl acids, polyoxins and nikkomycins
Nikkomycins (109–111) comprise a class of chitin synthase inhibitors produced by Streptomyces tendae and Streptomyces ansochromogenes that are biosynthetically and structurally related to polyoxins.143 Four nikkomycin homologues, nikkomycin Sz, Soz, Sx, and Sox isolated from S. tendae culture media contain the same bicyclic anhydrooctose uronic acid found in octosyl acid A (102).144 Therefore, the octosyl acids and nikkomycins Sz, Soz, Sx, and Sox have been regarded as the “shunt metabolites” of the biosynthetic pathways for polyoxin and nikkomycin biosynthesis, respectively.
Biosynthetic gene clusters involved in the production of polyoxins in S. cacaoi (pol) and nikkomycins in S. tendae (nik) and S. ansochromogenes (san) have recently been identified.145,146 The orthologous enzymes encoded by nikO147,148 and polA145 have been shown to catalyze the formation of 3′-enoylpyruvoyl-UMP from PEP (108) and UMP in assays with purified recombinant proteins. Gene knockout experiments demonstrated that nikO149 and sanX146 are required for the biosynthesis of nikkomycins and that polA is required for the biosynthesis of polyoxins.145 Presumably the corresponding orthologs are also involved in the biosynthesis of octosyl acids and nikkomycins Sz, Soz, Sx, and Sox. Although PEP was previously shown to be the source of C6′ in polyoxin C (105),141 the demonstrated activity of NikO and PolA came as a surprise given the hypothesis that PEP undergoes condensation with uridine 5′-aldehyde (90). The biosynthetic steps responsible for the conversion of 3′-enoylpyruvoyl-UMP (112) to the aminohexuronic acid moiety in nikkomycins and polyoxins remain to be demonstrated.150 Furthermore, it is also unclear whether the octosyl acids and nikkomycins Sz, Soz, Sx, and Sox are shunt metabolites or precursors to polyoxins and nikkomycins.
Ezomycins (113) comprise another set of antibiotics that contain octosyl acid.151–153 They were isolated from culture filtrates of a species similar to Streptomyces kitazawaensis, and are toxic to the phytopathogenic fungus Sclerotinia sclerotiorum. To date, no information is available regarding the biosynthesis of ezomycins.
4.4. Mildiomycin
Mildiomycin (114) is an antimicrobial peptidyl nucleoside antibiotic produced by Streptoverticilium rimofaciens that inhibits protein synthesis by blocking the peptidyltransferase site on the large ribosomal subunit in both eukaryotic and prokaryotic organisms.154,155 It is structurally similar to the antibiotics blasticidin S (115),156 arginomycin (116),157 gougerotin (117),158 and bagougeramines A (118) and B,159 which all share a common core structure. Mildiomycin, however, contains a unique decose carbon skeleton. The identification and subsequent sequencing of the blasticidin S biosynthetic gene cluster in Streptomyces griseochromogenes (blsA-S)160 laid the foundation for the discovery of the mildiomycin biosynthetic gene cluster (milA-Q).161 Several initial steps regarding the biosynthesis of mildiomycin have been demonstrated (see Scheme 16). MilA was previously shown to catalyze the hydroxymethylation of C-5 in CMP in the presence of formaldehyde and tetrahydrofolate to give hydroxymethylcytosine (119) and MilB could hydrolyze the N-glycosidic bond in CMP to give 120.162 Assays with purified, recombinant MilC further demonstrated its ability to catalyze the formation of cytosylglucuronic acid (CGA, 121) from cytosine and UDP-glucuronic acid (123).161 MilG, a member of the radical SAM enzyme superfamily, is thought to be involved in the formation of the 2,3-deoxy-4-keto-2,3-ene-sugar moiety (124) of mildiomycin, since the milG disruption mutant accumulated 5-hydroxymethyl-CGA (122) in the fermentation broth. In vitro labeling experiments using [U-13C]-arginine or [guanidino-13C]-4-hydroxyarginine showed that the former, but not the latter, was incorporated into mildiomycin. Therefore, the 5-guanidino-2,4-dihydroxyvalerate moiety in mildiomycin is derived from arginine (125). MilM and MilN are homologous to aspartate aminotransferase and dihydropicolinate synthetase, respectively. MilM is proposed to catalyze the deamination of arginine (125) to give α-keto-δ-guanidinovalerate (126), which is subsequently coupled to the nucleoside by MilN. Finally, the hydroxylation of mildiomycin at C-8′ may occur after the formation of the decose sugar (see Scheme 16).
Scheme 16.

Proposed pathway for the biosynthesis of mildiomycin
4.5. Griseolic acids
Griseolic acids A (127), B (128) and C (129) are cyclic AMP analogues that contain an unusual nine-carbon bicyclic sugar, dicarboxylate glycone.163,164 They were identified and isolated from culture media of Streptomyces griseoaurantiacus during a search for inhibitors of cyclic nucleotide phosphodiesterase. A ring-opened griseolic acid analogue (130) lacking the inhibitory activity was subsequently isolated from S. griseoaurantiacus media.165 Feeding experiments showed that adenosine (131) is a biosynthetic precursor for griseolic acid A.166 Incorporation of 13C from [2-13C]-acetate into C-6′, 7′, 8′, and 9′ of 127 suggests that these carbons could originate from a dicarboxylate intermediate derived from the citric acid cycle. Since cells fed [1,4-13C]-succinate incorporated 13C at the carboxylate carbons, oxaloacetate (132) has been proposed as a possible donor of the four-carbon dicarboxylic acid in griseolic acid A (see Scheme 17).
Scheme 17.

Proposed biosynthetic precursors for griseolic acid A
4.6. Sinefungin and dehydrosinefungin
The S-adenosyl-L-methionine analogues sinefungin (A9145) (133) and dehydrosinefungin (A9145C) (134), were first isolated from the culture media of Streptomyces griseolus.167,168 Sinefungin displays antifungal and antiviral activity as well as activity against trypanosomal pathogens including malaria.169–171 Both radiotracer feeding experiments and cell-free assays have provided preliminary clues regarding sinefungin biosynthesis. However, the genes and enzymes involved in the process remain unidentified.
Feeding experiments in which a panel of 14C-labeled metabolites was added to S. griseolus culture indicated that sinefungins are derived from ornithine (135) and an adenosine derivative.172 Cell-free assays with Streptomyces incarnatus NRRL 8089, which also produces sinefungin, led to the observation that radiolabel from ATP or adenosine and arginine (125), rather than ornithine, are incorporated most efficiently into sinefungin (the arginine guanidino group was not incorporated).173 Addition of pyridoxal 5′-phosphate, Mg2+, and dithiothreitol enhanced sinefungin production during this assay. Further S. griseolus feeding experiments with site-specifically labeled ornithine showed that the C-6′ and the 6′ amino group of sinefungin (133) are derived from the C-5 and the C-5 amino group of ornithine (135, see Scheme 18).174 In this study, the authors observed equal label incorporation with both arginine and ornithine as substrates and pointed out that ornithine is a precursor for arginine in vivo. Tritium from [5R-3H, 5-14C]-L-ornithine but not [5S-3H, 5-14C]-L-ornithine was incorporated into sinefungin during feeding experiments. Since the configuration at C-6′ of sinefungin is S, the C-C bond formation at C-5 of ornithine to form sinefungin must occur with inversion of configuration (Scheme 18). This conclusion places sinefungin biosynthesis among a small number of PLP-dependent processes that do not proceed with retention of configuration.
Scheme 18.

Proposed biosynthetic precursors for sinefungin (A9145)
When S. griseolus cell lysate was incubated with a blend of [5′ (RS)-5′-3H]-ATP and [8-14C]-ATP, together with Mg2+ and PLP, both labels were incorporated into sinefungin.175 The same result was obtained with a blend of [5′ (RS)-5′-3H]-adenosine and [U-14C]-adenosine or [1-14C]-ribose and [8-3H]-adenine. This suggests that adenosine is catabolized by a nucleoside hydrolase or nucleoside phosphorylase prior to sinefungin biosynthesis. When the assay was conducted with a blend containing a 4:1 ratio of [5′ (RS)-5′-3H]-adenosine to [1α-14C]-adenosine, the same ratio of 3H:14C was observed in the product. This result implies that the ribose moiety of adenosine is incorporated intact into sinefungin and that tritium is not lost from ribose C-5′ during sinefungin formation, thereby ruling out dehydrosinefungin (134) as a plausible biosynthetic intermediate. When cell-free assays were carried out with added PLP and either [1-14C]-ribose or [U-14C]-arginine, but neither ATP, adenosine, nor adenine, radiolabel was incorporated into an unknown compound designated “X”. Radiolabeled sinefungin was observed when compound “X” was added to cell lysate that had been supplemented with adenine, suggesting that “X” could be a sinefungin precursor (see Scheme 18).
More recently, a rifamycin-resistant mutant strain of S. incarnatus that overproduces sinefungin was generated by UV irradiation.176 The strain was designated rif-400. The production of sinefungin by rif-400 was further enhanced by adding L-arginine to the culture media, but not D-arginine or L-ornithine. Addition of urea diminished the yield of sinefungin. Addition of L-arginine, but not L-ornithine, to rif-400 culture media, resulted in the accumulation of ornithine-δ;-lactam in the media.176 An enzyme that catalyzes the conversion of L-arginine to ornithine-δ-lactam, with release of urea, was purified to homogeneity from rif-400 lysate. The enzyme did not utilize D-arginine or L-ornithine as substrates. It did, however, convert NAD+ to NADH in the presence of alanine, consistent with N-terminal amino acid sequencing results, which suggested that the enzyme is homologous to L-alanine dehydrogenase. It remains unclear whether or not ornithine-δ-lactam is truly an intermediate in sinefungin biosynthesis.
4.7. Lincosamide-containing antibiotics: lincomycin, celesticetin, and Bu-2545
The lincosamide-containing antibiotics were introduced earlier in section 3.2.3. Here, the currently available data regarding the construction of the octose core will be discussed. Feeding experiments were conducted in order to identify the biosynthetic origin of the octose sugar in lincomycin A (55). When S. licolnensis was grown with a blend of unlabeled and [13C6]-glucose as the sole carbon source, the 13C labeling pattern of the resulting α-methylthiolincosamide (MTL, 56) suggested that MTL is formed via the condensation of a three-carbon (C3) and a five-carbon (C5) unit derived from glucose.79 This led to a proposal that the C5 unit might be a pentose 5-phosphate from the pentose phosphate pathway and that the C3 unit might be derived from D-sedoheptulose 7-phosphate (137) and joined to the C5 unit by a transaldolase-catalyzed reaction analogous to that involved in the pentose phosphate pathway. Interestingly, both the lincomycin (55) and celesticetin (58) biosynthetic gene clusters encode a putative transaldolase (LmbR and CcbR) that may catalyze an intermolecular aldol condensation to produce the octose sugar precursor to lincosamine (56).47–76 This hypothesis has been substantiated by a recent experiment in which the octose intermediate is shown to be formed via a transaldol reaction catalyzed by LmbR using D-fructose 6-phosphate (136) or D-sedoheptulose 7-phosphate (137) as the C3 donor and D-ribose 5-phosphate (138) as the C5 acceptor (Scheme 19). Subsequent 1,2- isomerization catalyzed by LmbN converts the resulting octulose 8-phosphate (139) to octose 8-phosphate (140). These results provide, for the first time, in vitro evidence revealing the biosynthetic origin of the eight-carbon backbone of MTL (56).177
Scheme 19.

Reactions catalyzed by LmbR and LmbN in the biosynthesis of MTL
4.8. Albomycin
Albomycin (38) is first mentioned in section 3.2.1. Although currently there is no biochemical data regarding the biosynthesis of the 4-thioheptouronic acid moiety in albomycin, three possible pathways have been proposed based on sequence analysis of the albomycin biosynthetic gene cluster.54 As illustrated in Scheme 20, in pathway A the reaction is initiated by the nucleophilic addition of cysteine sulfur to C-3 of PEP (108) catalyzed by the cysteine desulfurase homologue AbmD to form a thioether adduct (141). This is then followed by cyclization, oxidative decarboxylation, and deamination via the combined actions of AbmM and J, both of which are members of the radical SAM superfamily, and AbmF and L, both of which are hypothetical proteins of unknown function, to give 1-phospho-4-thio-ribo-pentodialdose (142). The aldehyde would then undergo aldol condensation with glycine catalyzed by AmbH, which is a SHMT homologue, to yield 6-amino-6-deoxy-1-phospho-4-thio-heptofuranose uronic acid (47) as the product.135 The final step is similar to the condensation reaction proposed for the SHMT homologues LpmL, Cpz14, Mur17, and LipK in the biosynthesis of liposidomycin, caprazamycin, muraymycin, and A-90289, respectively (90 → 101, see Scheme 14). An analogous reaction sequence is also proposed for pathway B where homocysteine instead of cysteine is used as substrate in an AbmD-catalyzed reaction. In the third proposed pathway, either serine or glycine is joined to threose or ribose, respectively, via an intermolecular aldol condensation reaction catalyzed by AbmH. This is followed by sulfur insertion by AbmD and AbmJ, and cyclization by the combined actions of AbmF, M, and L to give 6-amino-6-deoxy-4-thio-heptofuranose uronic acid (143), which could then be phosphorylated at C-1 by the deoxynucleoside kinase homologue AbmG to give 47.
Scheme 20.

Proposed pathways for the biosynthesis of 4-thioheptouronic acid in albomycin
4.9. Apramycin
Apramycin (144) is an aminoglycoside antibiotic that contains an unusual bicyclic octose sugar (145).178,179 Labeling studies have indicated that the carbon atoms from C-1′ to C-6′ of the octose moiety are derived from glucose while C-7′ and C-8′ may originate from C-2 and C-3 of pyruvate.180 Gene clusters for the biosynthesis of apramycin in Streptoalloteichus tenebrarius DSM 40477T and Streptoalloteichus hindustanus DSM 44523T have been identified and sequenced (designated apr). During the biosynthesis of the aminoglycoside antibiotics neomycin and butirosin, the C-6′ hydroxyl group of the common biosynthetic intermediate, paromamine (146), is oxidized to an aldehyde (147) by a FAD-dependent dehydrogenase (Neo11 and BtrQ). The resulting aldehyde is then converted to an amine (148) by a PLP-dependent transaminase (Neo18 and BtrB, see Scheme 21).181 While the apramycin biosynthetic gene cluster encodes the homologous protein of paromamine 6′-dehydrogenase (AprQ), the gene encoding the potential transaminase is absent as expected. It is possible that the resulting C-6′ aldehyde (149) may undergo an aldol condensation with a two-carbon unit to give the octose sugar (150, Scheme 21).182 However, the precise origin of C-7′ and C-8′ and the mechanism of the octose formation remain unknown.
Scheme 21.

Proposed pathway for the biosynthesis of bicyclic octose moiety in apramycin
4.10. Quinocyclines
Quinocyclines A and B (151a and 151b) and isoquinocyclines A and B (152a and 152b) are a complex of antitumor antibiotics from Streptomyces aurofaciens.183 Each contains a branched octose sugar substituent.184 Quinocycline B (konosinostatin) and isoquinocycline B were also found in culture media of Micromonospora sp. TP-A0468.185 Feeding experiments demonstrated that [2-14C]-pyruvate was incorporated more efficiently into the branched octose than [1-14C]-acetate.186 Thus, a mechanism in which an NDP-linked 4-keto-2,6-dideoxy-L-erythropyranose (153) undergoes condensation at C-4 with the hydroxyethyl group from hydroxyethyl-TPP (154) was proposed for the production of the branched octose (155). A similar transformation has been observed in the biosynthesis of yersiniose A (171, see Scheme 23).187 Furthermore, the efficient reduction of quinocycline B to quinocycline A by cultured S. aurofaciens strongly suggests that (iso)quinocycline B is the biosynthetic precursor of (iso)quinocycline A.
Scheme 23.

Proposed pathway for the biosynthesis of yersiniose and mechanism of YerE
4.11. Other antibiotics with high-carbon sugars
Several additional antibiotic compounds carrying high-carbon sugars with no information regarding their biosynthetic pathways or the genes involved will only be briefly mentioned here. Amipurimycin (156), isolated from the culture media of Streptomyces novoguineensis, was shown to display antibiotic activity against Pyricularia oryzae, a fungal phytopathogen that causes sheath blight in rice plants.188 The structure of amipurimycin includes an unusual branched nonofuranose uronic acid sugar.189 Miharamycins A and B (157a and 157b) are adenine-containing nucleosides isolated from cultures of Streptomyces miharaensis SF-4890, and are active against rive blast disease.190 The structures of the miharamycins, reported 15 years after their initial discovery,191 include a branched, nonofuranose uronic acid similar to that found in amipurimycin. In miharamycins, however, the sugar is bicyclic, containing an ether linkage between C-2′ and C-9′. Hikizimycin (158), also named as anthelmycin, was isolated from the culture media of Streptomyces sp., strain A-5 and exhibits antibiotic activity against phytopathogenic fungi,192 which might be related with inhibition of transpeptidation during protein synthesis.193 Hikizimycin is a cytosine nucleoside analogue containing hikosamine (159), a 4-amino-4-deoxy-undecose sugar.194–196 The antitumor compounds septacidin (161) from Streptomyces fimbriatus197 and its C-2 epimer spicamycin (160) from Streptomyces alanosinicus 87-MT3198 were both isolated as mixtures of similar compounds carrying normal and iso-chain fatty acyl substituents of variable lengths. The structures of both septacidin and spicamycin contain 4-amino-4-deoxyheptose (162) as does anicemycin (163) from Streptomyces sp. TP-A0648, which was identified recently.199 Herbicidins A, B, C, E, F and G are antibiotics with herbicidal activity produced by Streptomyces sagamonensis.200–202 The herbicidins are adenosine nucleoside analogues that contain a tricyclic dodecose scaffold. The array of herbicidins produced by a series of S. sagamonensis mutants led to a proposal that herbicidin A (166) is derived from herbicidin F (165), of which the precursor is herbicidin G (164).203 Unfortunately, there is no information regarding the biosynthesis of the dodecose sugar.
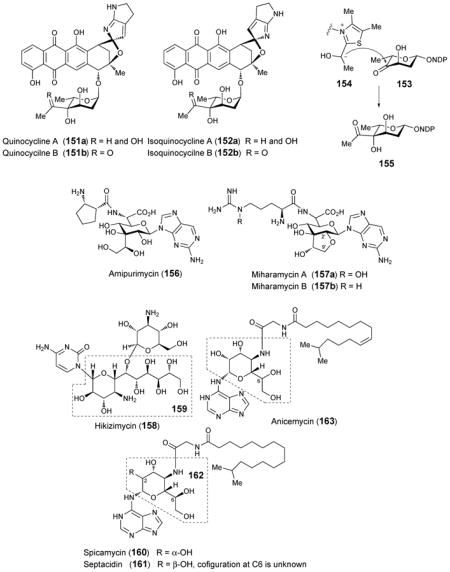
4.12. High-carbon sugars from bacterial cell walls
In addition to the high-carbon sugar antibiotics described above, a number of high-carbon sugars have been identified as components of bacterial cell walls. Perhaps the most widely recognized high-carbon sugar is N-acetyl-neuraminic acid (NeuNAc, 168). This nine-carbon sugar is a key component of the eukaryotic extracellular matrix, and is also found to a lesser extent in bacteria. In bacterial systems, it is formed via an aldol condensation between N-acetyl-mannosamine (ManNAc, 167) and either PEP (108) or pyruvate by N-acetyl-neuraminic acid synthase or aldolase, respectively (Scheme 22).204,205 KDO (170), 3-deoxy-D-manno-oct-2-ulosonic acid, is an eight-carbon sugar found in the lipopolysaccharides of Gram-negative bacteria.206 KDO is biosynthesized via an aldol condensation between arabinose-5-phosphate (169) and PEP (108) catalyzed by KDO-8-phosphate synthase (Scheme 22).207 In addition to neuraminic acid and KDO, heptoses, especially those have a glycero-D-manno configuration, are essential components of the lipopolysaccharides. These bacterial heptoses are biosynthetically derivatized from D-sedoheptulose 7-phosphate (137) and their biosynthetic pathways have been extensively studied and reviewed.208,209
Scheme 22.

Biosynthetic pathways for NeuNAc and KDO
Yersiniose A (171) is a branched chain octose found in the lipopolysaccharides of Yersinia pseudotuberculosis cultivar VI.210 Early feeding experiments suggested that the two-carbon branch is derived from pyruvate186,211 and a gene cluster dedicated to the biosynthesis of yersiniose in Y. pseudotuberculosis cultivar VI was identified.187 The yer gene cluster contains eight genes and the yerE and yerF gene products were proposed to be essential for the formation of the two-carbon branch in yersiniose A (Scheme 23). Further investigation using the purified YerE expressed heterologously in E. coli showed that YerE indeed catalyzes the TPP-dependent attachment of the two-carbon branch using pyruvate and CDP-3,6-dideoxy-4-hexulose (172) as substrates (Scheme 23). A similar reaction had been proposed in the biosynthesis of the quinocycline compounds (153 → 155)186. YerE is a flavoprotein and displays homology to the large subunit of acetolactate synthase, an FAD-binding TPP-dependent enzyme.212 When the YerE reaction is conducted under anaerobic conditions in the presence of sodium dithionite, the preservation of YerE activity suggest that flavin is not directly involved in the reaction mechanism. Thus, FAD might only play a structural role in reaction catalyzed by YerE.213
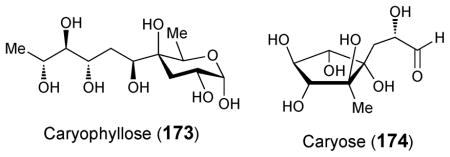
Caryophyllose (173), a branched 12-carbon sugar,214 and caryose (174), a nine-carbon sugar containing a carbocycle,215 were identified in the lipopolysaccharide fraction of the phytopathogenic bacterium Pseudomonas (Burkholderia) caryophylli. Caryophyllose, a 3,6,10-trideoxy-4-C-[(R)-1-hydroxyethyl]-D-erythro-D-gulo-decose, exists as an oligomer with α-(1→7)-linkages known as caryophyllan, and caryose, a 4,8-cyclo-3,9-dideoxy-L-erythro-D-ido-nonose, exists as an oligomer with β-(1→7)-linkages known as caryan.216 Although little is known about the biosynthesis of either sugar, two genes in Mycobacterium marinum, which also produces carryophyllose,217 have been implicated in caryophyllose incorporation into lipooligosaccharides. When the two genes, MMAR_2332 and MMAR_2333, were disrupted by transposon mutagenesis, caryophyllose was absent from M. marinum lipooligosaccharides.218 The former is thought to encode a glycosyltransferase that may be responsible for the incorporation of carryophyllose into lipooligopolysaccharides.219 The latter appears to encode a putative TPP-dependent carboxylase, suggesting that a TPP-dependent enzyme, which may be similar to that established for yersiniose formation, could be involved in the biosynthesis of this C-4 branched sugar (173) as well.
5. Conclusions
As described in this review, sugars with oxidized amines, C–S linkages, and high-carbon chain lengths, although rare, are broadly distributed throughout nature. Microbially-derived natural products with antibiotic activity are especially, although not exclusively, rich sources of these unusual sugars. P450-dependent aminosugar-N-oxygenases are involved in the biosynthesis of N-hydroxyaminosugars with nitrosugars as a minor product. Flavin-dependent aminosugar-N-oxygenases have been demonstrated to yield nitrososugars. It is thought that non-enzymatic oxidation is responsible for the conversion of the nitrososugar product to the corresponding nitrosugar.
Many questions surrounding the biosynthesis of thiosugars remain unanswered. There is no genetic or biochemical data available regarding the biosynthesis of rhodonocardins A (34) and B or 5-thio-D-glucose. Based on the predicted function of several enzymes encoded within the albomycin (38) biosynthetic gene cluster, it is possible that a radical-mediated mechanism is involved in sulfur incorporation during albomycin biosynthesis, but this remains pure speculation. The thiazole synthase (ThiG) homologue, BexX, encoded within the BE-7585A (48) biosynthetic gene cluster forms a covalent adduct with glucose 6-phosphate, in accordance with its predicted function. However, the remaining steps involved in sulfur incorporation into BE-7585A are unclear. Although the gene clusters for lincomycin (55) and celesticetin (58) biosynthesis have been identified, they provide no obvious clues regarding the mechanism of sulfur incorporation into this class of antibiotics. The gene cluster for the biosynthesis of calicheamicin (1) encodes a C–S lyase but its function and role remain to be demonstrated. The biosynthetic pathways for glucosinolates (67) are fairly well characterized, but it is unclear whether the incorporated sulfide is derived from cysteine or glutathione. Nothing is known about the biosynthesis of the sulfonium containing plant products salacinol (77), kotalanol (78), ponkolanol (79) and salaprinol (80), but based on structural considerations these compounds may be derived, in part, from methionine. SQD1 from Arabidopsis thaliana catalyzes the incorporation of inorganic sulfite into UDP-glucose during the biosynthesis of sulfolipids (81).
Elucidation of the various mechanisms involved in the biosynthesis of high-carbon sugars is an active research topic, and limited advances have been made in several systems. According to recent in vitro experiments, octulose-8-phosphate, an intermediate in lincosamine (56) biosynthesis, is formed via a transaldol reaction between D-ribose 5-phosphate and either D-fructose 6-phosphate or D-sedoheptulose 7-phosphate catalyzed by LmbR. LmbN then catalyzes the isomerization of octulose-8-phosphate to octose-8-phosphate. The 5′-C-glycyluridine moiety of liposidomycin (96), caprazamycin (97), muraymycin (98), A-90289A (99) and B, and FR-900493 (100) is probably formed in two sequential steps: an α-ketoglutarate dioxygenase homologue oxidizes UMP to uridine 5′-aldehyde, which then undergoes subsequent condensation with glycine via the PLP-dependent action of an enzyme homologous to serine hydroxymethyl transferase. The first step has been demonstrated but the second has not. The mechanism behind sugar chain elongation in octosyl acids (102–104) and ezomycins (113) remains to be elucidated. However, 3′-enoylpyruvoyl-UMP has been identified as a biosynthetic intermediate in polyoxin (106) and nikkomycin (109–111) production and it could, therefore, serve as an intermediate in the formation of octosyl acids and ezomycins as well. The TPP-dependent addition of a branched carbon chain during yersiniose (171) biosynthesis has been demonstrated in vitro and similar reactions have been proposed for the branched octose sugars in quinocyclines (155) and the branched dodecose sugar caryophyllose (173).
Despite of the above progress, there is much that remains undiscovered in this area. It has been proposed that the radical SAM enzyme TunB might catalyze the formation of a C–C bond between C-6 of UDP-6-deoxy-GalNAc-5,6-ene and 5′-uridine radical to give the contiguous C-11 chain in tunicamine (84) but this remains a hypothesis. The gene cluster for mildiomycin (114) biosynthesis has been identified and MilM, an aspartate aminotransferase homologue, is proposed to catalyze the deamination of arginine to give α-keto-δ-guanidinovalerate, which is subsequently coupled to the nucleoside by MilN, a dihydropicolinate synthetase homologue. Activity has yet to be demonstrated for either of these enzymes. Although radiotracer data regarding griseolic acid (127) biosynthesis has been reported, no additional genetic or biochemical data is available at present. Similarly, while cell-free assays have implied that sinefungin (133) is derived from ribose, and arginine, with subsequent addition of adenine, no genes or enzymes involved in this process have been identified. Several pathways have been proposed for the biosynthesis of the 4-thioheptouronic acid core (47) of albomycin (38) based on he albomycin gene cluster but they are purely hypothetical. The source and mechanism of incorporation of C-7 and C-8 into the bicyclic octose sugar moiety remain to be established despite the availability of sequence information on the apramycin (144) gene cluster from two organisms. Moreover, nothing is known regarding the biosynthesis of the high-carbon sugars in hikizimycin (158), amipurimycin (156), the miharamycins (157), spicamycin (160), septacidin (161), anicemycin (163), the herbicidins (164–166), or caryose (174).
This review is intended to provide an update of current research in the biosynthesis of several classes of “rare” unusual sugars along with a summary of work that remains to be done. It is our hope that this review will inspire more scientists to get involved in this underexplored field to study the biosynthesis of unusual sugars. The challenge is not only to elucidate the complex biosynthetic pathways but also to investigate the mechanisms of intriguing enzyme-catalyzed transformations. The opportunities to find new pathways and new enzymes are numerous, and the likelihood of discovering new and exciting chemistry is high. In addition, future research in this field offers tremendous potential to develop an array of glycosylated metabolites that possess useful biological activities.
Acknowledgments
This work is supported in part by grants from the National Institutes of Health (GM035906 and GM054346) and the Welch Foundation (F-1511). R.M.M is a recipient of the Ruth L. Kirschstein National Research Service Award (GM103181).
Footnotes
Part of the carbohydrate chemistry themed issue.
References
- 1.Thibodeaux CJ, Melançon CE, Liu H-w. Nature. 2007;446:1008–1016. doi: 10.1038/nature05814. [DOI] [PubMed] [Google Scholar]
- 2.Thibodeaux CJ, Melançon CE, Liu Hw. Angew Chem, Int Ed. 2008;47:9814–9859. doi: 10.1002/anie.200801204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nedal A, Zotchev SB. Appl Microbiol Biotechnol. 2004;64:7–15. doi: 10.1007/s00253-003-1535-9. [DOI] [PubMed] [Google Scholar]
- 4.Romo AJ, Liu Hw. Biochim Biophys Acta, Proteins Proteomics. 2011;1814:1534–1547. doi: 10.1016/j.bbapap.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Timmons SC, Thorson JS. Curr Opin Chem Biol. 2008;12:297–305. doi: 10.1016/j.cbpa.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee MD, Dunne TS, Chang CC, Ellestad GA, Siegel MM, Morton GO, McGahren WJ, Borders DB. J Am Chem Soc. 1987;109:3466–3468. [Google Scholar]
- 7.Golik J, Clardy J, Dubay G, Groenewold G, Kawaguchi H, Konishi M, Krishnan B, Ohkuma H, Saitoh K, Doyle TW. J Am Chem Soc. 1987;109:3461–3462. [Google Scholar]
- 8.Golik J, Dubay G, Groenewold G, Kawaguchi H, Konishi M, Krishnan B, Ohkuma H, Saitoh K, Doyle TW. J Am Chem Soc. 1987;109:3462–3464. doi: 10.7164/antibiotics.38.1605. [DOI] [PubMed] [Google Scholar]
- 9.Ahlert J, Shepard E, Lomovskaya N, Zazopoulos E, Staffa A, Bachmann BO, Huang K, Fonstein L, Czisny A, Whitwam RE, Farnet CM, Thorson JS. Science. 2002;297:1173–1176. doi: 10.1126/science.1072105. [DOI] [PubMed] [Google Scholar]
- 10.Liu W, Ahlert J, Gao Q, Wendt-Pienkowski E, Shen B, Thorson JS. Proc Natl Acad Sci U S A. 2003;100:11959–11963. doi: 10.1073/pnas.2034291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kelly WL, Townsend CA. J Am Chem Soc. 2002;124:8186–8187. doi: 10.1021/ja025926g. [DOI] [PubMed] [Google Scholar]
- 12.Johnson HD, Thorson JS. J Am Chem Soc. 2008;130:17662–17663. doi: 10.1021/ja807557a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parry R, Nishino S, Spain J. Nat Prod Rep. 2011;28:152–167. doi: 10.1039/c0np00024h. [DOI] [PubMed] [Google Scholar]
- 14.Mallams AK, Puar MS, Rossman RR, McPhail AT, Macfarlane RD, Stephens RL. J Chem Soc, Perkin Trans 1. 1983:1497–1534. [Google Scholar]
- 15.Tamaoki T, Kasai M, Shirahata K, Ohkubo S, Morimoto M, Mineura K, Ishii S, Tomita F. J Antibiot. 1980;33:946–950. doi: 10.7164/antibiotics.33.946. [DOI] [PubMed] [Google Scholar]
- 16.Tamaoki T, Kasai M, Shirahata K, Tomita F. J Antibiot. 1982;35:979–984. doi: 10.7164/antibiotics.35.979. [DOI] [PubMed] [Google Scholar]
- 17.Jiang ZD, Jensen PR, Fenical W. Bioorg Med Chem Lett. 1999;9:2003–2006. doi: 10.1016/s0960-894x(99)00337-6. [DOI] [PubMed] [Google Scholar]
- 18.Igarashi Y, Takagi K, Kan Y, Fujii K, Harada K-i, Furumai T, Oki T. J Antibiot. 2000;53:233–240. doi: 10.7164/antibiotics.53.233. [DOI] [PubMed] [Google Scholar]
- 19.Mizsak SA, Hoeksema H, Pschigoda LM. J Antibiot. 1979;32:771–772. doi: 10.7164/antibiotics.32.771. [DOI] [PubMed] [Google Scholar]
- 20.Hoeksema H, Mizsak SA, Baczynskyj L, Pschigoda L. J Am Chem Soc. 1982;104:5173–5181. [Google Scholar]
- 21.Brimacombe JS, Rahman KMM. J Chem Soc, Perkin Trans 1. 1985:1067–1072. [Google Scholar]
- 22.Brimacombe JS, Rahman KMM. J Chem Soc, Perkin Trans 1. 1985:1073–1079. [Google Scholar]
- 23.Ganguly AK, Sarre OZ, Reimann H. J Am Chem Soc. 1968;90:7129–7130. doi: 10.1021/ja01027a047. [DOI] [PubMed] [Google Scholar]
- 24.Ganguly AK, Sarre OZ, Greeves D, Morton J. J Am Chem Soc. 1975;97:1982–1985. doi: 10.1021/ja00840a078. [DOI] [PubMed] [Google Scholar]
- 25.Ganguly AK, Sarre OZ, McPhail AT, Onan KD. J Chem Soc, Chem Commun. 1977:313–314. [Google Scholar]
- 26.Ishii K, Kondo S, Nichimura Y, Hamada M, Takeuchi T, Umezawa H. J Antibiot. 1983;36:451–453. doi: 10.7164/antibiotics.36.451. [DOI] [PubMed] [Google Scholar]
- 27.Ishii K, Nichimura Y, Naganawa H, Kondo S, Umezawa H. J Antibiot. 1984;37:344–353. doi: 10.7164/antibiotics.37.344. [DOI] [PubMed] [Google Scholar]
- 28.Kawai H, Hayakawa Y, Nakagawa M, Imamura K, Tanabe K, Shimazu A, Seto H, take N. J Antibiot. 1983;36:1569–1571. doi: 10.7164/antibiotics.36.1569. [DOI] [PubMed] [Google Scholar]
- 29.Kawai H, Hayakawa Y, Nakagawa M, Furihata K, Seto H, take N. Tetrahedron Lett. 1984;25:1941–1944. [Google Scholar]
- 30.Ishigami K, Hayakawa Y, Seto H. J Antibiot. 1994;47:1219–1225. doi: 10.7164/antibiotics.47.1219. [DOI] [PubMed] [Google Scholar]
- 31.Hutter K, Baader E, Frobel K, Zeeck A, Bauer K, Gau W, Kurz J, Schroder T, Wunsche C, Karl W, Wendisch D. J Antibiot. 1986;39:1193–1204. doi: 10.7164/antibiotics.39.1193. [DOI] [PubMed] [Google Scholar]
- 32.King R, Hutter K, Zeeck A, Schmidt-Base K, Egert E. J Antibiot. 1989;42:7–13. doi: 10.7164/antibiotics.42.7. [DOI] [PubMed] [Google Scholar]
- 33.Winkler R, Hertweck C. ChemBio Chem. 2007;8:973–977. doi: 10.1002/cbic.200700042. [DOI] [PubMed] [Google Scholar]
- 34.Zhang H, White-Phillip JA, Melancon CE, Kwon H-j, Yu W-l, Liu H-w. J Am Chem Soc. 2007;129:14670–14683. doi: 10.1021/ja0744854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bannister B, Zapotocky BA. J Antibiot. 1992;45:1313–1324. doi: 10.7164/antibiotics.45.1313. [DOI] [PubMed] [Google Scholar]
- 36.Fang J, Zhang Y, Huang L, Jia X, Zhang Q, Zhang X, Tang G, Liu W. J Bacteriol. 2008;190:6014–6025. doi: 10.1128/JB.00533-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim CG, Lamichhane J, Song KI, Nguyen V, Kim DH, Jeong TS, Kang SH, Kim KW, Maharjan J, Hong YS, Kang J, Yoo JC, Lee JJ, Oh TJ, Liou K, Sohng J. Arch Microbiol. 2008;189:463–473. doi: 10.1007/s00203-007-0337-3. [DOI] [PubMed] [Google Scholar]
- 38.Hosted TJ, Wang TX, Alexander DC, Horan AC. J Ind Microbiol Biotechnol. 2001;27:386–392. doi: 10.1038/sj.jim.7000189. [DOI] [PubMed] [Google Scholar]
- 39.Hu Y, Al-Mestarihi A, Grimes CL, Kahne D, Bachmann BO. J Am Chem Soc. 2008;130:15756–15757. doi: 10.1021/ja8051415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vey JL, Al-Mestarihi A, Hu Y, Funk MA, Bachmann BO, Iverson TM. Biochemistry. 2010;49:9306–9317. doi: 10.1021/bi101336u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bruender NA, Thoden JB, Holden HM. Biochemistry. 2010;49:3517–3524. doi: 10.1021/bi100318v. [DOI] [PubMed] [Google Scholar]
- 42.Parry RJ. In: Comprehensive Natural Products Chemistry. Meth-Cohn O, Barton Sir D, Nakanishi K, editors. Pergamon; Oxford: 1999. pp. 825–863. [Google Scholar]
- 43.Yuasa H, Izumi M, Hashimoto H. ChemInform. 2002;33:250–250. [Google Scholar]
- 44.Witczak Z, Culhane J. Appl Microbiol Biotechnol. 2005;69:237–244. doi: 10.1007/s00253-005-0156-x. [DOI] [PubMed] [Google Scholar]
- 45.Etoh H, Iguchi M, Nagasawa T, Tani Y, Yamada H, Fukami H. Agric Biol Chem. 1987;51:1819–1824. [Google Scholar]
- 46.Whistler RL, Feather MS, Ingles DL. J Am Chem Soc. 1962;84:122–122. [Google Scholar]
- 47.Capon RJ, MacLeod JK. J Chem Soc, Chem Commun. 1987:1200–1201. [Google Scholar]
- 48.Benz G, Schröder T, Kurz J, Wünsche C, Karl W, Steffens G, Pfitzner J, Schmidt D. Angew Chem, Int Ed. 1982;21:1322–1335. [Google Scholar]
- 49.Benz G, Born L, Brieden M, Grosser R, Kurz J, Paulsen H, Sinnwell V, Webber B. Liebigs Ann Chem. 1984;1984:1408–1423. [Google Scholar]
- 50.Braun V, Pramanik A, Gwinner T, Köberle M, Bohn E. BioMetals. 2009;22:3–13. doi: 10.1007/s10534-008-9199-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Braun V, Günthner K, Hantke K, Zimmermann L. J Bacteriol. 1983;156:308–315. doi: 10.1128/jb.156.1.308-315.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stefanska AL, Fulston M, Houge-Frydrych CSV, Jones JJ, Warr SR. J Antibiot. 2000;53:1346–1353. doi: 10.7164/antibiotics.53.1346. [DOI] [PubMed] [Google Scholar]
- 53.Paulsen H, Brieden M, Benz G. Liebigs Ann Chem. 1987;1987:565–575. [Google Scholar]
- 54.Zeng Y, Kulkarni A, Yang Z, Patil PB, Zhou W, Chi X, Van Lanen S, Chen S. ACS Chem Biol. 2012;7:1565–1575. doi: 10.1021/cb300173x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jarrett JT. Arch Biochem Biophys. 2005;433:312–321. doi: 10.1016/j.abb.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 56.Booker SJ, Cicchillo RM, Grove TL. Curr Opin Chem Biol. 2007;11:543–552. doi: 10.1016/j.cbpa.2007.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frey PA, Hegeman AD, Ruzicka FJ. Crit Rev Biochem Mol Biol. 2008;43:63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- 58.Jpn. Kokai Tokkyo Koho JP 02–16894 A. Japan Pat. 1990
- 59.Sasaki E, Ogasawara Y, Liu Hw. J Am Chem Soc. 2010;132:7405–7417. doi: 10.1021/ja1014037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Begley TP. Nat Prod Rep. 2006;23:15–25. doi: 10.1039/b207131m. [DOI] [PubMed] [Google Scholar]
- 61.Begley TP, Ealick SE, McLafferty FW. Biochem Soc Trans. 2012;40:555–560. doi: 10.1042/BST20120084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sasaki E, Liu H-w. J Am Chem Soc. 2010;132:15544–15546. doi: 10.1021/ja108061c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rohr J. J Chem Soc, Chem Commun. 1989:492–493. [Google Scholar]
- 64.Mason DJ, Dietz A, DeBoer C. Antimicrob Agents Chemother. 1962:554–559. [Google Scholar]
- 65.Herr RR, Bergy ME. Antimicrob Agents Chemother. 1962:560–564. [Google Scholar]
- 66.Mason DJ, Lewis C. Antimicrob Agents Chemother. 1964:7–12. [PubMed] [Google Scholar]
- 67.Lewis C, Clapp HW, Grady JE. Antimicrob Agents Chemother. 1963:570–582. [Google Scholar]
- 68.Dunkle JA, Xiong L, Mankin AS, Cate JHD. Proc Natl Acad Sci U S A. 2010;107:17152–17157. doi: 10.1073/pnas.1007988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hoeksema H, Bannister B, Birkenmeyer RD, Kagan F, Magerlein BJ, MacKellar FA, Schroeder W, Slomp G, Herr RR. J Am Chem Soc. 1964;86:4223–4224. [Google Scholar]
- 70.Hanada M, Tsunakawa M, Tomita K, Tsukiura H, Kawaguchi H. J Antibiot. 1980;33:751–753. doi: 10.7164/antibiotics.33.751. [DOI] [PubMed] [Google Scholar]
- 71.Hoeksema H, Crum GF, DeVries WH. Antibiot Ann. 1955;2:837–841. [Google Scholar]
- 72.Hoeksema H. J Am Chem Soc. 1968;90:755–757. doi: 10.1021/ja01005a036. [DOI] [PubMed] [Google Scholar]
- 73.Smieja M. Can J Infect Dis Med Microbiol. 1998;9:22–28. doi: 10.1155/1998/538090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Peschke U, Schmidt H, Zhang HZ, Piepersberg W. Mol Microbiol. 1995;16:1137–1156. doi: 10.1111/j.1365-2958.1995.tb02338.x. [DOI] [PubMed] [Google Scholar]
- 75.Koběrská M, Kopecký J, Olšovská J, Jelínková M, Ulanova D, Man P, Flieger M, Janata J. Folia Microbiol. 2008;53:395–401. doi: 10.1007/s12223-008-0060-8. [DOI] [PubMed] [Google Scholar]
- 76.Čermák L, Novotná J, Ságová-Marečková M, Kopecký J, Najmanová L, Janata J. Folia Microbiol. 2007;52:457–462. doi: 10.1007/BF02932104. [DOI] [PubMed] [Google Scholar]
- 77.Neusser D, Schmidt H, Spizèk J, Novotnà J, Peschke U, Kaschabeck S, Tichy P, Piepersberg W. Arch Microbiol. 1998;169:322–332. doi: 10.1007/s002030050578. [DOI] [PubMed] [Google Scholar]
- 78.Hola K, Janata J, Kopecky J, Spizek J. Lett Appl Microbiol. 2003;37:470–474. doi: 10.1046/j.1472-765x.2003.01432.x. [DOI] [PubMed] [Google Scholar]
- 79.Brahme NM, Gonzalez JE, Mizsak S, Rolls JR, Hessler EJ, Hurley LH. J Am Chem Soc. 1984;106:7878–7883. [Google Scholar]
- 80.Spížek J, Řezanka T. Appl Microbiol Biotechnol. 2004;63:510–519. doi: 10.1007/s00253-003-1431-3. [DOI] [PubMed] [Google Scholar]
- 81.McDonald LA, Capson TL, Krishnamurthy G, Ding WD, Ellestad GA, Bernan VS, Maiese WM, Lassota P, Kramer RA, Ireland CM. J Am Chem Soc. 1996;118:10898–10899. [Google Scholar]
- 82.Oku N, Matsunaga S, Fusetani N. J Am Chem Soc. 2003;125:2044–2045. doi: 10.1021/ja0296780. [DOI] [PubMed] [Google Scholar]
- 83.US 6733998 B1. US Pat. 2004
- 84.Agerbirk N, Olsen CE. Phytochemistry. 2012;77:16–45. doi: 10.1016/j.phytochem.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 85.Fahey JW, Zalcmann AT, Talalay P. Phytochemistry. 2001;56:5–51. doi: 10.1016/s0031-9422(00)00316-2. [DOI] [PubMed] [Google Scholar]
- 86.Halkier BA, Gershenzon J. Ann Rev Plant Biol. 2006;57:303–333. doi: 10.1146/annurev.arplant.57.032905.105228. [DOI] [PubMed] [Google Scholar]
- 87.Sonderby IE, Geu-Flores F, Halkier BA. Trends Plant Sci. 2010;15:283–290. doi: 10.1016/j.tplants.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 88.Jones MG. In: The Chemistry and Biology of Volatiles. Herrmann A, editor. John Wiley & Sons; New York City: 2010. pp. 203–229. [Google Scholar]
- 89.Schlaeppi K, Bodenhausen N, Buchala A, Mauch F, Reymond P. Plant J. 2008;55:774–786. doi: 10.1111/j.1365-313X.2008.03545.x. [DOI] [PubMed] [Google Scholar]
- 90.Bednarek P, Piślewska-Bednarek M, Svatoš A, Schneider B, Doubsky J, Mansurova M, Humphry M, Consonni C, Panstruga R, Sanchez-Vallet A, Molina A, Schulze-Lefert P. Science. 2009;323:101–106. doi: 10.1126/science.1163732. [DOI] [PubMed] [Google Scholar]
- 91.Geu-Flores F, Nielsen MT, Nafisi M, Moldrup ME, Olsen CE, Motawia MS, Halkier BA. Nat Chem Biol. 2009;5:575–577. doi: 10.1038/nchembio.185. [DOI] [PubMed] [Google Scholar]
- 92.Dixon DP, Skipsey M, Edwards R. Phytochemistry. 2010;71:338–350. doi: 10.1016/j.phytochem.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 93.Shapiro SK, Schlenk F. Advances in Enzymology and Related Areas of Molecular Biology. John Wiley & Sons; New York City: 2006. pp. 237–280. [Google Scholar]
- 94.Reisch CR, Moran MA, Whitman WB. Front Microbiol. 2011:2. doi: 10.3389/fmicb.2011.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yoshikawa M, Murakami T, Shimada H, Matsuda H, Yamahara J, Tanabe G, Muraoka O. Tetrahedron Lett. 1997;38:8367–8370. [Google Scholar]
- 96.Yoshikawa M, Murakami T, Yashiro K, Matsuda H. Chem Pharm Bull. 1998;46:1339–1340. doi: 10.1248/cpb.46.1339. [DOI] [PubMed] [Google Scholar]
- 97.Yoshikawa M, Xu F, Nakamura S, Wang T, Matsuda H, Tanabe G, Muraoka O. Heterocycles. 2008;75:1397–1405. [Google Scholar]
- 98.Mohan S, Pinto BM. Carbohydr Res. 2007;342:1551–1580. doi: 10.1016/j.carres.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 99.Benson AA. Ann Rev Plant Biol. 2002;53:1–25. doi: 10.1146/annurev.arplant.53.091201.142547. [DOI] [PubMed] [Google Scholar]
- 100.Benson AA, Daniel H, Wiser R. Proc Natl Acad Sci. 1959;45:1582–1587. doi: 10.1073/pnas.45.11.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Block MA, Dorne AJ, Joyard J, Douce R. J Biol Chem. 1983;258:13281–13286. [PubMed] [Google Scholar]
- 102.Shimojima M. Prog Lipid Res. 2010;50:234–239. doi: 10.1016/j.plipres.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 103.Shibuya I, Yagi T, Benson AA. Japanese Society of Plant Physiology. University of Tokyo Press; Tokyo: 1963. pp. 627–636. [Google Scholar]
- 104.Benning C, Somerville CR. J Bacteriol. 1992;174:2352–2360. doi: 10.1128/jb.174.7.2352-2360.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Benning C, Somerville CR. J Bacteriol. 1992;174:6479–6487. doi: 10.1128/jb.174.20.6479-6487.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pugh CE, Roy AB, Hawkes T, Harwood JL. Biochem J. 1995;309:513–519. doi: 10.1042/bj3090513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sanda S, Leustek T, Theisen MJ, Garavito RM, Benning C. J Biol Chem. 2001;276:3941–3946. doi: 10.1074/jbc.M008200200. [DOI] [PubMed] [Google Scholar]
- 108.Shimojima M, Benning C. Arch Biochem Biophys. 2003;413:123–130. doi: 10.1016/s0003-9861(03)00112-7. [DOI] [PubMed] [Google Scholar]
- 109.Winn M, Goss RJM, Kimura Ki, Bugg TDH. Nat Prod Rep. 2010;27:279–304. doi: 10.1039/b816215h. [DOI] [PubMed] [Google Scholar]
- 110.Takatsuki A, Arima K, Tamura G. J Antibiot. 1971;24:215–223. doi: 10.7164/antibiotics.24.215. [DOI] [PubMed] [Google Scholar]
- 111.US 4237225. US Pat. 1980
- 112.Takatsuki A, Kawamura K, Okina M, Kodama Y, Ito T, Tamura G. Agric Biol Chem. 1977;41:2307–2309. [Google Scholar]
- 113.Eckardt K, Thrum H, Bradler G, Tonew E, Tonew M. J Antibiot. 1975;28:274–279. doi: 10.7164/antibiotics.28.274. [DOI] [PubMed] [Google Scholar]
- 114.Vogel P, Petterson DS, Berry PH, Frahn JL, Anderton N, Cockrum PA, Edgar JA, Jago MV, Lanigan GW, Payne AL, Culvenor CC. Aust J Exp Biol Med Sci. 1981;59:455–467. doi: 10.1038/icb.1981.39. [DOI] [PubMed] [Google Scholar]
- 115.Kenig M, Reading C. J Antibiot. 1979;32:549–554. doi: 10.7164/antibiotics.32.549. [DOI] [PubMed] [Google Scholar]
- 116.Nakamura S, Arai M, Karasawa K, Yonehara H. J Antibiot. 1957;10:248–253. [PubMed] [Google Scholar]
- 117.Mizuno M, Shimojim Y, Sugawara T, Takeda I. J Antibiot. 1971;24:896–899. doi: 10.7164/antibiotics.24.896. [DOI] [PubMed] [Google Scholar]
- 118.Brandish PE, Kimura KI, Inukai M, Southgate R, Lonsdale JT, Bugg TD. Antimicrob Agents Chemother. 1996;40:1640–1644. doi: 10.1128/aac.40.7.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Heifetz A, Keenan RW, Elbein AD. Biochemistry. 1979;18:2186–2192. doi: 10.1021/bi00578a008. [DOI] [PubMed] [Google Scholar]
- 120.Price NP, Momany FA. Glycobiology. 2005;15:29R–42R. doi: 10.1093/glycob/cwi065. [DOI] [PubMed] [Google Scholar]
- 121.Patterson SI, Skene JHP. J Cell Biol. 1994;124:521–536. doi: 10.1083/jcb.124.4.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tsvetanova BC, Kiemle DJ, Price NP. J Biol Chem. 2002;277:35289–35296. doi: 10.1074/jbc.M201345200. [DOI] [PubMed] [Google Scholar]
- 123.Wyszynski FJ, Hesketh AR, Bibb MJ, Davis BG. Chem Sci. 2010;1:581–589. [Google Scholar]
- 124.Chen W, Qu D, Zhai L, Tao M, Wang Y, Lin S, Price NP, Deng Z. Protein Cell. 2010;1:1093–1105. doi: 10.1007/s13238-010-0127-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wyszynski FJ, Lee SS, Yabe T, Wang H, Gomez-Escribano JP, Bibb MJ, Lee SJ, Davies GJ, Davis BG. Nat Chem. 2012;4:539–546. doi: 10.1038/nchem.1351. [DOI] [PubMed] [Google Scholar]
- 126.Isono K, Uramoto M, Kusakabe H, Kimura Ki, Izaki K, Nelson CC, McCloskey JA. J Antibiot. 1985;38:1617–1621. doi: 10.7164/antibiotics.38.1617. [DOI] [PubMed] [Google Scholar]
- 127.Igarashi M, Nakagawa N, Doi N, Hattori S, Naganawa H, Hamada M. J Antibiot. 2003;56:580–583. doi: 10.7164/antibiotics.56.580. [DOI] [PubMed] [Google Scholar]
- 128.Igarashi M, Takahashi Y, Shitara T, Nakamura H, Naganawa H, Miyake T, Akamatsu Y. J Antibiot. 2005;58:327–337. doi: 10.1038/ja.2005.41. [DOI] [PubMed] [Google Scholar]
- 129.McDonald LA, Barbieri LR, Carter GT, Lenoy E, Lotvin J, Petersen PJ, Siegel MM, Singh G, Williamson RT. J Am Chem Soc. 2002;124:10260–10261. doi: 10.1021/ja017748h. [DOI] [PubMed] [Google Scholar]
- 130.Funabashi M, Baba S, Nonaka K, Hosobuchi M, Fujita Y, Shibata T, Van Lanen SG. ChemBio Chem. 2010;11:184–190. doi: 10.1002/cbic.200900665. [DOI] [PubMed] [Google Scholar]
- 131.4950605, 1990.
- 132.Kaysser L, Siebenberg S, Kammerer B, Gust B. ChemBio Chem. 2010;11:191–196. doi: 10.1002/cbic.200900637. [DOI] [PubMed] [Google Scholar]
- 133.Kaysser L, Lutsch L, Siebenberg S, Wemakor E, Kammerer B, Gust B. J Biol Chem. 2009;284:14987–14996. doi: 10.1074/jbc.M901258200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cheng L, Chen W, Zhai L, Xu D, Huang T, Lin S, Zhou X, Deng Z. Mol Biosyst. 2011;7:920–927. doi: 10.1039/c0mb00237b. [DOI] [PubMed] [Google Scholar]
- 135.Makart S, Bechtold M, Panke S. J Biotechnol. 2007;130:402–410. doi: 10.1016/j.jbiotec.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 136.Yang Z, Chi X, Funabashi M, Baba S, Nonaka K, Pahari P, Unrine J, Jacobsen JM, Elliott GI, Rohr J, Van Lanen SG. J Biol Chem. 2011;286:7885–7892. doi: 10.1074/jbc.M110.203562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fujita Y, Murakami R, Muramatsu Y, Miyakoshi S, Takatsu T. J Antibiot. 2008;61:545–549. doi: 10.1038/ja.2008.72. [DOI] [PubMed] [Google Scholar]
- 138.Murakami R, Fujita Y, Kizuka M, Kagawa T, Muramatsu Y, Miyakoshi S, Takatsu T, Inukai M. J Antibiot. 2007;60:690–695. doi: 10.1038/ja.2007.88. [DOI] [PubMed] [Google Scholar]
- 139.Isono K, Crain PF, Mccloskey JA. J Am Chem Soc. 1975;97:943–945. [Google Scholar]
- 140.Hori M, Eguchi J, Kakiki K, Misato T. J Antibiot. 1974;27:260–266. doi: 10.7164/antibiotics.27.260. [DOI] [PubMed] [Google Scholar]
- 141.Kiyoshi I, Tsutomu S, Kiyoshi H, Shunji F, Saburo S. J Am Chem Soc. 1978;100:3937–3939. [Google Scholar]
- 142.Sato T, Hirasawa K, Uzawa J, Inaba T, Isono K. Tetrahedron Lett. 1979;20:3441–3444. [Google Scholar]
- 143.Dähn U, Hagenmaier H, Höhne H, König WA, Wolf G, Zähner H. Arch Microbiol. 1976;107:143–160. doi: 10.1007/BF00446834. [DOI] [PubMed] [Google Scholar]
- 144.Schuz TC, Fiedler HP, Zahner H, Rieck M, Konig WA. J Antibiot. 1992;45:199–206. doi: 10.7164/antibiotics.45.199. [DOI] [PubMed] [Google Scholar]
- 145.Chen W, Huang T, He X, Meng Q, You D, Bai L, Li J, Wu M, Li R, Xie Z, Zhou H, Zhou X, Tan H, Deng Z. J Biol Chem. 2009;284:10627–10638. doi: 10.1074/jbc.M807534200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Liao G, Li J, Li L, Yang H, Tian Y, Tan H. Microb Cell Fact. 2010;9:6. doi: 10.1186/1475-2859-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ginj C, Ruegger H, Amrhein N, Macheroux P. ChemBio Chem. 2005;6:1974–1976. doi: 10.1002/cbic.200500208. [DOI] [PubMed] [Google Scholar]
- 148.Oberdorfer G, Binter A, Ginj C, Macheroux P, Gruber K. J Biol Chem. 2012 doi: 10.1074/jbc.M112.352096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lauer B, Sussmuth R, Kaiser D, Jung G, Bormann C. J Antibiot. 2000;53:385–392. doi: 10.7164/antibiotics.53.385. [DOI] [PubMed] [Google Scholar]
- 150.Binter A, Oberdorfer G, Hofzumahaus S, Nerstheimer S, Altenbacher G, Gruber K, Macheroux P. FEBS J. 2011;278:4122–4135. doi: 10.1111/j.1742-4658.2011.08319.x. [DOI] [PubMed] [Google Scholar]
- 151.Kanzo S, Akira S, Saburo T. Agric Biol Chem. 1977;41:2033–2039. [Google Scholar]
- 152.Kanzo S, Akira S, Saburo T. Agric Biol Chem. 1976;40:1993–1999. [Google Scholar]
- 153.Kanzo S, Akira S, Saburo T. Agric Biol Chem. 1974;38:1883–1890. [Google Scholar]
- 154.Harada S, Kishi T. Journal of Antibiotics. 1978;31:519–524. doi: 10.7164/antibiotics.31.519. [DOI] [PubMed] [Google Scholar]
- 155.Feduchi E, Cosin M, Carrasco L. J Antibiot. 1985;38:415–419. doi: 10.7164/antibiotics.38.415. [DOI] [PubMed] [Google Scholar]
- 156.Otake N, Takeuchi S, Endo T, Yonehara H. Tetrahedron Lett. 1965:1411–1419. doi: 10.1016/s0040-4039(00)90080-7. [DOI] [PubMed] [Google Scholar]
- 157.Argoudelis AD, Baczynskyj L, Kuo MT, Laborde AL, Sebek OK, Truesdell SE, Shilliday FB. J Antibiot. 1987;40:750–760. doi: 10.7164/antibiotics.40.750. [DOI] [PubMed] [Google Scholar]
- 158.Fox JJ, Kuwada Y, Watanabe KA. Tetrahedron Lett. 1968:6029–6032. doi: 10.1016/s0040-4039(00)70784-2. [DOI] [PubMed] [Google Scholar]
- 159.Takahashi A, Ikeda D, Naganawa H, Okami Y, Umezawa H. J Antibiot. 1986;39:1041–1046. doi: 10.7164/antibiotics.39.1041. [DOI] [PubMed] [Google Scholar]
- 160.Cone MC, Petrich AK, Gould SJ, Zabriskie TM. J Antibiot. 1998;51:570–578. doi: 10.7164/antibiotics.51.570. [DOI] [PubMed] [Google Scholar]
- 161.Wu J, Li L, Deng Z, Zabriskie TM, He X. ChemBio Chem. 2012;13:1613–1621. doi: 10.1002/cbic.201200173. [DOI] [PubMed] [Google Scholar]
- 162.Li L, Xu Z, Xu X, Wu J, Zhang Y, He X, Zabriskie TM, Deng Z. ChemBio Chem. 2008;9:1286–1294. doi: 10.1002/cbic.200800008. [DOI] [PubMed] [Google Scholar]
- 163.Takahashi S, Nakagawa F, Kawazoe K, Furukawa Y, Sato S, Tamura C, Naito A. J Antibiot. 1985;38:830–834. doi: 10.7164/antibiotics.38.830. [DOI] [PubMed] [Google Scholar]
- 164.Takahashi S, Nakagawa F, Sato S. J Antibiot. 1988;41:705–706. doi: 10.7164/antibiotics.41.705. [DOI] [PubMed] [Google Scholar]
- 165.Cooper R, Mierzwa R, Patel M, Pramanik B, Puar MS, Troyanovich J, Gullo V. J Nat Prod. 1988;51:1207–1214. [Google Scholar]
- 166.Miyakoshi S, Haruyama H, Shioiri T, Takahashi S, Torikata A, Yamazaki M. J Antibiot. 1992;45:394–399. doi: 10.7164/antibiotics.45.394. [DOI] [PubMed] [Google Scholar]
- 167.Hamil RL, Hoehn MM. J Antibiot. 1973;26:463–465. doi: 10.7164/antibiotics.26.463. [DOI] [PubMed] [Google Scholar]
- 168.Nagarajan R, Chao B, Dorman DE, NSM, Occolowitz JL, Schabel A. Abstracts of the 17th interscience Conference on Antimicrobial Agents and Chemotherapy; 1977. p. Abstract 50. [Google Scholar]
- 169.Gordee RS, Butler TF. J Antibiot. 1973;26:466–470. doi: 10.7164/antibiotics.26.466. [DOI] [PubMed] [Google Scholar]
- 170.Pugh CS, Borchardt RT, Stone HO. J Biol Chem. 1978;253:4075–4077. [PubMed] [Google Scholar]
- 171.Dube DK, Mpimbaza G, Allison AC, Lederer E, Rovis L. Am J Trop Med Hyg. 1983;32:31–33. doi: 10.4269/ajtmh.1983.32.31. [DOI] [PubMed] [Google Scholar]
- 172.Berry DR, Abbott BJ. J Antibiot. 1978;31:185–191. doi: 10.7164/antibiotics.31.185. [DOI] [PubMed] [Google Scholar]
- 173.Malina H, Tempete C, Robert-Gero M. J Antibiot. 1987;40:505–511. doi: 10.7164/antibiotics.40.505. [DOI] [PubMed] [Google Scholar]
- 174.Parry RJ, Arzu IY, Ju S, Baker BJ. J Am Chem Soc. 1989;111:8981–8982. [Google Scholar]
- 175.Parry RJ, Ju SC. Tetrahedron. 1991;47:6069–6078. [Google Scholar]
- 176.Fukuda K, Tamura T, Ito H, Yamamoto S, Ochi K, Inagaki K. J Biosci Bioeng. 2010;109:459–465. doi: 10.1016/j.jbiosc.2009.10.017. [DOI] [PubMed] [Google Scholar]
- 177.Sasaki E, Lin C-I, Lin K-Y, Liu H-w. J Am Chem Soc. 2012 doi: 10.1021/ja308221z. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Thompson RQ, Presti EA. Nebramycin, a new broad-spectrum antibiotic complex. 3. Isolation and chemical-physical properties. 1967 [PubMed] [Google Scholar]
- 179.O’Connor S, Lam LK, Jones ND, Chaney MO. J Org Chem. 1976;41:2087–2092. doi: 10.1021/jo00874a003. [DOI] [PubMed] [Google Scholar]
- 180.Rinehart KL. Abstracts of Papers of the American Chemical Society. 1979:12. [Google Scholar]
- 181.Huang F, Spiteller D, Koorbanally NA, Li Y, Llewellyn NM, Spencer JB. ChemBio Chem. 2007;8:283–288. doi: 10.1002/cbic.200600371. [DOI] [PubMed] [Google Scholar]
- 182.Kudo F, Eguchi T. J Antibiot. 2009;62:471–481. doi: 10.1038/ja.2009.76. [DOI] [PubMed] [Google Scholar]
- 183.Celmer WD, Murai K, Rao KV, Tanner FW, Marsh WS. Antibiot Ann. 1957–1958:484. [PubMed] [Google Scholar]
- 184.Matern U, Grisebach H, Karl W, Achenbach H. Eur J Biochem. 1972;29:1–4. doi: 10.1111/j.1432-1033.1972.tb01949.x. [DOI] [PubMed] [Google Scholar]
- 185.Furumai T, Igarashi Y, Higuchi H, Saito N, Oki T. J Antibiot. 2003;56:B14–B14. [Google Scholar]
- 186.Matern U, Grisebach H. Eur J Biochem. 1972;29:5–11. doi: 10.1111/j.1432-1033.1972.tb01950.x. [DOI] [PubMed] [Google Scholar]
- 187.Chen HW, Guo ZH, Liu HW. J Am Chem Soc. 1998;120:11796–11797. [Google Scholar]
- 188.Harada S, Kishi T. J Antibiot. 1977;30:11–16. doi: 10.7164/antibiotics.30.11. [DOI] [PubMed] [Google Scholar]
- 189.Goto T, Toya Y, Ohgi T, Kondo T. Tetrahedron Lett. 1982;23:1271–1274. [Google Scholar]
- 190.Tsuruoka T, Yumoto H, Ezaki N, Niida A. Sci Rep Meiji Seika Kaisha. 1967;9:1967. [Google Scholar]
- 191.Seto H, Koyama M, Ogino H, Tsuruoka T, Inouye S, Otake N. Tetrahedron Lett. 1983;24:1805–1808. [Google Scholar]
- 192.Uchida K, Ichikawa T, Shimauch Y, Ishikura T, Ozaki A. J Antibiot. 1971;24:2593. doi: 10.7164/antibiotics.24.259. [DOI] [PubMed] [Google Scholar]
- 193.Gonzalez A, Vazquez D, Jimenez A. Biochim Biophys Acta. 1979;561:403–409. doi: 10.1016/0005-2787(79)90148-5. [DOI] [PubMed] [Google Scholar]
- 194.Uchida K. Agric Biol Chem. 1976;40:395–404. [Google Scholar]
- 195.Hamill RL, Hoehn MM. J Antibiot. 1964;17:100–103. [PubMed] [Google Scholar]
- 196.Ennifar S, Das BC, Nash SM, Nagarajan R. J Chem Soc, Chem Commun. 1977:41–42. doi: 10.1021/jo00440a019. [DOI] [PubMed] [Google Scholar]
- 197.Dutcher JD, Vonsaltza MH, Pansy FE. Antimicrob Agents Chemother. 1963;161:83–88. [PubMed] [Google Scholar]
- 198.Hayakawa Y, Nakagawa M, Kawai H, Tanabe K, Nakayama H, Shimazu A, Seto H, Otake N. Agric Biol Chem. 1985;49:2685–2691. [Google Scholar]
- 199.Igarashi Y, Ootsu K, Onaka H, Fujita T, Uehara Y, Furumai T. Journal of Antibiotics. 2005;58:322–326. doi: 10.1038/ja.2005.40. [DOI] [PubMed] [Google Scholar]
- 200.Arai M, Haneishi T, Kitahara N, Enokita R, Kawakubo K. J Antibiot. 1976;29:863–869. doi: 10.7164/antibiotics.29.863. [DOI] [PubMed] [Google Scholar]
- 201.Takiguchi Y, Yoshikawa H, Terahara A, Torikata A, Terao M. J Antibiot. 1979;32:857–861. doi: 10.7164/antibiotics.32.857. [DOI] [PubMed] [Google Scholar]
- 202.Takiguchi Y, Yoshikawa H, Terahara A, Torikata A, Terao M. J Antibiot. 1979;32:862–867. doi: 10.7164/antibiotics.32.862. [DOI] [PubMed] [Google Scholar]
- 203.Yoshikawa H, Takiguchi Y, Terao M. J Antibiot. 1983;36:30–35. doi: 10.7164/antibiotics.36.30. [DOI] [PubMed] [Google Scholar]
- 204.Komaki E, Ohta Y, Tsukada Y. Biosci Biotechnol Biochem. 1997;61:2046–2050. doi: 10.1271/bbb.61.2046. [DOI] [PubMed] [Google Scholar]
- 205.Comb DG, Roseman S. J Biol Chem. 1960;235:2529–2537. [PubMed] [Google Scholar]
- 206.Cipolla L, Gabrielli L, Bini D, Russo L, Shaikh N. Nat Prod Rep. 2010;27:1618–1629. doi: 10.1039/c004750n. [DOI] [PubMed] [Google Scholar]
- 207.Levin DH, Racker E. J Biol Chem. 1959;234:2532–2539. [PubMed] [Google Scholar]
- 208.Kosma P. Curr Org Chem. 2008;12:1021–1039. [Google Scholar]
- 209.Messner P, Steiner K, Zarschler K, Schaffer C. Carbohydr Res. 2008;343:1934–1951. doi: 10.1016/j.carres.2007.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Gorshkova RP, Zubkov VA, Isakov VV, Ovodov YS. Carbohydr Res. 1984;126:308–312. doi: 10.1016/0008-6215(84)85389-6. [DOI] [PubMed] [Google Scholar]
- 211.Schmid R, Grisebach H. Eur J Biochem. 1970;14:243–252. doi: 10.1111/j.1432-1033.1970.tb00283.x. [DOI] [PubMed] [Google Scholar]
- 212.Chipman D, Barak ZA, Schloss JV. Biochim Biophys Acta, Protein Struct Mol Enzymol. 1998;1385:401–419. doi: 10.1016/s0167-4838(98)00083-1. [DOI] [PubMed] [Google Scholar]
- 213.Mansoorabadi SO, Thibodeaux CJ, Liu HW. J Org Chem. 2007;72:6329–6342. doi: 10.1021/jo0703092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Adinolfi M, Corsaro MM, Decastro C, Lanzetta R, Parrilli M, Evidente A, Lavermicocca P. Carbohydr Res. 1995;267:307–311. [Google Scholar]
- 215.Adinolfi M, Corsaro MM, DeCastro C, Evidente A, Lanzetta R, Molinaro A, Parrilli M. Carbohydr Res. 1996;284:111–118. [Google Scholar]
- 216.Adinolfi M, Corsaro MM, DeCastro C, Evidente A, Lanzetta R, Lavermicocca P, Parrilli M. Carbohydr Res. 1996;284:119–133. [Google Scholar]
- 217.Rombouts Y, Burguiere A, Maes E, Coddeville B, Elass E, Guerardel Y, Kremer L. J Biol Chem. 2009;284:20975–20988. doi: 10.1074/jbc.M109.011429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Sarkar D, Sidhu M, Singh A, Chen JM, Lammas DA, van der Sar AM, Besra GS, Bhatt A. J Bacteriol. 2011;193:2336–2340. doi: 10.1128/JB.00065-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Stinear TP, Seemann T, Harrison PF, Jenkin GA, Davies JK, Johnson PDR, Abdellah Z, Arrowsmith C, Chillingworth T, Churcher C, Clarke K, Cronin A, Davis P, Goodhead I, Holroyd N, Jagels K, Lord A, Moule S, Mungall K, Norbertczak H, Quail MA, Rabbinowitsch E, Walker D, White B, Whitehead S, Small PLC, Brosch R, Ramakrishnan L, Fischbach MA, Parkhill J, Cole ST. Genome Res. 2008;18:729–741. doi: 10.1101/gr.075069.107. [DOI] [PMC free article] [PubMed] [Google Scholar]