

Abstract
Mitochondria have been largely considered as ‘end-function’ organelles, servicing the cell by producing energy and regulating cell death in response to complex signals. Being cellular entities with vital roles, mitochondria communicate back to the cell and actively engage in determining major cellular policies. These signals, collectively referred to as retrograde signals, are encoded in the nuclear genome, or are secondary products of mitochondrial metabolism. Here, we discuss humanin, the first small peptide of a putative set of mitochondrial-derived peptides, which exhibits strong cytoprotective actions against various stress and disease models. The study of humanin and other mitochondrial-derived retrograde signal peptides will aid in the identification of genes and peptides with therapeutic and diagnostic potential in treating human diseases.
Keywords: Humanin, mitochondria, retrograde signaling, protection, stress-resistance, mitochondrial-derived peptide
Mitochondrial biology and function
It is widely accepted that the endosymbiotic origin of mitochondria was derived from α-proteobacteria that have been engulfed by and integrated into eukaryotic host cells. The incorporation of the mitochondrion (proto-mitochondrion) has changed the course of eukaryotic evolution through a monumental metabolic upgrade by employing oxygen to mass-produce energy and biosynthetic precursors [1]. Owing to their prokaryotic origin, mitochondria are also unique among intracellular organelles in that they contain their own genome. This small genome is thought to be the result of gradual loss or transfer of the great majority of the original bacterial genes to the nucleus, leaving only 13 full-size protein encoding genes that are essential for oxidative phosphorylation. This component of the human genome is beginning to be explored for its potential role in conditions such as aging, cancer, diabetes, deafness, and neurodegeneration [2].
The mitochondrion is responsible for several crucial cellular activities including energy production, regulation of programmed cell death (apoptosis), biosynthetic precursor production, heme synthesis, Fe-S cluster production, ion homeostasis, and ROS production [3]. Bestowed with such critical responsibilities, it has been known that mitochondria actively participate in determining cellular processes; however, little is understood about how mitochondria transmit information to the host cell. In this article, we will discuss a newly emerging concept regarding a novel class of communication conveyed by the mitochondrion to regulate cellular processes and cell fate (Fig 1).
Figure 1. Mitochondrial-Derived Peptides are Retrograde Signaling Molecules.

Mitochondria communicate with the cell through a process largely known as retrograde signaling. Traditionally described means of communication from the mitochondria include Ca2+, cytochrome C (cyt C), and ROS. MDP are recently identified retrograde signals, which are unique in that they are encoded within the mitochondrial genome sequences. MDPs such as humanin are thought to act as endocrine as well as intracellular factors with several biological roles regulating cell survival and metabolism.
The concept of retrograde signaling from the mitochondria to the nucleus and beyond
Mitochondria have traditionally been perceived as “end-function” organelles that receive cellular signals and in response regulate energy production and apoptosis. However, a coordinated regulation of mitochondrial and nuclear gene expression is critical for cellular homeostasis, which requires constant and active exchange of information [4]. Mitochondria-initiated communication events that signal and regulate various cellular aspects under normal, stress, and pathological states are referred to en masse as retrograde signaling [4], a biological process well conserved from the simple yeast to humans (Fig 1).
To date, a relatively limited number of retrograde signaling molecules and signaling cascades have been explored. Some of the molecules described in this regard are cytochrome C, reactive oxygen species (ROS), Ca2+, Fe2+, nitric oxide (NO), and carbon monoxide (CO) [5–7] (Fig 2). Notably, an interesting study in C. elegans described non-canonical signals exported from the mitochondria in response to proteotoxic insults [8]. However, these were not peptides encoded within the mitochondria but rather damaged matrix protein fragments generated by proteolysis that are encoded in the nuclear genome and imported later into the mitochondria. In a way, they are in a comparable class of retrograde signals with cytochrome C, in that they are nuclear-encoded protein products released back to the cell from the mitochondria. In addition, mitochondrial DNA (mtDNA) itself can act as a signal. A recent study demonstrated that inhibiting autophagy causes mitochondrial dysfunction and subsequent mtDNA export to the cytosol, via increased mitochondrial membrane permeability transition (MPT), resulting in the activation of caspase-1 inflammasome [9].
Figure 2. Cellular Actions of Humanin.
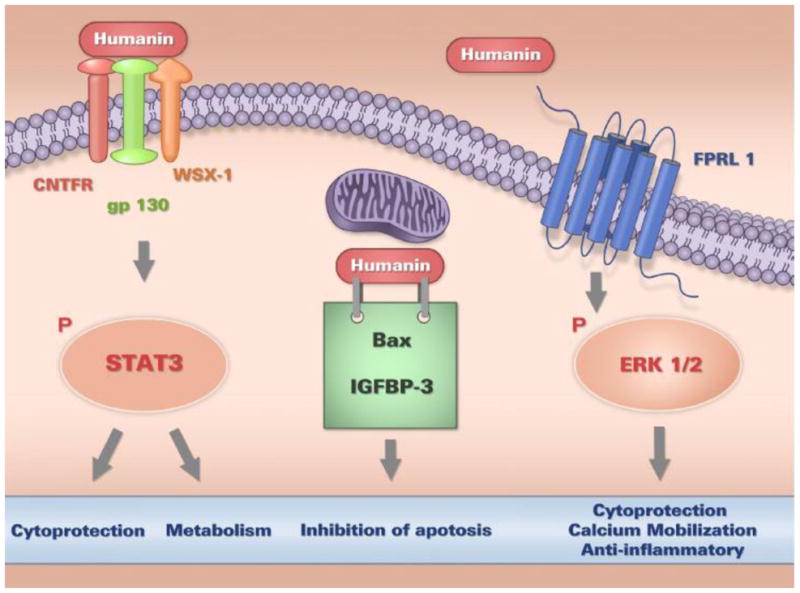
Humanin has been shown to possess both intra- and extra-cellular modes of action. Within a cell, humanin interacts with pro-apoptotic proteins such as Bax and IGFBP-3, thereby preventing apoptosis. Extracellular humanin also regulates important cellular processes such as survival, metabolism, and inflammation via two types of cell surface receptors; a trimeric receptor involving CNTFR/WSX-1/gp130, which relays signals through the STAT3 signaling pathway, and the formyl-peptide receptor-like-1 (FPRL1), which relays signals through the ERK 1/2 signaling cascade.
Is humanin the first mitochondria-derived peptide?
Humanin is a polypeptide that is highly conserved across species. It was originally discovered in 2001 by screening a cDNA library created from the surviving fraction of an Alzheimer’s disease (AD) patient brain [10, 11]. Unbiased functional screening for clones that protect neuronal cells from cell death induced by amyloid precursor protein (APP) mutants, which are associated with early-onset familial AD (FAD) [10], identified multiple clones whose sequences were identical to a part of the mitochondrial 16S rRNA. Further functional and positional screening of the clones carrying various fragments of the 16S rRNA revealed a 75bp open reading frame (ORF) sequence that yields a 24 amino acid peptide, which was designated as humanin [10, 12]. However, it is not clear whether humanin is translated in the mitochondrion or the cytoplasm. Mitochondria use a slightly different genetic code from that used in cytoplasmic translation to produce a distinct peptide from the same sequence. Humanin has been shown to be biologically effective when synthesized using both mitochondrial and cytoplasmic codes, and thus the translational site remains undetermined [11]. When the humanin ORF was cloned and expressed in neuronal cells, it was again able to provide protection from several AD-related toxicities. The term humanin was coined by its discoverer, Professor Nishimoto, prior to his premature passing from cancer, to denote the potential of this molecule to restore the “humanity” of patients with Alzheimer’s disease.
A second group, using a yeast two-hybrid screen, also cloned humanin, as a partner of the insulin-like growth factor-binding protein-3 (IGFBP-3), and demonstrated that humanin binds to IGFBP-3 with high affinity and specificity. IGFBP-3 regulates IGF bioactivity, and also induces apoptosis and inhibits cell growth independently of IGFs, and humanin protects against IGFBP-3 induced apoptosis, discussed in detail below. However, humanin does not interfere with IGF-I/IGFBP-3 binding, but appears to bind to the heparin-binding domain (HBD) of IGFBP-3 [13]. Humanin has been also cloned by a third group as a binding partner of Bax, a central pro-apoptotic protein that translocates to the mitochondria to initiate a cell death signal. The Humanin-bax complex is retained in the cytoplasm and this interaction protects from apoptosis. [11].
The humanin peptide sequence has been proposed to contain a “pseudo-signal peptide”, and when transfected into neuronal cells it is detected in the culture medium, indicating secretory capacity which is blocked by inhibition of the endoplasmic reticulum-golgi transport processes [10]. In animals models and humans, humanin is found in tissues as wells as in plasma. Cell surface receptors for humanin have been proposed by several groups as noted below (Fig 2).
Mechanisms of action of humanin: receptors and signaling pathways
Humanin is secreted from cells and found in circulation (plasma), as well as bound to cellular membranes [10, 14], and its actions are thought to be mediated by specific receptors. To date, humanin has been shown to act as a ligand to two different types of receptors; the seven-transmembrane, G-protein-coupled receptor formyl-peptide receptor-like-1 (FPRL1), and a trimeric receptor consisting of ciliary neurotrophic factor receptor (CNTFR), the cytokine receptor WSX-1 and the transmembrane glycoprotein gp130 (CNTFR/WSX-1/gp130) (Fig 2).
The first humanin receptor to be described was FPRL1, which has been linked to Alzheimer’s disease [15]. In neuroblasts, both the 42-aa form of the β-amyloid peptide (Aβ42) and humanin were able to activate FPRL1, but only Aβ42 was cytotoxic, and this cytotoxicity was reversed by humanin, suggesting that the neuroprotective effects of humanin might be accomplished by competitively binding to FPRL1 [15]. Although this study did not directly demonstrate the humanin binds to FPRL1, it did successfully show that humanin acts as an agonist for FPRL1 by inducing Ca2+ mobilization and rapid activation of ERK1/2 signaling, a commonly induced downstream pathway of G-protein-coupled receptors, which might contribute to its cytoprotective aspects [15].
The second reported humanin receptor is the trimeric CNTFR/WSX-1/gp130 complex [16]. Hashimoto et al previously showed that humanin induces STAT3 activation, which was required for its neuroprotective effects [16]. Glycoprotein 130 (gp130) is a common element of receptors that belong to the interleukin-6 (IL-6) receptor family, and thus may be responsible for the signaling pathways mediated by Janus kinase (JAK)/STAT and ERK1/2 [16]. CNTF is a known IL-6 family cytokine. WSX-1 was found while testing gp130-coupling proteins that co-overexpressed with human gp130 [16]. Using in vitro pulldown assays, the study shows that humanin coprecipitated with the ciliary neurotrophic factor receptor α (CNTFR) and the IL-27 subunit WSX-1, but not IL-6 receptor α [16]. Furthermore, overexpression of CNTFR and/or WSX-1 results in enhanced humanin binding to neuronal cells, whereas siRNA-mediated knockdown of both or either component reduces binding. In addition, knockdown of CNTFR or WSX-1 abolishes humanin-dependent cytoprotection against AD-related insults [16]. Humanin also appears to promote the hetero-oligomerization of CNTFR, WSX-1, and pg130 [16].
Considering the diverse set of actions the humanin peptide elicits in vitro, it is possible that both types of receptors described above are important in mediating its various activities, however, specific experiments using genetic ablation of receptor subtypes need to performed before more definitive roles of these pathways are identified.
Cyto-protective activities of humanin
The cytoprotective effects of humanin were first tested and confirmed in vitro against mutant forms of amyloid precursor protein (APP) and presenilin 1 and 2 (PS1/2) which cause FAD [10]. F11 neuronal cells were challenged by transfecting APP, or PS1 or 2 cDNA, or by treating with Aβ1–43 (25μM), and humanin was co-treated by either transfecting its cDNA or using synthesized peptides. Using trypan blue exclusion and WST-8 dyes, the study showed that humanin effectively protects neuronal cells from FAD-related toxicities.
As mentioned above, humanin also interacts with IGFBP-3 and Bax to antagonize their pro-apoptotic activity. Humanin also prevents Bax translocation, and activation, in response to the proapoptotic alkaloid staurosporine (STS) and subsequent release of cytochrome C into the cytoplasm [11]. Conversely, STS-treated Bax-null cells did show improved survival with humanin treatment. Humanin also binds to Bax-like BH3 proteins Bid and BimEL [17, 18], and inhibits their proapoptotic activity. Endogenous humanin, produced by transfecting expression vectors, suppresses tBID- and BimEL-induced apoptosis in vitro [18, 19]. These studies have broadened the scope of humanin activity by suggesting a crucial intracellular role in addition to the previously shown extracellular effects.
Following suit, several laboratories have tested the protective effect of humanin on various types of stress or disease states and have shown that humanin protects against them. Table 1 shows the type of stress or disease model used to test the protective effect of humanin. Although the specific molecular mechanism of cytotoxicity is unclear for some of these models, e.g. Aβ and 3xTg-AD, many have oxidative stress as a common mechanistic denominator suggesting a specific oxidative insult-related cytoprotective role of humanin.
Table 1. Cyto-protective Activities of Humanin.
Various studies have shown cyto-protective effects in a wide-spectrum of stressors. Many of the stressors/models have oxidative stress as a common mechanism of action.
| Type of Stress/Disease Model | Description | Ref |
|---|---|---|
| Scopolamine | Acetylcholine muscarinic receptor antagonist-induced memory loss in mice. | [47 |
| Dentatorubral-pallidoluysian atrophy (DRPLA) | An autosomal-dominant neurodegenerative disorder caused by expansion of CAG repeats (polyQ stretch) which form intracellular aggregates and confer neurotoxic activity | [48] |
| β-amyloid (Aβ25–35) | Cell death | [10, 13, 49] |
| Behavioral deficits in mice | [50] | |
| Memory impairment in mice | [51] | |
| Dexamethasone | Dexamethasone-induced apoptosis in rat Leydig cells. | [52] |
| Familial amyotrophic lateral sclerosis (ALS) | Mice expressing a mutant superoxide dismutase (G93A-SOD1) (protection by Colivelin, a potent humanin derivative). | [53] |
| Intratesticular androgen deprivation | Intratesticular hormone deprivation-induced germ cell apoptosis (intratesticular HN administration). | [54] |
| Oxidized LDL | Oxidized LDL-induced oxidative stress in human aortic endothelial cells (HAEC) | [28] |
| Myocardial ischemia/reperfusion (IR) injury | Mouse model of mouse cardiac IR injury. 45 minutes of left coronary artery occlusion followed by a 24-hour reperfusion. | [14] |
| Cerebral ischemia/reperfusion (IR) injury | Mouse model of cerebral IR injury.75 minutes of cerebral artery occlusion followed by a 24-hour reperfusion. | [55, 56] |
| Oxygen-Glucose Deprivation (OGD) | Mouse primary cortical neurons under OGD for 60 minutes. | [56] |
| Hypoxia (in vitro) | CoCl2-induced apoptosis in transformed rat retinal ganglion cells. | [57] |
| Triple transgenic AD mouse model (3xTg-AD) | Memory deficit and β-amyloid accumulation in a triple transgenic AD mouse model (3xTg-AD) harboring APPswe, tauP310L, and PS-1M146V | [58] |
| Double transgenic AD mouse model | Middle-aged double transgenic AD mouse model harboring APPswe/PS1dE9 | [59] |
Metabolic protection by humanin
Humanin appears to affect a number of metabolic disorders. Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) is a mitochondrial disorder strongly associated with A3243G mutant mtDNA, a location that is immediately downstream of the 16S rRNA locus. MELAS patients have defective ATP synthesis which is a major cause of tissue damage, especially those with high energy demands. Humanin restores ATP levels in lymphocytes from MELAS patients preventing them from falling into energy deficiency [20]. Notably, humanin levels are considerably elevated in skeletal muscles of patients with MELAS [21] and chronic progressive external ophthalmoplegia (CPEO), a mitochondrial disease associated with mtDNA abnormalities [22]. It is likely that the stress conferred by these pathological states could induce elevated levels of humanin.
Peripheral intravenous and central intra-cerebro-ventricular (ICV) infusion of humanin and its derivatives, F6AHN (a single amino acid substitution of alanine for phenylalanine in position 6; this substitution abrogates IGFBP-3 binding), HNG (a single amino acid substitution at position 14 (glycine for serine) that increases the biologic potency by ~1000 fold), and HNGF6A (contains both of these substitutions at positions 6 and 14, resulting in a potent non-IGFBP-3 binding peptide), significantly improve overall insulin sensitivity, mediated in part, by hypothalamic STAT3 activation in rats [23]. This effect has been suggested to be, in part, mediated by the CNTFR/WSX-1/gp130 trimeric humanin receptor. The enhanced insulin sensitivity following ICV HN administration in rats was also coupled with a significant reduction of hepatic glucose production (indicator of hepatic insulin sensitivity), which together with increased peripheral glucose uptake, raised the rate of glucose infusion in the euglycemic clamp study [23]. Furthermore, a single treatment with a highly-potent humanin analog (HNGF6A) normalizes blood glucose levels in Zucker diabetic fatty rats, a model of type 2 diabetes.
In addition, humanin improves glucose tolerance and onset of diabetes in NOD mice, a model of type 1 diabetes that involves progressive accumulation of lymphocytes within the pancreatic islets of Langerhans [24, 25]. In NOD mouse pancreata, humanin treatment was shown to dramatically suppress inflammation and lymphocyte infiltration. Furthermore, humanin treatment decreases serum starvation- and cytokine-induced apoptosis in the pancreatic β-cell line NIT-1 [24], suggesting that it is islet protective and thus might be considered for a new therapeutic target for type 1 diabetes (T1D), which is a chronic autoimmune disease which destruction of the β-cells in the islets of Langerhans [26]. In addition, cytokines including interleukin (IL)-1β, tumor necrosis factor (TNF)α, and interferon (IFN)γ, released by T cells and activated macrophages, have a role β-cell destruction; humanin was able to protect NIT-1 cells from these cytokine-induced apoptosis in an ERK and STAT3-depedent manner [24].
Humanin modulates oxidative stress and apoptosis in the developing plaque to protect endothelial function and prevent atherosclerosis. Lerman and colleagues treated ApoE-deficient mice fed a high cholesterol diet with the humanin analogue HNGF6A for 16 weeks and showed a dramatic decrease of the atherosclerotic plaque size, preserved endothelial dysfunction, and improved expression of endothelial nitric oxide (NO) synthase in aorta [27]. This study suggests that anti-oxidative stress, antiapoptosis, and preservation of eNOS are mechanisms partially responsible for the protection against atherosclerosis by humanin.
Intrestingly, humanin levels appear to decline with age [23, 28] suggesting that a relative deficiency of humanin may be involved in metabolic diseases during aging. In one study, older mice had significantly reduced levels of circulating humanin levels (2 vs. 13 months-old), and rats had decreased levels of hypothalamic and skeletal muscle humanin (3 vs. 24 months-old). The study also reported that circulating levels of humanin also gradually decline with age in humans (45–65 vs. 66–80 vs. 81–100 years-old) [23]. In another study, plasma humanin levels also significantly decreased with age in healthy volunteers (39 ± 4 vs. 60 ± 6 years-old) [28].
The emerging concept of mitochondrial derived peptides – evidence from model organisms
Humanin may be the first small peptide of its kind representing a putative set of mitochondrial-derived peptides (MDPs), a novel concept that can radically change the traditional thinking about retrograde mitochondrial signaling as well as mitochondrial gene expression. However, polyadenylated mitochondrial RNA transcripts have been previously cloned from gene libraries. In the early 1980s, a cDNA library was constructed from interferon-induced human myeloblast cells, to be used for finding interferon-induced-specific sequences [29], and the majority of the sequences with strong induction mapped to the mitochondrial 12S and 16S rRNA regions.
A recent report supports the concept of specific mitochondrial-derived signaling with endocrine properties. In the nematode C. elegans, a stress-induced tissue-specific signal originating from mitochondria, can act on distant organs to regulate life span. Neuron-specific disruption of mitochondria induces mitochondrial unfolded-protein response (UPR) in the worm intestine, and leads to an extension of life span, most likely mediated by an unidentified circulating signal(s) that allows the mitochondria of neuronal and intestinal cells to communicate stress conditions [30, 31]. These studies strongly support the novel concept of specific and active mitochondria-initiated signals that play significant roles in cellular stress response and longevity. However, these studies did not provide the specific identity or function of such signals. Although direct evidence has yet to be shown, the involvement of mitochondrial-derived peptides should be considered because, as mentioned above, currently identified stress induced mitochondrial retrograde signals are cell-autonomous (e.g. fragmented mitochondrial proteins [8], or mtDNA [9]), whereas humanin is also an endocrine factor that can act non-autonomously.
The mitochondrial transcriptome and its implications
Compared to the human nuclear genome, mitochondria have a modest sized circular genome of ~16,570 bp, which includes only 13 protein coding genes, which are all structural components of the electron transport chain system [32]. Yet, despite its small size, our understanding of its gene regulation and protein expression is very crude.
mtDNA replication and transcription starts is regulated by nuclear-encoded proteins. The entire genome is thought to be transcribed as a single polycistronic precursor that is processed into individual genes by excising the strategically positioned 22 tRNAs (tRNA punctuation model), giving rise to two rRNAs and 13 mRNA [32]. Also, owing to cutting-edge technology such as near-exhaustive deep sequencing that allows single base resolution, a comprehensive map of the human mitochondrial transcriptome is available, that gives access to previously unseen short RNA species [32].
The human mitochondrion has two promoters in the heavy strand (major and minor) in close proximity, and one in the light strand, thereby giving rise to three different single polycistronic transcripts; the heavy major promoter is responsible for 80% of all mtRNA transcripts. Yet, although the entire gene is thought to be transcribed as a single transcript, the abundance of individual rRNA, tRNA, and mRNA transcripts varies greatly, and the rRNAs are the most abundant, indicating an unexplored class of posttranscriptional processing in the mitochondria [32]. Importantly, many of the mRNA species identified from the mitochondria are discrete smaller length ones that do not map to the traditional mitochondrial protein encoding genes. In particular, multiple such mRNAs are observed from the 16S rRNA region including the site of the humanin ORF. Considering its bacterial ancestry, it is not entirely surprising to find that such a compact genome evolved with a polycistronic strategy that includes “genes-with-genes” [32]. Furthermore, recent evidence shows that polycistronic gene arrangements are not exclusive to prokaryotes but also used by eukaryotes [33, 34].
Using parallel analysis of RNA ends (PARE) to map the transcript cleavage sites, a plethora of expected and unexpected cleavage sites have been discovered for the mitochondria [32]. The majority of tRNAs and mRNA had distinct dominant cleavage site at the 5′ termini, but intragenic cleavage sites are especially abundant in rRNAs.
The latter stages of precursor modification include 3′ polyadenylation of mRNAs, with the exception of the mitochondrial ND6 gene, and rRNAs, in agreement with previous reports [32, 35]. Although the precise role of rRNA polyadenylation is still unknown, the humanin cDNA clone was found to be polyadenylated. As mentioned above, the mitochondrion evolved to maintain a polycistronic transcript which requires a sophisticated regulatory machinery to process each gene separately. Notably, there is compelling evidence from the emerging field of small peptides showing biologically active peptides of 11–32 amino acids in length which are encoded by small open reading frames (sORFs) from a polycistronic mRNA [33, 36, 37]. In addition, mitochondrial rRNAs are transported and found in the cytoplasm with significant biological roles in, for instance, Drosophila germ cell establishment [38], and the 16S rRNA was found localized in the nucleus of human spermatogenic cells [39]. It is possible that the polyadenylated humanin transcript is exported to the cytoplasm where it can be translated.
Curiously, the mitochondrial transcriptome includes small regulatory RNAs. Furthermore, multiple transcription binding sites determined by high-throughput in vivo footprinting assay, and higher order organization of the mitochondrial genome were shown [32]. Shuttling of RNA to and from the mitochondria is evident and may involve mRNA binding proteins such as PNPase [40]. Current understanding is insufficient to explain the more recent observations and is in urgent need of revision and amendments.
The concept of NUMTS and evidence for the mitochondrial origin of humanin
The incorporation of the ancient α-proteobacterium has immensely accelerated the evolutionary course of metazoan cells. During the prolonged transition from endosymbiotic residence to organelle status in host cells, mitochondria are thought to have transferred most of its genome to the host nucleus leaving chromosomal “doppelgangers”, through the process of Nuclear Mitochondrial DNA-Transfer or nuclear insertions of mitochondrial origin (NUMTs) [41, 42].
NUMTS come in various sizes from all parts of the mtDNA with various degrees of homology with the original sequences, and the “partial ghosts” of entire mtDNA can be found in the nuclear genome, although in most cases with substantial sequence degeneration [43]. In humans, there are an estimated 755 insertions dispersed throughout the nuclear genome with chromosome two having the most insertions [42] and chromosome 18 having the least [42]. Most NUMTs are small insertions of <500 bp and only 12.85% are >1500 bp [42]. The percentage identity is inversely correlated with size and the mean percentage between NUMTs and mtDNA is 79.2% with a range of 63.5% to 100% identity [43].
To date, the mitochondrial origin of humanin is still debated. NUMTs that reflect humanin sequences are found in chromosomes 1–13 in varying degrees of homology, but none of the peptide sequence deduced from NUMTs are fully identical to the original cDNA clone from the Alzheimer’s patient brain cloned by Nishimoto and colleagues, or the HeLa cell library cloned by Ikonen, both of which were 100% identical to the mitochondrial sequence [44].
Furthermore, northern blot analysis using the humanin ORF (antisense) as probe, showed hybridization to total RNA, from HeLa cells, which was identical in size to mitochondrial 16S rRNA, whereas total RNA from ρ0 HeLa cells, which lack mtDNA, did not show any significant hybridization [45]. Nonetheless, synthetic peptides corresponding to a NUMT origin that have 90% amino-acid homology to the mitochondrial humanin, retain some (incomplete) biological activity [44]. On the other hand, formylated humanin has enhanced activity compared to humanin by increasing the affinity to FPRL1, a humanin receptor, supporting a mitochondrial origin as N-termini formylation is a hallmark of mitochondrially encoded proteins [46]. Thus, strong circumstantial data supports a mitochondrial origin for humanin, but a NUMT-derived humanin has not been ruled out yet.
Concluding remarks
Humanin may be a harbinger of additional mitochondrial-derived peptides (MDPs), as numerous putative ORFs disperse throughout the mitochondrial genome. Humanin is the first described MDP shown to be a neuroprotective peptide, and has rapidly expanded its protective repertoire to various types of stress and disease models. It should be noted that many of these models have oxidative stress as a common denominator, begging the question if humanin is a more general cytoprotective factor against oxidative stress. Interestingly, humanin levels decline with age, which is a major risk factor for many of the diseases in which humanin is protective, including Alzheimer’s disease, atherosclerosis, myocardial and cerebral ischemia, and type 2 diabetes, all of which involve an oxidative stress component.
The emerging biology of mitochondrial-derived peptides represents the foreground of novel mitochondrial regulation of cellular activity. Deciphering the details of these processes and identifying further related genes and peptides holds therapeutic and diagnostic potential in a wide spectrum of human diseases.
Highlights.
Humanin represents a putative set of mitochondrial-derived peptides
Humanin is secreted from cells and found in plasma, as well as bound to cellular membranes
Humanin acts as a ligand to two different types of receptors
Humanin has cytoprotective effects and also improves glucose tolerance and onset of Type 1 diabetes
Glossary
- Membrane permeability transition (MPT)
An increase in mitochondrial permeability via the formation of a non-specific pore across the inner mitochondrial membrane permitting the free distribution of ions and low molecular weight molecules (<1500 Da) across the membrane that can lead to mitochondrial swelling and cell death
- Mitochondrial-derived peptides (MDPs)
Small peptides that are encoded within the mitochondrial genome in addition to the canonical 13 protein-coding mitochondrial genes
- Nuclear Mitochondrial DNA-Transfer (NUMTs)
a copy of mitochondrial DNA (mtDNA) that is integrated into the nuclear genome that are generally considered pseudogenes; also known as nuclear DNA sequences of mitochondrial origin
- Parallel analysis of RNA ends (PARE)
A technique that combines 5′-rapid amplification of cDNA ends (RACE) with high-throughput deep sequencing (SBS) and bioinformatics tools that can reveal RNA cleavage sites
- Retrograde signaling
A pathway of communication from the mitochondria to the cell that influences cellular and organismal activities
- Retrograde signaling molecules
these are ions, proteins, and molecules that act as messengers of retrograde signaling (e.g. Ca2+, reactive oxygen species, cytochrome C, etc)
- tRNA punctuation model
A model describing mitochondrial RNA processing, whereby individual genes are released from a long polycistronic transcript by excising the 22 interspersed tRNAs that are strategically encoded in the mtDNA
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gray MW. Mitochondrial evolution. Cold Spring Harbor perspectives in biology. 2012;4:a011403. doi: 10.1101/cshperspect.a011403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wallace DC. Bioenergetic origins of complexity and disease. Cold Spring Harbor symposia on quantitative biology. 2011;76:1–16. doi: 10.1101/sqb.2011.76.010462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallace DC. Mitochondria and cancer. Nature reviews Cancer. 2012;12:685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verdin E, et al. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends in biochemical sciences. 2010;35:669–675. doi: 10.1016/j.tibs.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ganta VC, Alexander JS. Focus on carbon monoxide: a modulator of neutrophil oxidants and elastase spatial localization? American journal of physiology Heart and circulatory physiology. 2009;297:H902–904. doi: 10.1152/ajpheart.00587.2009. [DOI] [PubMed] [Google Scholar]
- 7.Ichikawa Y, et al. Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:4152–4157. doi: 10.1073/pnas.1119338109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haynes CM, et al. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Molecular cell. 2010;37:529–540. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakahira K, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature immunology. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hashimoto Y, et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Abeta. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6336–6341. doi: 10.1073/pnas.101133498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo B, et al. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature. 2003;423:456–461. doi: 10.1038/nature01627. [DOI] [PubMed] [Google Scholar]
- 12.Hashimoto Y, et al. Mechanisms of neuroprotection by a novel rescue factor humanin from Swedish mutant amyloid precursor protein. Biochemical and biophysical research communications. 2001;283:460–468. doi: 10.1006/bbrc.2001.4765. [DOI] [PubMed] [Google Scholar]
- 13.Ikonen M, et al. Interaction between the Alzheimer’s survival peptide humanin and insulin-like growth factor-binding protein 3 regulates cell survival and apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:13042–13047. doi: 10.1073/pnas.2135111100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muzumdar RH, et al. Acute humanin therapy attenuates myocardial ischemia and reperfusion injury in mice. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:1940–1948. doi: 10.1161/ATVBAHA.110.205997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ying G, et al. Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. J Immunol. 2004;172:7078–7085. doi: 10.4049/jimmunol.172.11.7078. [DOI] [PubMed] [Google Scholar]
- 16.Hashimoto Y, et al. Humanin inhibits neuronal cell death by interacting with a cytokine receptor complex or complexes involving CNTF receptor alpha/WSX-1/gp130. Molecular biology of the cell. 2009;20:2864–2873. doi: 10.1091/mbc.E09-02-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi J, et al. Mapping the specific cytoprotective interaction of humanin with the pro-apoptotic protein bid. Chemical biology & drug design. 2007;70:383–392. doi: 10.1111/j.1747-0285.2007.00576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luciano F, et al. Cytoprotective peptide humanin binds and inhibits proapoptotic Bcl-2/Bax family protein BimEL. The Journal of biological chemistry. 2005;280:15825–15835. doi: 10.1074/jbc.M413062200. [DOI] [PubMed] [Google Scholar]
- 19.Zhai D, et al. Humanin binds and nullifies Bid activity by blocking its activation of Bax and Bak. The Journal of biological chemistry. 2005;280:15815–15824. doi: 10.1074/jbc.M411902200. [DOI] [PubMed] [Google Scholar]
- 20.Kariya S, et al. Effect of humanin on decreased ATP levels of human lymphocytes harboring A3243G mutant mitochondrial DNA. Neuropeptides. 2005;39:97–101. doi: 10.1016/j.npep.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Kariya S, et al. Humanin detected in skeletal muscles of MELAS patients: a possible new therapeutic agent. Acta neuropathologica. 2005;109:367–372. doi: 10.1007/s00401-004-0965-5. [DOI] [PubMed] [Google Scholar]
- 22.Kin T, et al. Humanin expression in skeletal muscles of patients with chronic progressive external ophthalmoplegia. Journal of human genetics. 2006;51:555–558. doi: 10.1007/s10038-006-0397-2. [DOI] [PubMed] [Google Scholar]
- 23.Muzumdar RH, et al. Humanin: a novel central regulator of peripheral insulin action. PloS one. 2009;4:e6334. doi: 10.1371/journal.pone.0006334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoang PT, et al. The neurosurvival factor Humanin inhibits beta-cell apoptosis via signal transducer and activator of transcription 3 activation and delays and ameliorates diabetes in nonobese diabetic mice. Metabolism: clinical and experimental. 2010;59:343–349. doi: 10.1016/j.metabol.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wicker LS, et al. Genetic control of autoimmune diabetes in the NOD mouse. Annual review of immunology. 1995;13:179–200. doi: 10.1146/annurev.iy.13.040195.001143. [DOI] [PubMed] [Google Scholar]
- 26.Bluestone JA, et al. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. 2010;464:1293–1300. doi: 10.1038/nature08933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oh YK, et al. Humanin preserves endothelial function and prevents atherosclerotic plaque progression in hypercholesterolemic ApoE deficient mice. Atherosclerosis. 2011;219:65–73. doi: 10.1016/j.atherosclerosis.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bachar AR, et al. Humanin is expressed in human vascular walls and has a cytoprotective effect against oxidized LDL-induced oxidative stress. Cardiovascular research. 2010;88:360–366. doi: 10.1093/cvr/cvq191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maeda S, et al. Construction and identification of bacterial plasmids containing nucleotide sequence for human leukocyte interferon. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:7010–7013. doi: 10.1073/pnas.77.12.7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Durieux J, et al. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woo DK, Shadel GS. Mitochondrial stress signals revise an old aging theory. Cell. 2011;144:11–12. doi: 10.1016/j.cell.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 32.Mercer TR, et al. The human mitochondrial transcriptome. Cell. 2011;146:645–658. doi: 10.1016/j.cell.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Savard J, et al. A segmentation gene in tribolium produces a polycistronic mRNA that codes for multiple conserved peptides. Cell. 2006;126:559–569. doi: 10.1016/j.cell.2006.05.053. [DOI] [PubMed] [Google Scholar]
- 34.Tautz D. Polycistronic peptide coding genes in eukaryotes--how widespread are they? Briefings in functional genomics & proteomics. 2009;8:68–74. doi: 10.1093/bfgp/eln054. [DOI] [PubMed] [Google Scholar]
- 35.Baserga SJ, et al. Polyadenylation of a human mitochondrial ribosomal RNA transcript detected by molecular cloning. Gene. 1985;35:305–312. doi: 10.1016/0378-1119(85)90009-5. [DOI] [PubMed] [Google Scholar]
- 36.Kondo T, et al. Small peptides switch the transcriptional activity of Shavenbaby during Drosophila embryogenesis. Science. 2010;329:336–339. doi: 10.1126/science.1188158. [DOI] [PubMed] [Google Scholar]
- 37.Galindo MI, et al. Peptides encoded by short ORFs control development and define a new eukaryotic gene family. PLoS biology. 2007;5:e106. doi: 10.1371/journal.pbio.0050106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amikura R, et al. Presence of mitochondria-type ribosomes outside mitochondria in germ plasm of Drosophila embryos. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:9133–9138. doi: 10.1073/pnas.171286998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villegas J, et al. Localization of the 16S mitochondrial rRNA in the nucleus of mammalian spermatogenic cells. Molecular human reproduction. 2002;8:977–983. doi: 10.1093/molehr/8.11.977. [DOI] [PubMed] [Google Scholar]
- 40.Wang G, et al. PNPASE regulates RNA import into mitochondria. Cell. 2010;142:456–467. doi: 10.1016/j.cell.2010.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bensasson D, et al. Mitochondrial pseudogenes: evolution’s misplaced witnesses. Trends in ecology & evolution. 2001;16:314–321. doi: 10.1016/s0169-5347(01)02151-6. [DOI] [PubMed] [Google Scholar]
- 42.Ramos A, et al. Nuclear insertions of mitochondrial origin: Database updating and usefulness in cancer studies. Mitochondrion. 2011;11:946–953. doi: 10.1016/j.mito.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 43.Mourier T, et al. The Human Genome Project reveals a continuous transfer of large mitochondrial fragments to the nucleus. Molecular biology and evolution. 2001;18:1833–1837. doi: 10.1093/oxfordjournals.molbev.a003971. [DOI] [PubMed] [Google Scholar]
- 44.Bodzioch M, et al. Evidence for potential functionality of nuclearly-encoded humanin isoforms. Genomics. 2009;94:247–256. doi: 10.1016/j.ygeno.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 45.Tajima H, et al. Evidence for in vivo production of Humanin peptide, a neuroprotective factor against Alzheimer’s disease-related insults. Neuroscience letters. 2002;324:227–231. doi: 10.1016/s0304-3940(02)00199-4. [DOI] [PubMed] [Google Scholar]
- 46.Harada M, et al. N-Formylated humanin activates both formyl peptide receptor-like 1 and 2. Biochemical and biophysical research communications. 2004;324:255–261. doi: 10.1016/j.bbrc.2004.09.046. [DOI] [PubMed] [Google Scholar]
- 47.Mamiya T, Ukai M. [Gly(14)]-Humanin improved the learning and memory impairment induced by scopolamine in vivo. British journal of pharmacology. 2001;134:1597–1599. doi: 10.1038/sj.bjp.0704429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kariya S, et al. Humanin attenuates apoptosis induced by DRPLA proteins with expanded polyglutamine stretches. Journal of molecular neuroscience: MN. 2005;25:165–169. doi: 10.1385/JMN:25:2:165. [DOI] [PubMed] [Google Scholar]
- 49.Jin H, et al. Protective effects of [Gly14]-Humanin on beta-amyloid-induced PC12 cell death by preventing mitochondrial dysfunction. Neurochemistry international. 2010;56:417–423. doi: 10.1016/j.neuint.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 50.Miao J, et al. S14G-Humanin ameliorates Abeta25–35-induced behavioral deficits by reducing neuroinflammatory responses and apoptosis in mice. Neuropeptides. 2008;42:557–567. doi: 10.1016/j.npep.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 51.Tajima H, et al. A humanin derivative, S14G-HN, prevents amyloid-beta-induced memory impairment in mice. Journal of neuroscience research. 2005;79:714–723. doi: 10.1002/jnr.20391. [DOI] [PubMed] [Google Scholar]
- 52.Colon E, et al. Anti-apoptotic factor humanin is expressed in the testis and prevents cell-death in leydig cells during the first wave of spermatogenesis. Journal of cellular physiology. 2006;208:373–385. doi: 10.1002/jcp.20672. [DOI] [PubMed] [Google Scholar]
- 53.Chiba T, et al. Colivelin prolongs survival of an ALS model mouse. Biochemical and biophysical research communications. 2006;343:793–798. doi: 10.1016/j.bbrc.2006.02.184. [DOI] [PubMed] [Google Scholar]
- 54.Lue Y, et al. Opposing roles of insulin-like growth factor binding protein 3 and humanin in the regulation of testicular germ cell apoptosis. Endocrinology. 2010;151:350–357. doi: 10.1210/en.2009-0577. [DOI] [PubMed] [Google Scholar]
- 55.Xu X, et al. Humanin is a novel neuroprotective agent against stroke. Stroke; a journal of cerebral circulation. 2006;37:2613–2619. doi: 10.1161/01.STR.0000242772.94277.1f. [DOI] [PubMed] [Google Scholar]
- 56.Xu X, et al. Neuroprotective effect of humanin on cerebral ischemia/reperfusion injury is mediated by a PI3K/Akt pathway. Brain research. 2008;1227:12–18. doi: 10.1016/j.brainres.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Men J, et al. An AD-Related Neuroprotector Rescues Transformed Rat Retinal Ganglion Cells from CoCl(2)-Induced Apoptosis. Journal of molecular neuroscience: MN. 2012 doi: 10.1007/s12031-011-9701-5. [DOI] [PubMed] [Google Scholar]
- 58.Niikura T, et al. A humanin derivative reduces amyloid beta accumulation and ameliorates memory deficit in triple transgenic mice. PloS one. 2011;6:e16259. doi: 10.1371/journal.pone.0016259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang W, et al. S14G-humanin improves cognitive deficits and reduces amyloid pathology in the middle-aged APPswe/PS1dE9 mice. Pharmacology, biochemistry, and behavior. 2012;100:361–369. doi: 10.1016/j.pbb.2011.09.012. [DOI] [PubMed] [Google Scholar]