

Abstract
Both basic and clinical research indicates that females are more susceptible to stress-related affective disorders than males. One of the mechanisms by which stress induces depression is via inflammatory signaling in the brain. Stress during adolescence, in particular, can also disrupt the activation and continued development of both the hypothalamic–pituitary–adrenal (HPA) and –gonadal (HPG) axes, both of which modulate inflammatory pathways and brain regions involved in affective behavior. Therefore, we tested the hypothesis that adolescent stress differentially alters brain inflammatory mechanisms associated with affective-like behavior into adulthood based on sex. Male and female Wistar rats underwent mixed-modality stress during adolescence (PND 37–48) and were challenged with lipopolysaccharide (LPS; 250 μg/kg, i.p.) or saline 4.5 weeks later (in adulthood). Hippocampal inflammatory marker gene expression and circulating HPA and HPG axes hormone concentrations were then determined. Despite previous studies indicating that adolescent stress induces affective-like behaviors in female rats only, this study demonstrated that adolescent stress increased hippocampal inflammatory responses to LPS in males only, suggesting that differences in neuroinflammatory signaling do not drive the divergent affective-like behaviors. The sex differences in inflammatory markers were not associated with differences in corticosterone. In females that experienced adolescent stress, LPS increased circulating estradiol. Estradiol positively correlated with hippocampal microglial gene expression in control female rats, whereas adolescent stress negated this relationship. Thus, estradiol in females may potentially protect against stress-induced increases in neuroinflammation.
Keywords: Variable stress, Cytokines, iNOS, NF-κB, Estradiol, Progesterone, Testosterone, Microglia, Depression, Anxiety
1. Introduction
The prevalence of depression in women is approximately twice that of men (Frackiewicz et al., 2000; Noble, 2005; Solomon and Herman, 2009). Adolescence marks a period of vast physical and mental changes and the emergence of many sex differences, including this female-biased prevalence of depression (Angold et al., 1998; Lenroot and Giedd, 2010; Nolen-Hoeksema and Girgus, 1994). Significant stress exposure during adolescence is particularly associated with emergence of the increased prevalence of depression in females (Angold et al., 1998; Becker and Grilo, 2007; Conley and Rudolph, 2009) suggesting that females may be more susceptible to stress-induced depression than men (Kudielka et al., 2004; Young, 1998). Non-human animal models support these hypotheses: females may be more susceptible to depressive-like behavior (Dalla et al., 2005) and specifically more susceptible to adolescent stress-induced depressive-like behavior (Bourke and Neigh, 2011; Desbonnet et al., 2008).
Mounting evidence suggests that stress induces these emotional and behavioral changes through inflammatory signaling in the brain (Anisman, 2009; Maier, 2003). Rapidly after stress exposure, proinflammatory cytokines are transiently induced in brain regions that regulate affective-like and cognitive behaviors in rodents (e.g., hypothalamus, hippocampus, and frontal cortex (Nguyen et al., 1998)). Longer-lasting or “priming” effects of stressors (i.e., those that occur after the initial inflammatory response subsides; >24 h) are also observed, but may require a subsequent challenge, like lipopolysaccharide (LPS) treatment, to be detectable (Frank et al., 2007; Gibb et al., 2011; Girotti et al., 2011; Johnson et al., 2003). This relationship between stress and neuroinflammation is complex, however, as some stressors instead attenuate or result in mixed neuroinflammatory responses to LPS (Audet et al., 2011; Barnum et al., 2012; Gibb et al., 2008). Little work has been done to determine whether adolescence is a critical period during which stress confers similar changes in neuroinflammatory processes as predicted by human studies or how this may occur. Additionally, the longevity of these changes has not been determined, as they are usually examined within 2–4 days post-stressor.
Among other changes, the hypothalamic–pituitary–adrenal (HPA) and –gonadal (HPG) axes continue to develop during adolescence. Therefore, their hormone effectors may be partly responsible for the concomitant emergence of sex differences in mental health disorders (McCormick and Mathews, 2007; Naninck et al., 2011). In support of this hypothesis, the sex differences in rates of depression that emerge during adolescence subside following reproductive senescence when HPG axis hormone production wanes (Bebbington et al., 2003). In rodents, chronic stress during adolescence disrupts HPA glucocorticoid responses, an effect that is sexually dimorphic (Bourke and Neigh, 2011, 2012). Estradiol, progesterone, and glucocorticoids are also known modulators of neuroinflammation (Kipp and Beyer, 2009; Schweingruber et al., 2012). For example, ovariectomy inhibits LPS-induced brain microglial expression of cytokines in mice, whereas estradiol treatment rescues this function (Soucy et al., 2005). Glucocorticoids also regulate inflammatory cytokine production and function in the brain (Dantzer et al., 1999), and therefore, dysregulation of glucocorticoids due to adolescent stress may be one mechanism by which brain inflammatory signaling is altered.
In this study, we predicted that the previously observed female-biased effects of adolescent stress on affective-like behaviors (Bourke and Neigh, 2011) would be mediated by increased brain inflammatory responses in rats. To measure brain inflammatory response, hippocampal gene expression of various markers that have been implicated in inflammation-induced depression (Leonard, 2007; Pace and Miller, 2009) was quantified following a peripheral LPS challenge: proinflammatory cytokines (IL-1β, TNFα, a microglial cell marker (CD11b), an inflammatory mediator (iNOS), and the canonical inflammatory transcription factor and target of stress-induced glucocorticoid signaling (NF-κB indirectly via IκBα). HPA and HPG axes hormones were also measured as potential modulators of these inflammatory mechanisms.
2. Materials and methods
2.1. Animals
Timed pregnant Wistar rats (Charles River, Wilmington, MA) arrived on gestational day 12 (n = 24). This timing of shipping stress is not associated with changes in developmental outcomes (Ogawa et al., 2007), whereas shipping stress during puberty can have enduring effects on behavior (Laroche et al., 2009a, b). Rats were housed on a 14:10 reverse light:dark cycle in a facility controlled for humidity (60%) and temperature (20–23 °C). Rodent diet 5001 chow (Purina Mills, Richmond, IN) and water were available ad libitum throughout the study. Three days after birth, rat pups were sexed and litters were culled to four male and four female pups. Littermates were assigned to control and stress groups with no more than two pups per litter assigned to each group. Pups were weighed and weaned on postnatal day (PND) 21 and housed in same-sex pairs. All animal experiments were approved by Emory University’s Institutional Animal Care and Use Committee and carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (National Research Council, 2011). All efforts were made to minimize animal suffering and to reduce the number of rats used.
2.2. Experimental design
Rats of both sexes were exposed to either a mixed modality chronic stress paradigm during adolescence (see details below) or remained pair-housed as controls. Four and a half weeks after the cessation of the stress paradigm, neuroinflammatory responses were assessed in these adult rats following an i.p. lipopolysaccharide (LPS; Escherichia coli strain 127:B8; 250 μg/kg) or saline injection. Four hours post-injection, rats were deeply anesthetized with sodium pentobarbital (1 mg/kg; i.p.). Blood was sampled into heparin-coated syringes via cardiac puncture followed by brief (~1 min) cold saline perfusion to flush circulating macrophages out of the brain. Blood was centrifuged at 1800 rcf and plasma was collected and stored at −80 °C until used for hormone analyses. Brains were removed and hippocampi were dissected out, flash frozen, and stored at −80 °C until used for quantitative RT-PCR analyses.
2.3. Mixed modality chronic stress paradigm
This mixed modality chronic stress paradigm has previously been used to elicit sex-specific behavioral changes in adolescent rats (Bourke and Neigh, 2011, 2012). Here, adolescent stress was defined as individual housing beginning at PND 36 and continuing throughout the study combined with randomly alternating daily exposure to either social defeat or restraint for 12 days (PND 37–48; 6 days each of social defeat and restraint). This period corresponds to the middle of adolescence in rodents as defined by Eiland and Romeo (2012). The control groups remained pair-housed with a same sex littermate throughout the study. The study was not designed to assess the specific effects of individual housing, restraint, or social defeat, but was designed to use this combination of established stressors to induce chronic stress during adolescence.
Social defeat stress was performed during the light phase in the home cage of a same-sex resident: a mature, territorial Long-Evans rat (retired male breeders or ovariectomized females) selected based on their likelihood to pin an intruder (Bourke and Neigh, 2011). For social defeat, a mesh barrier was placed in the middle of the resident’s cage. The resident remained on one side of the barrier and the same-sex intruder (experimental rat) was placed on the opposite side. After two minutes of acclimation, the barrier was removed for 5 min or until the intruder was attacked by the resident (without physical injury) five times on the first day, three times on the second day, and once each day thereafter. At the conclusion of the physical criteria, the wire mesh screen was replaced. Intruders and residents remained separated by the wire mesh screen, which allowed for continued visual and olfactory contact for an additional 25 min. Subsequently the intruder was returned to its home cage. Intruder and resident pairings were randomly assigned each day to prevent stabilization of a dominance hierarchy.
For the restraint portion of the mixed-modality stressor, animals were restrained during the light phase for 60 min in clear acrylic rat restraints (BrainTree Scientific, Braintree, MA, USA) that prevented head-to-tail turns but did not compress the rat.
2.4. Endocrine analyses
Plasma hormones were assayed via ELISA: corticosterone (sensitivity: 27 pg/ml, Enzo Life Sciences, Farmingdale, NY, USA), progesterone (sensitivity: 8.57 pg/ml, Enzo Life Sciences), estradiol using 50 μl samples (sensitivity: 19 pg/ml, Cayman Chemical, Ann Arbor, MI, USA). Samples were run in duplicate for all endocrine assays.
2.5. Quantitative RT-PCR
Hippocampal RNA was extracted using a RNeasy Mini kit (Qiagen, Valencia, CA, USA) and RNA integrity was assessed by a Nano-Drop 2000 spectrophotometer (ThermoScientific, Wilmington, DE, USA) and an Agilent Bioanalyzer 2100 (Agilent, Santa Clara, CA). RNA was reverse transcribed with the High Capacity RNA to cDNA kit (Applied Biosystems, Foster City, CA). cDNA was quantified with the PicoGreen Assay (Invitrogen, Carlsbad, CA). A rat endogenous control plate (Applied Biosystems, Foster City, CA) was run in order to determine the optimal endogenous control, which was found to be Tfrc (transferrin receptor) based on the lowest deviation between treatment groups. Rat TaqMan Gene Expression Assays were purchased from Applied Biosystems (Carlsbad, CA, USA) with probes labeled with 6-FAM and MGB (non-fluorescent quencher) at the 5′ and 3′ ends, respectively: IL-1β (Rn00580432_m1), TNFα (Rn00562055_m1), CD11b (Rn01506864_m1), IκBa (Rn01473657_g1), iNOS (Rn00561646_m1), Tfrc (Rn01474701_m1). The universal two-step RT-PCR cycling conditions used on the 7900HT Sequence Detection System (Applied Bio-systems) were: 50 °C (2 min), 95 °C (10 min), 40 cycles of 95 °C (15 s) and 60 °C (1 min). Relative gene expression of individual samples run in triplicate (with coefficient of variation cut-off set to 4%) was calculated by the comparative CT quantification method relative to the same-sex no stress saline control group (2−ΔΔCT) with a Grubbs correction for outliers.
2.6. Statistical analysis
Statistical comparisons of differences in hippocampal gene expression and hormone concentrations were analyzed using 2-way ANOVAs and t-tests using SPSS v.19.0 software (Chicago, IL). Separate linear regression analyses were used to determine the extent to which estradiol concentrations predicted microglial gene expression in the hippocampus in each of the treatment groups. Data were determined to be statistically significant when p < 0.05 and are presented as mean ± standard error of the mean (SEM).
3. Results
3.1. Adolescent stress decreased body mass in males only
As predicted, males weighed significantly more than females throughout the experiment (F1,288 = 769.7; p < 0.001, Table 1). In males, adolescent stress significantly decreased body mass on the last day of the adolescent stress paradigm (F1,36 = 12.8; p = 0.001). No significant differences in body mass were observed in either sex at the initiation of the stress paradigm or one week before LPS treatment and tissue collection (p > 0.05 in all cases).
Table 1.
Mean ( ± SEM) body mass (g) throughout experiment in rats n = 10/groups.
| Beginning of stress (PND 36) | End of stress (PND 48) | 1 week before collection (PND 73) | ||
|---|---|---|---|---|
| Males | Control | 176.0 ± 12.9 | 267.7 ± 17.7 | 405.7 ± 28.3 |
| Adol. stress | 174.5 ± 10.7 | 249.0 ± 14.5* | 396.3 ± 25.1 | |
| Females | Control | 144.4 ± 8.3 | 186.5 ± 11.2 | 256.7 ± 27.1 |
| Adol. stress | 142.3 ± 9.7 | 181.6 ± 15.8 | 283.6 ± 19.6 |
p < 0.05, Compared with same sex controls.
3.2. Adult neuroinflammatory responses to LPS challenge following adolescent stress exposure
Our previous work indicating that adolescent stress induced depressive- and anxiety-like behavior during adulthood in females only led us to examine sex differences in potential underlying neuroinflammatory responses. The prolonged influence of adolescent stress on the acute hippocampal neuroinflammatory responses to an LPS injection was sex-dependent. Four hours after LPS administration, hippocampal IL-1β, TNFα, and IκBα gene expression in male rats that were stressed 4.5 weeks earlier during adolescence surpassed that of LPS treatment in males that were not exposed to adolescent stress (p < 0.05 for each gene; t17 = 2.2, t18 = 1.9, t16 = 3.6, respectively; Fig. 1A, B and D). Similar trends were observed for CD11b and iNOS, although statistical significance was not reached (p > 0.05 for both; Fig. 1C and E). In contrast, gene expression of the inflammatory markers examined remained unchanged by stress experience in female rats; unstressed female rats displayed the same magnitude of elevations in neuroinflammatory markers as stressed females (p > 0.05 for each gene). No differences in inflammatory gene expression were observed between stressed and non-stressed rats of either sex in the absence of the LPS stimulation (i.e., following a saline injection). Likewise, no statistically significant differences were detected between the sexes of the same treatment groups (p > 0.05 for each gene).
Fig. 1.

Stress during adolescence increased hippocampal inflammatory response to LPS in adulthood in male rats only. Fold differences (relative to a transferrin receptor endogenous control) in hippocampal IL-1β (A), TNFα (B), IκBα (C), CD11b (D), and iNOS (E) gene expression relative to the mean expression of the same-sex, no stress, saline control (−2ΔΔCT) 4 h after LPS (250 μg/kg, i.p.) or saline treatment in adult male or female rats that previously (4.5 weeks earlier) experienced a mixed modality stress paradigm during adolescence (PND 37–48). n = 7–10/group; *p < 0.05 within the same stress treatment between SALINE vs. LPS; #p < 0.05 within LPS treatment between ADOL. STRESS and CONTROL.
3.3. Adult endocrine responses to LPS challenge following adolescent stress exposure
Four hours after the injection, LPS increased plasma corticosterone concentrations in all rats compared with saline-injected controls (Fig. 2A; p < 0.05). There were no sex differences in these corticosterone responses (p > 0.05).
Fig. 2.

Adolescent stress does not alter corticosterone, progesterone, or testosterone concentrations. Circulating plasma corticosterone (A), progesterone (B), and testosterone (C) concentrations 4 h post-LPS (250 μg/kg, i.p.) or saline treatment in adult rats that experienced prior mixed-modality adolescent stress (or no stress). n = 8–10/group; *p < 0.05 within the same stress treatment between SALINE vs. LPS.
No differences in progesterone concentrations among any female treatment groups were observed (Fig. 2B; p > 0.05). LPS treatment comparably reduced testosterone concentrations among males, regardless of stress treatment (Fig. 3C; p < 0.5 in both cases). Saline-injected males that experienced adolescent stress had slightly elevated testosterone concentrations relative to the non-stress controls, although this increase was not statistically significant (p = 0.2). Adolescent stress exposure paired with an LPS challenge significantly increased female plasma estradiol concentrations compared with all other females (Fig. 3A; p < 0.05).
Fig. 3.
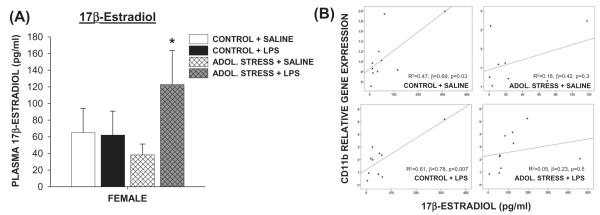
Stress during adolescence increased circulating estradiol response to LPS in female rats and attenuated the correlation between estradiol and Cd11b gene expression. (A) Circulating plasma 17β-estradiol concentrations 4 h post-LPS (250 μg/kg, i.p.) or saline treatment in adult female rats that experienced prior mixed-modality adolescent stress (or no stress). *p < 0.05 within the same stress treatment between SALINE vs. LPS. (B) Linear regressions between circulating estradiol concentrations and Cd11b (microglial) gene expression in the hippocampus. Regression coefficients only for stress-free rats were significant. n = 8–10/group.
3.4. Estradiol correlates with hippocampal microglial gene expression
Given the reported modulating effects of estradiol on microglia activation (Dimayuga et al., 2005; Loram et al., 2012; Soucy et al., 2005), a multiple linear regression analysis was used to determine the extent to which circulating estradiol, stress, and LPS treatment predicted hippocampal CD11b gene expression among females. Including all three predictors in the model produced R2 = 0.41, F3,37 = 8.0, p < 0.001. Both estradiol and LPS treatment had positive significant regression weights (β = 0.42 and 0.38, respectively) indicating that female rats that received LPS or had higher circulating estradiol were expected to have higher hippocampal CD11b expression, whereas stress did not contribute to this model. When females were divided into experimental groups (no stress + saline, no stress + LPS, stress + saline, stress + LPS), however, the significant predictability of estradiol on CD11b gene expression was restricted to those that did not experience adolescent stress (Fig. 3B). Therefore, adolescent stress attenuated the contribution of estradiol in terms of its value as a predictive factor for hippocampal CD11b gene expression.
4. Discussion
To our knowledge, this is the first study to test the potential for biological sex to modulate the effects of adolescent stress on brain inflammation. In contrast to our prediction based on previous behavioral studies in which adolescent stress increased depressive- and anxiety-like behavior in females (Bourke and Neigh, 2011), an LPS trigger injected four and a half weeks after chronic adolescent stress unmasked long-term priming of various neuroinflammatory mediators (IL-1β, TNFα, CD11b, NF-κB, iNOS) in males only. These data suggest that the mechanisms by which adolescent stress confer sexually dimorphic changes in adult affective-like behavior may not be inflammatory in nature. In support of this hypothesis, without LPS stimulation, neuroinflammatory marker gene expression was minimal and did not differ between the sexes around the same age that affective-like behaviors have been shown to diverge (Bourke and Neigh, 2011). In fact, neuroinflammation was only apparent when LPS treatment was added to the model. A similar lack of stress-induced constitutive brain cytokine production has been reported in some studies in adult male rodents (Espinosa-Oliva et al., 2011; Frank et al., 2007) but not in other studies (Maier, 2003; Minami et al., 1991; Nguyen et al., 1998).
The stress-induced exacerbation of LPS-triggered neuroinflammation only manifested in males. Therefore, sex differences in provoked neuroinflammatory signaling may confer subsequent behavioral differences yet to be determined, but unprovoked affective-like behavioral differences appear to be independent from sex differences in neuroinflammatory responses. Alternative mechanisms may include structural changes to the brain and autonomic or endocrine mechanisms (Saveanu and Nemeroff, 2012). Notably, the observed sex differences were significant in relation to their respective saline-injected controls and not in direct comparison between the sexes. Therefore, the magnitude of neuroinflammatory responses did not significantly differ by sex, which has been reported elsewhere (Santos-Galindo et al., 2011; Tonelli et al., 2008).
In addition, increases in neuroinflammatory gene expression in males are in direct opposition to the decreases in central inflammatory responses to peripheral LPS in mice two weeks following either adolescent chronic unpredictable stress or adolescent predatory stress (Barnum et al., 2012). It is possible that these contrasting effects of adolescent stress may be due to species or stress paradigm differences or differences in the time of assessment. Indeed, sensitization of central responses to immune perturbations changes over time for sickness behaviors in response to TNFα reexposure (Hayley et al., 1999). Regardless, stress during adolescence appears to consistently affect neuroinflammatory signaling, which endures for an extensive period of time and depends upon sex, species, and type of stressor.
Similar to previous results from our lab (Bourke and Neigh, 2011), exposure to this stress paradigm reduced body weight during the stress paradigm in male rats only. However, male body mass in the present study recovered in adulthood (e.g., one week before tissue collection), whereas it surpassed that of controls in the previous study. This discrepancy is likely due to differences in the timing of these body mass measurements. Taken together, these data indicate that metabolic consequences of adolescent stress primarily affect the males. The neuroinflammatory and metabolic impact of chronic adolescent stress on males, in contrast to the behavioral and HPA axis effects of chronic adolescent stress in females, has been hypothesized to represent a divergence in adaptations in response to developmental stress (Bourke et al., 2013).
Disruption of endocrine systems due to adolescent stress may be partly responsible for the prolonged neuroinflammatory responses in males and the previously observed behavioral changes in females (Bourke and Neigh, 2011). In general, most reported sex differences in HPA axis output are characterized by greater baseline corticosterone or corticosterone responses in females compared with males (Daneva et al., 1993; Iwasaki-Sekino et al., 2009; Tonelli et al., 2008; Weintraub et al., 2010). Here, sex differences in circulating corticosterone concentrations were absent. The lack of stress effects on either basal corticosterone or LPS-induced corticosterone responses in males substantiates previous data collected after homotypic (Bourke and Neigh, 2011) and heterotypic acute stressors (Bourke et al., 2013). However, female rats exposed to chronic stress during adolescence demonstrated an altered HPA axis response to both homotypic (Bourke and Neigh, 2011) and heterotypic (Bourke et al., 2013) stressors, but no alteration in the HPA axis response to LPS challenge in the present study. It is likely that the nature of the HPA axis stimulation (LPS vs. restraint or forced swim) accounts for this discrepancy. Based on these data, we conclude that prolonged modulation of glucocorticoid release by adolescent stress does not account for the sex differences in neuroinflammatory responses. Certainly, adolescent stress may still modify HPA axis function at a more molecular level (Binder, 2009).
Other potential mediators of adolescent stress-induced neuroinflammatory factors are the gonadal hormones of the developing HPG axis. The spike of circulating estradiol following LPS treatment in females that experienced adolescent stress is consistent with previous findings in unstressed male and female rodents (Castrogiovanni et al., 2008; Daneva et al., 1993), and is thought to be due to increased aromatization of testosterone to estradiol following LPS exposure (Christeff et al., 1992). In turn, estradiol has been shown to reduce microglial-derived inflammatory markers (Bruce-Keller et al., 2000; Dimayuga et al., 2005; Smith et al., 2011; Tapia-Gonzalez et al., 2008). Therefore, the LPS-induced increase in estradiol in females with a history of chronic stress exposure may have mitigated the LPS-induced increase in neuroinflammation that was present in males with a history of chronic stress exposure (Grossman, 1984). Further studies will be necessary to directly test this hypothesis. Notably, stress without LPS treatment did not affect estradiol concentrations. This is consistent with the previous findings that estradiol concentrations, estrous cycles, and uterine weights were not affected by this adolescent stress paradigm (Bourke and Neigh, 2011) and suggests that the possibility that the stress paradigm disrupted the timing or rate of puberty is unlikely. Furthermore, the estradiol spike observed in the stress-LPS group was not accompanied by an increase in progesterone as would be predicted if a disproportionate number of females were in estrus in this treatment group. The phase of the estrous cycle coincident with these endocrine and brain measures and the potential for the stress paradigm to alter the onset of puberty should be investigated in future studies.
Findings on the relationship between estradiol and microglia, however, are mixed and confined primarily to in vitro studies. Consistent with this complex relationship, a positive correlation between circulating estradiol and CD11b gene expression was observed among the non-stressed females. Likewise, estradiol has been shown, in some cases, to potentiate a proinflammatory response in microglia both in vitro and in vivo (Loram et al., 2012; Soucy et al., 2005). In contrast to estradiol, progesterone concentrations remained unaffected by stress or LPS in the present study even though progesterone is characterized as having anti-inflammatory effects on the brain in disease models (Garcia-Segura and Balthazart, 2009; Giatti et al., 2012).
In summary, these data demonstrate that adolescence is a sensitive period of time during which stress can confer sexually dimorphic, long-lasting effects on brain neuroinflammatory pathways. These inflammatory differences are unlikely to be responsible for the sex differences in adult affective-like behavior, but elucidate potential sexually dimorphic mechanisms by which adolescent stress can alter the adult brain.
Acknowledgments
The authors thank Emily Hardy for technical assistance. This work was supported by an American Cancer Society fellowship PF-08-086-01-TBE (L.M.P) and NIH grant MH091312 (G.N.N.).
References
- Angold A, Costello EJ, Worthman CM. Puberty and depression: the roles of age, pubertal status and pubertal timing. Psychol. Med. 1998;28:51–61. doi: 10.1017/s003329179700593x. [DOI] [PubMed] [Google Scholar]
- Anisman H. Cascading effects of stressors and inflammatory immune system activation: implications for major depressive disorder. J. Psychiatry Neurosci. 2009;34:4–20. [PMC free article] [PubMed] [Google Scholar]
- Audet MC, Jacobson-Pick S, Wann BP, Anisman H. Social defeat promotes specific cytokine variations within the prefrontal cortex upon subsequent aggressive or endotoxin challenges. Brain. Behav. Immun. 2011;25:1197–1205. doi: 10.1016/j.bbi.2011.03.010. [DOI] [PubMed] [Google Scholar]
- Barnum CJ, Pace TW, Hu F, Neigh GN, Tansey MG. Psychological stress in adolescent and adult mice increases neuroinflammation and attenuates the response to LPS challenge. J. Neuroinflammation. 2012;9:9. doi: 10.1186/1742-2094-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebbington P, Dunn G, Jenkins R, Lewis G, Brugha T, Farrell M, Meltzer H. The influence of age and sex on the prevalence of depressive conditions: report from the National Survey of Psychiatric Morbidity. Int. Rev. Psychiatry. 2003;15:74–83. doi: 10.1080/0954026021000045976. [DOI] [PubMed] [Google Scholar]
- Becker DF, Grilo CM. Prediction of suicidality and violence in hospitalized adolescents: comparisons by sex. Can. J. Psychiatry. 2007;52:572–580. doi: 10.1177/070674370705200905. [DOI] [PubMed] [Google Scholar]
- Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. 2009;34(Suppl. 1):S186–195. doi: 10.1016/j.psyneuen.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Bourke CH, Neigh GN. Behavioral effects of chronic adolescent stress are sustained and sexually dimorphic. Horm. Behav. 2011;60:112–120. doi: 10.1016/j.yhbeh.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourke CH, Neigh GN. Exposure to repeated maternal aggression induces depressive-like behavior and increases startle in adult female rats. Behav. Brain Res. 2012;227:270–275. doi: 10.1016/j.bbr.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourke CH, Raees MQ, Malviya S, Bradburn CA, Binder EB, Neigh GN. Glucocorticoid sensitizers Bag1 and Ppid are regulated by adolescent stress in a sex-dependent manner. Psychoneuroendocrinology. 2013;38:84–93. doi: 10.1016/j.psyneuen.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Keeling JL, Keller JN, Huang FF, Camondola S, Mattson MP. Antiinflammatory effects of estrogen on microglial activation. Endocrinology. 2000;141:3646–3656. doi: 10.1210/endo.141.10.7693. [DOI] [PubMed] [Google Scholar]
- Castrogiovanni D, Gaillard RC, Giovambattista A, Spinedi E. Neuroendocrine, metabolic, and immune functions during the acute phase response of inflammatory stress in monosodium l-glutamate-damaged, hyperadipose male rat. Neuroendocrinology. 2008;88:227–234. doi: 10.1159/000124131. [DOI] [PubMed] [Google Scholar]
- Christeff N, Carli A, Benassayag C, Bleichner G, Vaxelaire JF, Nunez EA. Relationship between changes in serum estrone levels and outcome in human males with septic shock. Circ. Shock. 1992;36:249–255. [PubMed] [Google Scholar]
- Conley CS, Rudolph KD. The emerging sex difference in adolescent depression: interacting contributions of puberty and peer stress. Dev. Psychopathol. 2009;21:593–620. doi: 10.1017/S0954579409000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalla C, Antoniou K, Drossopoulou G, Xagoraris M, Kokras N, Sfikakis A, Papadopoulou-Daifoti Z. Chronic mild stress impact: are females more vulnerable? Neuroscience. 2005;135:703–714. doi: 10.1016/j.neuroscience.2005.06.068. [DOI] [PubMed] [Google Scholar]
- Daneva T, Spinedi E, Hadid R, Jacquier MC, Giacomini M, Gaillard RC. Transient sex-related changes in the mice hypothalamo–pituitary–adrenal (HPA) axis during the acute phase of the inflammatory process. Mediators Inflamm. 1993;2:123–127. doi: 10.1155/S0962935193000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, Wollman EE, Vitkovic L, Yirmiya R. Cytokines, stress, and depression. Conclusions and perspectives. Adv. Exp. Med. Biol. 1999;461:317–329. doi: 10.1007/978-0-585-37970-8_17. [DOI] [PubMed] [Google Scholar]
- Desbonnet L, Garrett L, Daly E, McDermott KW, Dinan TG. Sexually dimorphic effects of maternal separation stress on corticotrophin-releasing factor and vasopressin systems in the adult rat brain. Int. J. Dev. Neurosci. 2008;26:259–268. doi: 10.1016/j.ijdevneu.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Dimayuga FO, Reed JL, Carnero GA, Wang C, Dimayuga ER, Dimayuga VM, Perger A, Wilson ME, Keller JN, Bruce-Keller AJ. Estrogen and brain inflammation: effects on microglial expression of MHC, costimulatory molecules and cytokines. J. Neuroimmunol. 2005;161:123–136. doi: 10.1016/j.jneuroim.2004.12.016. [DOI] [PubMed] [Google Scholar]
- Eiland L, Romeo RD. Stress and the developing adolescent brain. Neuroscience. 2012 doi: 10.1016/j.neuroscience.2012.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Oliva AM, de Pablos RM, Villaran RF, Arguelles S, Venero JL, Machado A, Cano J. Stress is critical for LPS-induced activation of microglia and damage in the rat hippocampus. Neurobiol. Aging. 2011;32:85–102. doi: 10.1016/j.neurobiolaging.2009.01.012. [DOI] [PubMed] [Google Scholar]
- Frackiewicz EJ, Sramek JJ, Cutler NR. Gender differences in depression and antidepressant pharmacokinetics and adverse events. Ann. Pharmacother. 2000;34:80–88. doi: 10.1345/aph.18465. [DOI] [PubMed] [Google Scholar]
- Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain. Behav. Immun. 2007;21:47–59. doi: 10.1016/j.bbi.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Garcia-Segura LM, Balthazart J. Steroids and neuroprotection: new advances. Front. Neuroendocrinol. 2009;30:v–ix. doi: 10.1016/j.yfrne.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giatti S, Boraso M, Melcangi RC, Viviani B. Neuroactive steroids, their metabolites, and neuroinflammation. J. Mol. Endocrinol. 2012;49:R125–134. doi: 10.1530/JME-12-0127. [DOI] [PubMed] [Google Scholar]
- Gibb J, Hayley S, Gandhi R, Poulter MO, Anisman H. Synergistic and additive actions of a psychosocial stressor and endotoxin challenge: circulating and brain cytokines, plasma corticosterone and behavioral changes in mice. Brain. Behav. Immun. 2008;22:573–589. doi: 10.1016/j.bbi.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Gibb J, Hayley S, Poulter MO, Anisman H. Effects of stressors and immune activating agents on peripheral and central cytokines in mouse strains that differ in stressor responsivity. Brain. Behav. Immun. 2011;25:468–482. doi: 10.1016/j.bbi.2010.11.008. [DOI] [PubMed] [Google Scholar]
- Girotti M, Donegan JJ, Morilak DA. Chronic intermittent cold stress sensitizes neuro-immune reactivity in the rat brain. Psychoneuroendocrinology. 2011;36:1164–1174. doi: 10.1016/j.psyneuen.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman CJ. Regulation of the immune system by sex steroids. Endocr. Rev. 1984;5:435–455. doi: 10.1210/edrv-5-3-435. [DOI] [PubMed] [Google Scholar]
- Hayley S, Brebner K, Lacosta S, Merali Z, Anisman H. Sensitization to the effects of tumor necrosis factor-alpha: neuroendocrine, central monoamine, and behavioral variations. J. Neurosci. 1999;19:5654–5665. doi: 10.1523/JNEUROSCI.19-13-05654.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki-Sekino A, Mano-Otagiri A, Ohata H, Yamauchi N, Shibasaki T. Gender differences in corticotropin and corticosterone secretion and corticotropin-releasing factor mRNA expression in the paraventricular nucleus of the hypothalamus and the central nucleus of the amygdala in response to footshock stress or psychological stress in rats. Psychoneuroendocrinology. 2009;34:226–237. doi: 10.1016/j.psyneuen.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Johnson JD, O’Connor KA, Hansen MK, Watkins LR, Maier SF. Effects of prior stress on LPS-induced cytokine and sickness responses. Am. J Physiol. Regul. Integr. Comp. Physiol. 2003;284:R422–R432. doi: 10.1152/ajpregu.00230.2002. [DOI] [PubMed] [Google Scholar]
- Kipp M, Beyer C. Impact of sex steroids on neuroinflammatory processes and experimental multiple sclerosis. Front. Neuroendocrinol. 2009;30:188–200. doi: 10.1016/j.yfrne.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Kudielka BM, Buske-Kirschbaum A, Hellhammer DH, Kirschbaum C. HPA axis responses to laboratory psychosocial stress in healthy elderly adults, younger adults, and children: impact of age and gender. Psychoneuroendocrinology. 2004;29:83–98. doi: 10.1016/s0306-4530(02)00146-4. [DOI] [PubMed] [Google Scholar]
- Laroche J, Gasbarro L, Herman JP, Blaustein JD. Enduring influences of peripubertal/adolescent stressors on behavioral response to estradiol and progesterone in adult female mice. Endocrinology. 2009a;150:3717–3725. doi: 10.1210/en.2009-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laroche J, Gasbarro L, Herman JP, Blaustein JD. Reduced behavioral response to gonadal hormones in mice shipped during the peripubertal/adolescent period. Endocrinology. 2009b;150:2351–2358. doi: 10.1210/en.2008-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenroot RK, Giedd JN. Sex differences in the adolescent brain. Brain Cogn. 2010;72:46–55. doi: 10.1016/j.bandc.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard BE. Inflammation, depression and dementia: are they connected? Neurochem. Res. 2007;32:1749–1756. doi: 10.1007/s11064-007-9385-y. [DOI] [PubMed] [Google Scholar]
- Loram LC, Sholar PW, Taylor FR, Wiesler JL, Babb JA, Strand KA, Berkelhammer D, Day HE, Maier SF, Watkins LR. Sex and estradiol influence glial pro-inflammatory responses to lipopolysaccharide in rats. Psychoneuroendocrinology. 2012;37:1688–1699. doi: 10.1016/j.psyneuen.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SF. Bi-directional immune-brain communication: implications for understanding stress, pain, and cognition. Brain. Behav. Immun. 2003;17:69–85. doi: 10.1016/s0889-1591(03)00032-1. [DOI] [PubMed] [Google Scholar]
- McCormick CM, Mathews IZ. HPA function in adolescence: role of sex hormones in its regulation and the enduring consequences of exposure to stressors. Pharmacol. Biochem. Behav. 2007;86:220–233. doi: 10.1016/j.pbb.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Minami M, Kuraishi Y, Yamaguchi T, Nakai S, Hirai Y, Satoh M. Immobilization stress induces interleukin-1 beta mRNA in the rat hypothalamus. Neurosci. Lett. 1991;123:254–256. doi: 10.1016/0304-3940(91)90944-o. [DOI] [PubMed] [Google Scholar]
- Naninck EF, Lucassen PJ, Bakker J. Sex differences in adolescent depression: do sex hormones determine vulnerability? J. Neuroendocrinol. 2011;23:383–392. doi: 10.1111/j.1365-2826.2011.02125.x. [DOI] [PubMed] [Google Scholar]
- National Research Council, N. Guide for the Care and Use of Laboratory Animals. Eighth ed The National Academies Press; 2011. [Google Scholar]
- Nguyen KT, Deak T, Owens SM, Kohno T, Fleshner M, Watkins LR, Maier SF. Exposure to acute stress induces brain interleukin-1beta protein in the rat. J. Neurosci. 1998;18:2239–2246. doi: 10.1523/JNEUROSCI.18-06-02239.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble RE. Depression in women. Metabolism. 2005;54:49–52. doi: 10.1016/j.metabol.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Nolen-Hoeksema S, Girgus JS. The emergence of gender differences in depression during adolescence. Psychol. Bull. 1994;115:424–443. doi: 10.1037/0033-2909.115.3.424. [DOI] [PubMed] [Google Scholar]
- Ogawa T, Kuwagata M, Hori Y, Shioda S. Valproate-induced developmental neurotoxicity is affected by maternal conditions including shipping stress and environmental change during early pregnancy. Toxicol. Lett. 2007;174:18–24. doi: 10.1016/j.toxlet.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Pace TW, Miller AH. Cytokines and glucocorticoid receptor signaling. Relevance to major depression. Ann. N. Y. Acad. Sci. 2009;1179:86–105. doi: 10.1111/j.1749-6632.2009.04984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Galindo M, Acaz-Fonseca E, Bellini MJ, Garcia-Segura LM. Sex differences in the inflammatory response of primary astrocytes to lipopolysaccharide. Biol. Sex Differ. 2011;2:7. doi: 10.1186/2042-6410-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saveanu RV, Nemeroff CB. Etiology of depression: genetic and environmental factors. Psychiatr. Clin. North. Am. 2012;35:51–71. doi: 10.1016/j.psc.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Schweingruber N, Reichardt SD, Luhder F, Reichardt HM. Mechanisms of glucocorticoids in the control of neuroinflammation. J. Neuroendocrinol. 2012;24:174–182. doi: 10.1111/j.1365-2826.2011.02161.x. [DOI] [PubMed] [Google Scholar]
- Smith JA, Das A, Butler JT, Ray SK, Banik NL. Estrogen or estrogen receptor agonist inhibits lipopolysaccharide induced microglial activation and death. Neurochem. Res. 2011;36:1587–1593. doi: 10.1007/s11064-010-0336-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon MB, Herman JP. Sex differences in psychopathology: of gonads, adrenals and mental illness. Physiol. Behav. 2009;97:250–258. doi: 10.1016/j.physbeh.2009.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucy G, Boivin G, Labrie F, Rivest S. Estradiol is required for a proper immune response to bacterial and viral pathogens in the female brain. J. Immunol. 2005;174:6391–6398. doi: 10.4049/jimmunol.174.10.6391. [DOI] [PubMed] [Google Scholar]
- Tapia-Gonzalez S, Carrero P, Pernia O, Garcia-Segura LM, Diz-Chaves Y. Selective oestrogen receptor (ER) modulators reduce microglia reactivity in vivo after peripheral inflammation: potential role of microglial ERs. J. Endocrinol. 2008;198:219–230. doi: 10.1677/JOE-07-0294. [DOI] [PubMed] [Google Scholar]
- Tonelli LH, Holmes A, Postolache TT. Intranasal immune challenge induces sex-dependent depressive-like behavior and cytokine expression in the brain. Neuropsychopharmacology. 2008;33:1038–1048. doi: 10.1038/sj.npp.1301488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub A, Singaravelu J, Bhatnagar S. Enduring and sex-specific effects of adolescent social isolation in rats on adult stress reactivity. Brain Res. 2010;1343:83–92. doi: 10.1016/j.brainres.2010.04.068. [DOI] [PubMed] [Google Scholar]
- Young EA. Sex differences and the HPA axis: implications for psychiatric disease. J. Gend. Specif. Med. 1998;1:21–27. [PubMed] [Google Scholar]