

Abstract
γ-MSH (γ-melanocyte-stimulating hormone: H-Tyr-Val-Met-Gly-His-Phe-Arg-Trp-Asp-Arg-Phe-Gly-OH), with its exquisite specificity and potency, has recently created much excitement as a drug lead. However, this peptide like most peptides susceptible to proteolysis in vivo which potentially decreases its beneficial activities. In our continued effort to design a proteolytically stable with specific receptor binding ligand, we have engineered peptides by cyclizing γ-MSH using a thioether bridge. A number of novel cyclic truncated γ-MSH analogues were designed and synthesized, in which a thioether bridge was incorporated between a cysteine side chain and an N-terminal bromoacyl group. One of these peptides, cyclo-[(CH2)3CO-Gly1-His2-D-Phe3-Arg4-D-Trp5-Cys(S-)6]-Asp7-Arg8-Phe9-Gly10-NH2, demonstrated potent antagonist activity and receptor selectivity for the human melanocortin 1 receptor (hMC1R) (IC50 = 17 nM). This novel peptide is the most selective antagonist for the human hMC1R to date. Further pharmacological studies have shown that this peptide can specifically target melanoma cells. The NMR analysis of this peptide in a membrane–like environment revealed a new turn structure, specific to the hMC1R antagonist, at the C terminal, wherein the side chain and backbone conformation of D-Trp5 and Phe9 of the peptide are contributors to the hMC1R selectivity. Cyclization strategies represent an approach for stabilizing bioactive peptides while keeping their full potencies and should boost applications of peptide-based drugs in human medicine.
Keywords: α-MSH, γ-MSH, melanocortin receptors, melanotropin antagonist, hMC1R, steric constraints, selective ligands, melanoma
Introduction
The melanocortin system is involved in numerous physiological functions and may have great potential for the novel drug discovery.1-10 The human melanocortin receptor 1 (hMC1R), mainly present in the periphery for example, in the skin of mammals, is involved in control of the relative amounts of pheomelanin and photoprotective eumelanin.11-13 Appropriate selective hMC1R ligands may offer protection against damaging and mutagenic effects of UV radiation, or be useful in the diagnosis and treatment of certain types of skin cancer, such as melanoma. The hMC1R is also present on the surface of various immune cells, macrophages, fibroblasts, monocytes, mast cells and neutrophils and is a potential target for the development of agents for the treatment of acute and chronic inflammatory diseases, neurodegenerative diseases and systemic host reactions.5, 11-14 In the CNS the MC1 receptor is present only on neurons in the periaqueductal grey matter of the midbrain where it is thought to have a role in pain control.15-17 Despite the multiple physiological functions and the great potential for medical application, there are only a few reports of selective antagonists for the hMC1R available in the literature. Thus design of selective hMC1R agonists and antagonists warrants special interest in the melanocortin research. Therefore, in our continued effort to produce selective melanocortin ligands, we devoted a great deal of time to obtain ligands with selective antagonist activity for the hMC1R and to develop the pharmacological properties that are suitable for medical applications of these compounds.
Structure based drug design is one of the major strategies in drug discovery.18-21 In peptide-based drug development, truncations and amino acid scans have been used to find the relevant pharmacophore. 19,22-25 The same method is important for the rational design of melanocortin receptors ligands. Conformational constraints and β-turn mimetics were applied to develop stable and selective melanotropins; the 3D structure of ligands (NMR) combined with computational aided drug design helped in obtaining several selective compounds.26,27 Recently, N-methylation of the amide bonds has been introduced into our novel drug design strategy to obtain the most selective agonist of the hMC1R.28 In this study we have designed a selective antagonist of the hMC1R by using a peptide fragment conformation from agouti signaling protein (ASIP)29 as a template combined with the pharmacophore of γ-MSH. ASIP is an endogenous antagonist of the hMC1R. The key feature of the design is a cyclic constraint via a thioether bridge as shown in Scheme 1. Utilizing the thioether bond constraints, γ-MSH leads to improved receptor (subtype) selectivity with enhanced pharmacological and chemical-physical properties that include metabolic stability, efficacy, lipophilicity, potency and bioavailability. Further, the newly discovered cyclic hMC1R peptide antagonist also was examined for its conformational properties and tested on melanoma cells to evaluate its potential for in vivo applications.
Scheme 1.

Synthesis of a cyclic analogue of γ-MSH Reagents: (i) assembly of linear peptide by Fmoc strategy; (ii) 20% piperidine in DMF: (iii), 4-bromo butyric acid, DIC, DMF (iv) 2%TFA/5% Et3SiH/CH2Cl2; (v) 5% DIEA/DMF; (vi) 95%TFA/5% H2O
Design
The thioether bond has been previously used as a stable disulfide replacement in bioactive cyclic peptides to increase the metabolic stability of peptides.30-32 The thioether linkage has also been used to prepare conformationally constrained peptides that are analogues of normally linear peptides.18,30,32-36 Cyclic peptides with thioether bonds have been found in nature, especially in a family of antibiotics and lantibiotics 37-39.
Previouly, cyclic thioether peptides have been used as more stable alternatives to disulfide bridges. Thioethers are usually prepared by alkylation of thiols. Thiols are powerful nucleophiles and they react with electrophiles in an SN2 manner to produce thioethers. Nucleophilic substitutions with thiols do not require the use of strong bases and generally proceeds at room temperature. In favorable cases, thiol alkylation can even be performed at room temperature in an acidic environment.
We have considered the synthesis of macrocyclic thioethers to be a good way for global conformational constraint of the natural ligand γ-MSH, since γ-MSH analogues tend to be more selective than α-MSH from our previous studies20. The new ligands were designed to contain an array of rings with sizes from 21, 22, 23 atoms to form a spectrum of cyclic peptides from less flexible to more flexible analogues. Intramolecular thioether bond formation was proposed to be a good synthetic method for peptide cyclization and a convenient approach for probing peptide conformations. Published data suggest that a 23-membered ring (e.g. MTII, SHU9119)41-43 is an optimal size for melanotropin ligand binding to the melanocortin receptors. So far, the majority of published work is based on α-MSH cyclic analogues with either lactam or disulfide bridges. The application of thioethers for cyclization of γ-MSH derivatives is a novel structural modification for melanotropin ligands. In order to form a thioether we need to introduce a thiol group into a peptide sequence and a choice for the thiol source was a cysteine residue. The next issue was where to incorporate a cysteine residue in γ-MSH and for that we examined two general ways. We can either replace an amino acid with cysteine or we can insert a cysteine between two amino acids in the natural sequence. Finally, we need to consider a suitable site for an electrophilic moiety. A very simple solution was to apply a bromoacetyl group at the amino terminus of the peptide. We then have two very reactive species, which can form intramolecular thioether bridged structures(Figure 1). The synthetic procedure is shown in Scheme 1. In this approach we used bromoacetic as well as 3-bromopropionic acid and 4-bromobutyric acid so as to obtain cyclic structures of different ring size. (Table 1)
Figure 1.
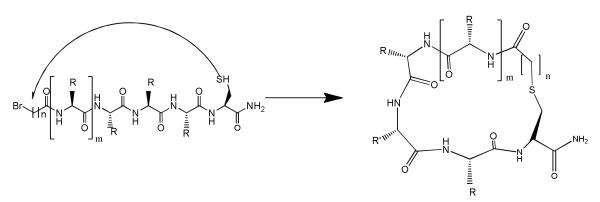
General scheme for peptide cyclization using sulfide bridge where n=1, 2 & 3 and m=1,2
Table 1.
Novel designed cyclic γ-MSH analogues
| LIGAND | SEQUENCE | RING SIZE |
|---|---|---|
| Peptide 1 | c(CH2-CO-Gly-His-D-Phe-Arg-Trp-Cys)-Asp-Arg-Phe-Gly-NH2 | 21 |
| Peptide 2 | c(CH2-CO-Gly-His-D-Phe-Arg-D-Trp-Cys)-Asp-Arg-Phe-Gly-NH2 | 21 |
| Peptide 3 | c(C2H4-CO-Gly-His-D-Phe-Arg-Trp-Cys)-Asp-Arg-Phe-Gly-NH2 | 22 |
| Peptide 4 | c(C2H4-CO-Gly-His-D-Phe-Arg-D-Trp-Cys)-Asp-Arg-Phe-Gly-NH2 | 22 |
| Peptide 5 | c(C3H6-CO-Gly-His-D-Phe-Arg-Trp-Cys)-Asp-Arg-Phe-Gly-NH2 | 23 |
| Peptide 6 | c(C3H6-CO-Gly-His-D-Phe-Arg-D-Trp-Cys)-Asp-Arg-Phe-Gly-NH2 | 23 |
Experimental Section
Nα-Fmoc-amino acids were obtained from Bachem (Torrance, CA), Nova Biochem (Gibbstown, NJ) and AdvancedChemTech (Louisville, KY). Fmoc Rink amide resin was purchased from Polymer Laboratories (Santa Clara, CA). Organic solvents and reagents were purchased from Aldrich and used without further purification. All cyclic peptides were synthesized according to a general protocol outline below.
Rink amide resin (100 mg, 0.065 mmol/g) was placed into a 5 mL polypropylene syringe with the frit on the bottom and swollen in DCM (2mL) for 30 min and in DMF (2 mL) for 30 min. Fmoc protecting group on the Rink linker was removed by 50 % piperidine in DMF. After 20 min the solution of piperidine was removed and the resin was washed with DMF (2 mL, 10 times). Fmoc amino acid (3 equiv., 0.195 mmol) and HOBt (3 equiv., 0.195 mmol) were dissolved in 700 μL of DMF and then DIC (3 equiv., 0.195 mmol) was added. The coupling mixture was transferred into the syringe with the resin and shaken for 1-3 hours. Coupling completion was monitored with bromophenol blue and ninhydrin tests 40,58. Coupling mixture was removed and the resin was washed with DMF (2 mL, 5 times). The Fmoc group was cleaved with 50% piperidine in DMF. Each coupling and deprotection step was repeated until a linear peptide was assembled. Bromoacetic acid (3 equiv. 0.195 mmol) was dissolved in 700 μL of DMF and DIC (3 equiv., 0.195 mmol) was added. The acylation reaction was usually complete after 20 min. The reaction mixture was removed and resin was washed with DMF (2 mL, 5 times) and DCM (2 mL, 5 times). The monomethoxytrityl group was cleaved by treatment with 2%TFA / 5%TIPS / DCM (2 mL, 5 times, 5 min each treatment). The resin was washed with DCM (2 mL, 10 times) and DMF (2 mL, 5 times). Peptide cyclization was performed in a solution of 5 % DIEA in DMF during 10-12 hours at room temperature. The final wash of the resin was done with DMF (2 mL, 5 times) and DCM (2 mL, 5 times). The product was cleaved from the resin with a mixture of 95% TFA, 2.5 % TIPS and 2.5% water in 1.5 hours. Side chain protecting groups were removed during the cleavage step. The cleavage mixture was evaporated on a rotary evaporator. A crude cyclic peptide was dissolved in acetic acid and purified by HPLC.
TLC was performed using two solvent systems. System I was composed of n-butanol-water-acetic acid-pyridine (4 : 2 : 1 : 2 ) and system II was n-butanol-water-acetic acid (4 :1 : 2 ). The Rf values are listed in the Table 2.
Table 2.
Analytical characterization of cyclic peptides 1-6
|
Peptide
No. |
Molecular Formula |
m/z (M+H+) | HPLC retention time, min[A] |
TLC Rf[B] | |||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| Calculated | Observed (ESI) |
System Ia | System IIb | System Ic Rf |
System Id Rf |
||
| 1 | C60H78N20O13S | 1319.58562 | 1319.5855 | 13.71 | 19.97 | 0.52 | 0.17 |
| 2 | C60H78N20O13S | 1319.58562 | 440.5299a | 13.14 | 18.59 | 0.51 | 0.15 |
|
| |||||||
| 3 | C61H80N20O13S | 1333.60127 | 1333.6080 | 13.45 | 18.25 | 0.48 | 0.13 |
|
| |||||||
| 4 | C61H80N20O13S | 1333.60127 | 1333.5981 | 13.09 | 18.43 | 0.54 | 0.20 |
|
| |||||||
| 5 | C62H82N20O13S | 1347.61692 | 1347.6152 | 13.86 | 18.93 | 0.49 | 0.13 |
|
| |||||||
| 6 | C62H82N20O13S | 1347.61692 | 1347.6187 | 13.47 | 18.73 | 050 | 0.13 |
MS: the theoretical and experimental masses for peptides 1-6 obtained by FT-MS analysis. a[M+3H]3+ HPLC: gradient: 10-90% B in A over 40 min, flow rate 1.0 mL/min;
System I: Buffer A: 0.1% TFA in H2O, Buffer B: 0.08%TFA in ACN (pH 3);
System II: Buffer A: 20mM Na2HPO4 in H2O, Buffer B: 20% H2O in ACN (pH 6.3) TLC:
System I: n-butanol / water / acetic acid / pyridine (4:2:1:2) pH 6;
System II: n-butanol / water / acetic acid (4:1:2) pH 2.5
For HPLC samples were dissolved in 10-20% of methanol in water (1 mg/mL) and 5 μL of that solution was injected onto the analytical HPLC column (YMC-Pack ODS-AM 150 × 4.6 mm, 3 μm, 120Å). Two buffer systems were used for peptide purity assessment. Solvent system I consists of buffers (A) 0.1% TFA in H2O and (B) 0.08% TFA in ACN. Solvent system II consists of buffers (A) 20 mM Na2HPO4 in H2O and (B) 20 % ACN in H2O. The retention times (RT) of cyclic peptides are listed in the Table 2. Peptide purification was achieved using preparative HPLC. A crude product was dissolved in 50% of acetic acid in water (10 mg/mL), filtrated trough a minidisk filter and 1 mL of clear solution was loaded onto the semi-preparative column (YMC-Pack ODS-AM, 100 × 10 mm, 5 μm, 120Å). The solvent system for HPLC purification consists of buffers (A) 0.1% TFA in H2O and (B) 0.08% TFA in ACN. The solvent gradient was usually 2 % (A) to 40 % (B) over 15 minutes with the flow rate of 10 mL/min. Collected fractions of the purified products were lyophilized.
We used high-resolution mass spectrometry for evaluation of molecular formulas of our novel macrocyclic peptide ligands (Table 2). Both ESI and MALDI were used to generate protonated peptides (Peptides 1-6), respectively. ESI experiments were carried out on an IonSpec 4.7 T Fourier ion cyclotron resonance (ICR) instrument with peak matching technique by using peptide reference standards. MALDI measurements have been carried out on a Bruker Reflex III MALDI-TOF instrument.
Biological Activity Assays
Receptor Binding Assays
Competition binding experiments were carried out using both of cloned cell line and melanoma cells (A375, ATCC). HEK293 cells stably expressing the human MC1, MC3, MC4, and MC5 receptors were described before44,45. HEK293 cells transfected with hMCRs44,45 were seeded on 96-well plates 48 hours before assay (50,000 cells/well). For the assay, the cell culture medium was aspirated and the cells were washed once with a freshly prepared MEM buffer containing 100% minimum essential medium with Earle’s salt (MEM, GIBCO), and 25 mM sodium bicarbonate. Next, the cells were incubated for 40 min at 37°C with different concentrations of unlabeled peptide and labeled [125I]-[Nle4,D-Phe7]-α-MSH(Perkin-Elmer Life Science, 20,000 cpm/well, 33.06 pM) diluted in a 125 μL of freshly prepared binding buffer containing 100% MEM, 25 mM HEPES (pH 7.4), 0.2% bovine serum albumin, 1mM 1,10-phenanthrolone, 0.5 mg/L leupeptin, 200 mg/L bacitracin. The assay medium was subsequently removed, the cells were washed once with basic medium, and then lysed by the addition of 10 μL of 0.1M NaOH and 10 μL of 1% Triton X-100. The radioactivity of lysed cells was measured by a Wallac MicroBeta TriLux 1450 LSC and Luminescence Counter (PerkinElmer Life Science, Boston, MA).
Adenylate Cyclase Assays
Both melanoma as well as cloned cells were grown to confluence in Dulbecco’s Modified Eagle’s Medium (DMEM) and MEM medium (GIBCO) respectively containing 10% fetal bovine serum, 100 units/mL penicillin and streptomycin, and 1 mM sodium pyruvate. The cells were seeded on 96-well plates 48 hours before assay (50,000 cells/well). For the assay, the cell culture medium was removed and the cells were rinsed with 100 μL of MEM buffer (GIBCO). An aliquot (100 μL) of the Earle’s balanced salt solution with 5 nM isobutylmethylxanthine (IBMX) was placed in each well along for 1 min at 37°. Next, aliquots (25 μL) of melanotropin peptides of varying concentration were added, and the cells were incubated for 3 min at 37°C. The reaction was stopped by aspirating the assay buffer and adding 60 μL ice-cold Tris/EDTA buffer to each well, then placing the plates in a boiling water bath for 7 min. The cell lysates were then centrifuged for 10 min at 2,300 × g. A 50 μL aliquot of the supernatant was transferred to another 96-well plate and placed with 50 μL [3H]cAMP and 100 μL protein kinase A (PKA) buffer in an ice bath for 2-3 hours. The PKA buffer consisted of Tris/EDTA buffer with 60 μg/mL PKA and 0.1% bovine serum albumin by weight. The incubation mixture was filtered through 1.0 μm glass fiber filters in MultiScreen™-FB 96-well plates (Millipore, Billerica, MA). The total [3H]cAMP was measured by a Wallac MicroBeta TriLux 1450 LSC and Luminescence Counter (PerkinElmer Life Science, Boston, MA). The cAMP accumulation data for each peptide analogue was determined with the help of a cAMP standard curve generated by the same method as described above. IC50 and EC50 values represent the mean of two experiments performed in triplicate. IC50 and EC50 values and their associated standard errors were determined for binding and cAMP assays by fitting the data using a nonlinear least squares analysis, with the help of GraphPad Prism 4 (GraphPad Software, San Diego, CA). The maximal cAMP produced at 10 μM concentration of each ligand was compared to the amount of cAMP produced at 10 μM concentration of the standard agonist MT-II, and is expressed in percent (as % max effect) in Table 3. The pA2 is 8.1.
Table 3.
Binding and cAMP assays of novel cyclized peptides towards human melanocortin receptors
| Code | hMC1R | hMC3R | hMC4R | hMC5R | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (nM) | EC50(nM) | Act% | IC50(nM) | EC50(nM) | Act% | IC50(nM) | EC50(nM) | Act% | IC50(nM) | EC50(nM) | Act% | |
| 1 | 2000 | >1000 | / | 1630±300 | 195±23 | 140 | N.B. | 800±90 | 37 | 510±60 | 86±10 | 31 |
| 2 | 1800 | N.A. | / | NB | >1000 | 30 | N.B. | N.A. | / | N.B. | >1000 | 0 |
| 3 | 330±60 | N.A. | 10 | 76±10 | 170±30 | 52 | N.B. | N.A. | / | 160±20 | 2500 | 100 |
| 4 | 30±4 | N.A. | / | 17000 | 1100±120 | 50 | N.B. | N.A. | NA | 115±10 | 1000 | 0 |
| 5 | 29±3 | N.A. | / | 8500 | 100±14 | 50 | N.B. | N.A. | / | 210±30 | 873±100 | 91 |
| 6 | 17±2 | N.A. | / | 3900 | 59±6 | 75 | N.B. | N.A. | / | 1000±110 | 1300 | 65 |
| Nle3 γ-MSH | 60±5 | 80±7 | 100 | 62±8 | 1000±100 | 100 | 45±5 | 70±7 | 100 | 640±100 | 82±8 | 100 |
| Nle3 γ-MSH-NH2 | -- | -- | -- | 45±7 | 1.6±0.2 | 100 | 64±7 | 34±4 | 105 | 200±23 | 99±10 | 114 |
| γ-MSH | 1000±20 | 300±30 | 100 | 71±10 | 700±89 | 100 | 760±80 | 710±70 | 100 | 2200±200 | 550±60 | 100 |
IC50 = concentration of peptide at 50% specific binding (N=4). NB = 0% of 125I-NDP-α-MSH displacement observed at 10 μM.. EC50 = Effective concentration of peptide that was able to generate 50% maximal intracellular cAMP accumulation (N=4). Act% = % of cAMP produced at 10 μM ligand concentration, in relation to MT-II. NA = 0% cAMP accumulation observed at 10 μM. The peptides were tested at a range of concentration from 10−10 to 10−5 M; NB = no binding observed at 10−5M.
Data Analysis
IC50 and EC50 values represent the mean of two experiments performed in triplicate. IC50 and EC50 estimates and their associated standard errors were determined by fitting the data using a nonlinear least squares analysis, with the help of GraphPad Prism 4 (GraphPad Software, San Diego, CA).
NMR spectroscopy methods
All NMR spectra were recorded on a Bruker DRX600 600MHz spectrometer at 298°K. Peptide concentration for the NMR experiments is 4.7 mM. The micelle samples were prepared by dissolving the peptide and 50 equivalents of perdeuterated SDS in 0.6 ml of acetate buffer (10 mM) containing 10% D2O. The pH of the each sample was adjusted to 5.5 by using DCl or NaOD as necessary. Deuterated 3-(Trimethylsilyl)propionic acid (TSP) was added as internal standard for referencing. Two-dimensional nuclear Overhauser enhancement (NOESY) and total correlation spectra (TOCSY) were acquired using standard pulse sequences and processed using XWINNMR (Bruker Inc) and FELIX2000 (Accelrys Inc, San Diego, CA). Mixing time for TOCSY spectra were 60 ms, and for NOESY spectra was 300 ms. All experiments were 750 increments in t1, 16 or 32 scans each, 1.5 s relaxation delay, size 2 or 4K and the spectral processing was with shifted sine bell window multiplications in both dimensions. The water suppression was achieved by using the WATERGATE pulse sequence. Coupling constants (3JαH-NH) were measured from DQF-COSY spectrum.
Structure calculation methods
Distance constraints for the structure calculation were obtained from integral volumes of the NOESY peaks. The NOE integral volumes were classified into strong, medium and weak with 3.0, 4.0 and 5.0 Å as upper bound distance. Molecular dynamics simulation was done with the INSIGHT/DISCOVER package (Accelrys Inc, San Diego, CA) with consistent valency force field (CVFF). All the calculations were done in vacuo. A distance dependent dielectric constant (2.5r where r is the distance in Å) was used. All peptide bonds were constrained to trans conformation by a 100kcal mol−1 energetic penalty. Distance restraints with a force constant of 25 kcal mol−1 were applied in the form of a flat-bottom potential well with a common lower bound of 1.8 Å and an upper bound of 3.0, 4.0 and 5.0 Å, respectively, in accordance with observed weak, moderate or strong NOE intensities. Only the distance restraints from inter-residue NOEs were included in the calculation. Dihedral angle restraints based on CαH chemical shift index (CSI) were imposed on the residues displaying negative deviation. Thus for a CSI of >−0.10 ppm, the ϕ and ψ restraints were in the range −90° to −30° and −60° to 0°, respectively while for a CSI of ≤−0.10 ppm, the corresponding ranges were −150° to −30° for ϕ and −90° to 150° for ψ. For D-chiral amino acids the opposite dihedral angle values were used.
Results and Discussions
Bioassay Results
It has been shown that D-phenylalanine is a critical amino acid in the tetrapeptide (Pharmacophore) sequence of γ-MSH to enhance potency, whereas a D-tryptophan improves selectivity41-47. We have prepared three peptides with the same sequence containing D-Phe, but with different ring sizes (peptides 1, 3, 5) and three other peptides which have D-Phe and D-Trp (Peptides 2, 4 and 6) (Table 1) in their sequences with systematic differences in the size of their rings. The binding affinities and the in vitro cAMP accumulation data for these analogues are summarized in Table 3. Analogue 1 with a 21 member ring was found to be a very weak antagonist (IC50 = 2000 nM) at the hMC1R, a full agonist at the hMC3R (EC50 = 190 nM), inactive at the hMC4R, and a partial agonist at the hMC5R (EC50 = 86 nM, 31% activation). Similar observations were made for analogue 2 which also has a 21 membered ring size. This analogue exhibited weak antagonist activity (IC50 = 1800 nM) at the hMC1R and was inactive at the hMC3R, hMC4R and hMC5R. It is interesting to note that peptide 2 which has a D-Trp residue, though not potent at the hMC1R, is nonetheless very selective. For analogues 3 and 4, the ring size was increased by one carbon atom to 22 and this improves binding affinity. Analogue 3 had partial antagonist activity towards the hMC3R (IC50 = 76 nM, 52% activation). Also, this analogue was found to have no binding to the hMC4R and to be a full but weak agonist at the hMC5R (EC50 = 2500 nM). Analogue 4 in which Trp was replaced with D-Trp had much improved binding affinity for the hMC1R (IC50 = 30 nM) with no cAMP accumulation activity (antagonist). This analogue was found to be a very weak partial agonist (EC50 = 1100 nM, 50% activation) at the hMC3R, modest antagonist activity at the hMC5R, and totally inactive towards the hMC4R. Analogue 5 was very similar to analogue 4 with antagonist activity at the hMC1R (IC50=29 nM) but with partial agonist activity at the hMC3R (EC50=100 nM). This analogue was inactive at the hMC4R but exhibited full agonist activity towards the hMC5R (91% activation). Analogue 6 (Table 3) was the most interesting analogue in this series. It demonstrated potent antagonist activity for the hMC1R (IC50 = 17 nM). This analogue was inactive at the hMC4R and was a very weak partial agonist at the hMC3R (EC50 = 3900 nM, 75% activation) and weak partial agonist at the hMC5R (EC50 = 1300, 65% activation). It is interesting to note that the hMC1R selectivity is very much dependent on the conformational constraint. Thus, cyclic analogues of γ-MSH obtained by incorporating a thioether bridge between a cysteine side chain and an N-terminal bromoacyl group were found to be very critical in forming the right conformation constraint for selectivity and in this study the 23 membered ring was most favorable for hMC1R selectivity (peptide 6).
NMR studies
To explore the conformation underlying the selective antagonist activity of the hMC1R, we have analyzed the structure of peptide 6 using NMR conformational studies. The spin-system of each amino acid present in the peptide was identified using TOCSY spectra. This information was used with NOESY spectra to determine the sequential assignment of the peptide. The finger-print regions of the TOCSY and NOESY spectra are shown in Figure 2. The dαN(i, i+1) and dβN(i, i+1) were helpful in unambiguous sequential assignment of the peptide. The chemical shift values along with 3JαNH and temperature coefficient values are given in Table 4. The observed NOEs are summarized in Figure 3. The peptide displayed sequential strong dαN(i, i+1) and dβN(i, i+1) NOEs throughout the sequence along with relatively weaker dNN(i, i+1) NOEs. Observation of strong consecutive dαN(i, i+1) and relatively weaker dNN(i, i+1) NOEs suggests that peptide undergoes rapid interconversion between extended and folded conformations. 48-49 Absence of helix diagnostic dαN(i, i+3) and dαN(i, i+4) NOEs and the observation of four dαN(i, i+2) NOEs and one dNN(i, i+2) NOE suggest multiple turns or a 310-type helical region in the middle segment of the peptide.
Figure 2.

a. Finger-print region TOCSY (left panel) and NOESY (right panel) of peptide peptide 6.
b. Amide (NH-NH) region of NOESY spectrum showing sequential dNN(i, i+1) NOEs of peptide peptide 6.
Table 4.
Chemical shift values (ppm) of peptide 6 as measured in SDS micelles at 6.11 mM concentration, pH=5.5 and at 25 °C.
| Residue | NH | CαH | CβH | Δδ ppm | dδ/dT (−ppb/°K) |
|---|---|---|---|---|---|
| Gly (1) | 8.088 | 3.782 | −0.228 | 5.40 | |
| His (2) | 7.946 | 4.637 | −0.133 | 2.82 | |
| Phe (3) | 8.235 | 4.551 | 2.922 3.097 |
−0.069 | 4.90 |
| Arg (4) | 7.734 | 4.118 | −0.222 | 4.94 | |
| Trp (5) | 8.034 | 4.729 | 3.243 3.455 |
0.059 | 3.88 |
| Cys (6) | 8.120 | 4.348 | 2.847 2.972 |
−0.232 | 4.26 |
| Asp (7) | 8.398 | 4.606 | 2.979 2.860 |
−0.024 | 5.02 |
| Arg (8) | 7.894 | 4.057 | −0.283 | 3.08 | |
| Phe (9) | 7.862 | 4.675 | 2.976 3.291 |
0.055 | 4.64 |
| Gly (10) | 8.039 | 3.866 3.949 |
−0.103 | 3.54 |
Figure 3.

Summary of observed NOEs and temperature coefficient values measured for peptide peptide 6 in SDS micelles at 25 °C and pH 5.5. Lower case letters denotes D chiral amino acids. The thickness of the lines is approximately proportional to the intensity of the NOE cross-peaks.
The hydrogen bond information of the peptide can be obtained from the temperature coefficient (dδ/dT) values of the amide protons. 50 The temperature coefficient values were obtained by recording TOCSY spectrum at different temperatures with 5 °C increments starting from 20 °C because the 1D proton NMR spectrum showed lot of overcrowding in the amide proton region. The observation of diminished temperature coefficient values (i.e. less than −4 ppb/K) for amide protons of His5, Trp8, Arg11 and Gly12 indicate that those amide protons are involved in hydrogen bonding or are shielded from the solvent, whereas the other amide protons are exposed to solvent.
It is possible to obtain qualitative structural information from CαH chemical shift values as they are sensitive to the secondary structure of the peptide.51 It is well established that consecutive negative deviations of CαH chemical shift value from random coil values indicate a helical fold and the positive deviation indicates a β-sheet structure. The chemical deviation plot (aka CSI plot) is shown in Figure 4. Negative deviation was observed for all the residues with interruptions at Trp8 and Phe11 suggesting that the residues at the N-terminal possibly having conformational preferences in the helical domain. The positive deviation at Trp8 and Phe11 implies a turn type fold around those residues.
Figure 4.
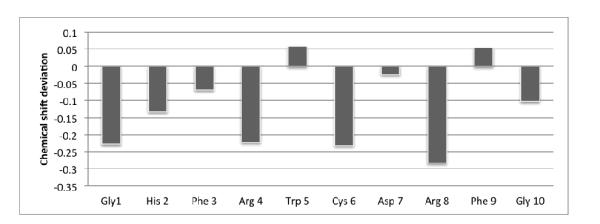
Chemical shift deviation of CαH chemical shift values from random coil values.
Structure calculations
In order to obtain the three-dimensional structure of peptide 6, a simulated annealing molecular dynamics calculation was carried out by applying distance as well as dihedral angle constraints as described in the methods section. The stereochemical quality of the final energy minimized structures from the calculation was checked by the distribution of backbone dihedral angles phi and psi in the Ramachandran plot 52 as shown in Figure 5. The distribution range is very narrow for all the amino acids except Asp7 and Phe9. This clearly suggests that all the residues except Asp7 and Phe9 are in a rigid conformation. Two distinct populations, namely extended and helical, are experienced by the Asp7 and Phe9. Interestingly, the dihedral space for the D-Trp5 falls in the negative quadrant of the Ramachandran plot which is unexpected for a D-chiral amino acid. The same result was obtained even after the calculation was repeated with strong chiral constraints in place. This may be due to the global constraint introduced by the cyclization.
Figure 5.

Phi (Φ) and Psi (ψ) dihedral angle distribution (i.e. Ramachandran Plot) of each amino acid in peptide 6 for the resultant hundred structures from the simulated annealing molecular dynamics calculations.
The lowest energy structure is shown in Figure 6. Consecutive type III β-turns are observed for the middle segment -Arg4-DTrp5-Cys6-Asp7-Arg8- as indicated by the consecutive dαN(i, i+2) NOEs in this region. As the 310- type helical dihedral angle values are similar to single type III β-turn dihedral values (ϕ=−60, ψ=−30), this region containing consecutive turns can be described as a 310 helical region. Both termini are flexible in nature despite the cyclization. Previous NMR analysis of shorter analogs related to α-MSH suggested a β-turn like structure at -His6-D-Phe7-Arg8-Trp9 but the exact type and position of the β-turn depended on the nature of amino acid substitution in the core pharmacophore region. 53 The main difference between the peptide discussed here and those peptides are the C-terminal extension by four additional residues. The C-terminal extension was favorable for the formation of consecutive β-turns, i.e. the short 310 helical type fold. This conformation implies that the C-terminal of the peptide could be a new binding site. The analysis of the backbone dihedral angle values is given in Table 5. Overlay of the five lowest energy structures from the calculation is shown in Figure 7. The backbone atoms aligned very well, but the side-chains of all the amino acids except His2 are flexible. This observation parallels our earlier studies in which the His residue in cyclized peptides can be replaced with different constrained amino acids without interfering with the binding.54 The stereo view of the potential new binding site of peptide 6 is shown in Figure 8.
Figure 6.
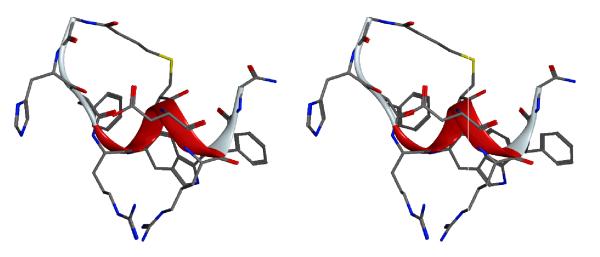
Stereo-view representation of lowest energy structure from the simulated annealing molecular dynamics calculations of peptide 6. The hydrogen atoms are not shown for clarity. The ribbon diagram shows the secondary structure of the peptide.
Table 5.
Phi and Psi angle distribution of peptide 6 from simulated annealing molecular dynamics study. The standard deviation from the average value is given in parenthesis and deviations greater than 30 are highlighted by bold.
| Amino acid |
All hundred structures | Lowest energy structure |
||
|---|---|---|---|---|
| Phi (ϕ) | Psi (ψ) | Phi (ϕ) | Psi(ψ) | |
| Gly 1 | −72 (7) | −64(3) | −75 | −67 |
| His 2 | −83(7) | −68(2) | −91 | −69 |
| DPhe 3 | 34(21) | 92(2) | 34 | 90 |
| Arg 4 | −47(8) | −60(3) | −55 | −65 |
| DTrp 5 | −26(3) | −60(3) | −28 | −61 |
| Cys 6 | −42(7) | −50(4) | −49 | −46 |
| Asp 7 | −87(33) | −45(65) | −70 | −59 |
| Arg 8 | −66(9) | −43(9) | −68 | −33 |
| Phe 9 | −107(21) | 70(52) | −116 | 82 |
| Gly 10 | −88(3) | −62(1) | −82 | −62 |
Figure 7.
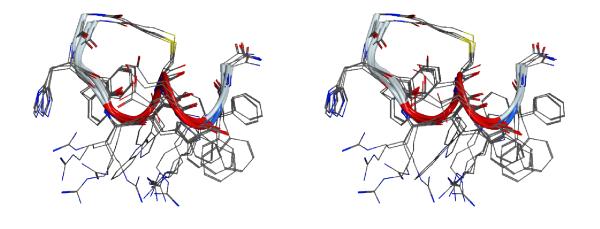
Stereo-view of overlay of five lowest energy structures resulting from the simulated annealing molecular dynamics calculations of peptide 6. The hydrogen atoms are removed for clarity. The ribbon diagram shows the secondary structure of the peptide.
Figure 8.

Stereo-view of the lowest energy structure showing the new binding site of the peptide 6 with the broken circle.
To further explore the structural features of the hMC1R antagonist selectivity of peptide 6, we have systematically compared the backbone and the side chain conformations of peptide 6 with a recently reported selective hMC1R agonist28 as well as the selective endogenous hMC1R antagonist Agouti Signal Protein: ASIP (PDB: 2KZA)55 in detail. The compared dihedral angles are given in Table 6. It is interesting to note that the backbone dihedral angles of the lowest energy conformation of peptide 6 for -D-Trp5-Cys6-Asp7-Arg8-Phe9-Gly10- matched the NMR derived pharmacophore of ASIP (117-122) -Arg117-Phe118-Phe119-Arg120-Ser121-Ala122- (Table 6) except for the pair of Asp47/Arg120. These dihedral angle data demonstrate that the backbone conformation of ASIP (117-122) and the C-terminus of the Peptide 6 are similar. This observation imply that there is a new binding site of the peptide 6 in the region of -D-Trp5-Cys6-Asp7-Arg8-Phe9-Gly10- instead of -His-D-Phe-Arg-Trp-. This result is consistent with our earlier work47 where a new binding site was observed based on the SAR study of γ-MSH. Comparing the backbone dihedral angles of hMC1R agonist (NMe)MTII28 and peptide 6, one can see that the major difference is at D-Trp5(Peptide 6) vs Trp8 (NMeMTII). Both the Phi and Psi angles of D-Trp5/Trp8 have a difference of 90-180 deg. Therefore, we conclude that the conformation of D-Trp5/Trp8 in MSHs is critical for selective agonist/antagonist activity at the hMC1R. Comparison of the Ramachandran plot of Peptide 6, NMe-MTII and ASIP (117-122) (Figure 9) shows the critical conformation properties of the constrained peptide 6. The D-Trp configuration falls in the negative quadrant of the Ramachandran plot which is unusual for a D-chiral amino acid because of the topographic constraint, but that is not true in the case of NMe-MTII. The Ramachandran plot of ASIP has dihedral angles on both sides of the L- and D-like configuration, even though there is no D amino acid in this natural protein. The D-like configuration of Phe in the ASIP protein is confined by the disulfide bond and the topographic constraint via the whole protein. Thus, the topographic constraint of ASIP (117-122) directs Phe to play its role for the hMC1R antagonist selectivity. This SAR study is an example of conformational based ligand design that may be generally useful in peptide drug design.
Table 6.
Comparison of the pharmacophore of the NMR structures of (NMe)-MTII, Peptide 6 and ASIP (117-122): Backbone torsion angles (deg) for the NMR structures of the peptide backbone conformation
| Compound | Nle3 | Asp4/Gly 1 | His5 /His2 | DPhe6 /DPhe3 /DPhe4 |
Arg7 /Arg4/Arg117 |
Trp8 /DTrp5/Phe118 |
Lys9 /Cys6/Phe119 |
Asp7/Arg120 | Arg8/Ser121 | Phe9/Ala122 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| φ | ψ | φ | ψ | φ | ψ | φ | ψ | φ | ψ | φ | ψ | φ | ψ | φ | ψ | φ | ψ | φ | ψ | |
| (NMe)MTII | −101 | 109.1 | 71.4 | 143.7 | −97.8 | 78.4 | 96.1 | −125.9 | −135.2 | 80.4 | −119.8 | 62.6 | −113.7 | 0.4 | ||||||
| Peptide 6 | −75.3 | −67.3 | −90.9 | −69.5 | 34.1 | 90.1 | −54.8 | −64.8 | −28.4 | −61.1 | −45.8 | −45.8 | −70.3 | −59.5 | −68.4 | −33.5 | −115.6 | 82.0 | ||
| ASIP 117-122 | −115.4 | −171.7 | −41.4 | −56.5 | −48.8 | −45.9 | 63.5 | 18.4 | −118.2 | −24.5 | −119.5 | 64.6 | ||||||||
Note: Number labeled for α-MSH analogues shown as red N(Me)MTII (Ac-Nle3-c[N(Me)Asp4- N(Me)His5-DPhe6-N(Me)Arg7- N(Me)Trp8-Lys9]- NH2; Peptide 6 (cyclo-[(CH2)3CO-Gly1-His2-D-Phe3-Arg4-D-Trp5-Cys(S-)6]-Asp7-Arg8-Phe9-Gly10-NH2) shown as blue; ASIP loop -Arg117-Phe118-Phe119-Arg120-Ser121-Ala122- shown as green.
Figure 9.
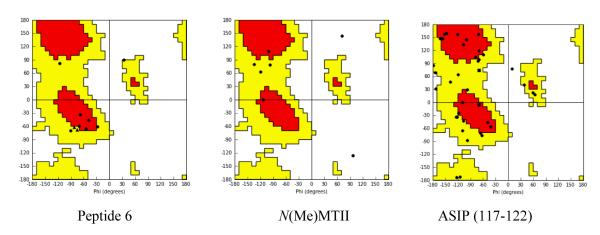
Ramachandran Plot of lowest energy conformation of the selective antagonist of hMC1R (peptide 6) and hMC1R selective agonist N(Me)MTII, and ASIP (117-122)
Biological Activity at the melanoma cancer cells
To further investigate the potential application of peptide 6 towards melanoma cells, we did both binding as well as cAMP assays using the A375 melanoma cell lines (ATCC). Table 7 shows that the peptide 6 binds to the melanoma cell receptor in the nano-molar range. It is interesting to note that peptide 6 and SHU9119 act as antagonists towards the melanoma cells. On the contrary, MTII kept its agonist activity, but the cAMP efficacy (EC50) is reduced by two orders of magnitude compared to the cloned hMC1R cells. This may be due to the lower density of the hMC1R in the cancer cell line. Peptide 6 is an excellent probe to target specifically melanoma cells for skin cancer diagnosis and treatment in the future. This observation consistence with the latest report of ASIP that is lack of natural antagonist of hMC1R (ASIP) will have risk of melanoma, ASIP pigmentation variants are associated with cutaneous melanoma and basal cell carcinoma57. The new result can also explain why the conformation of peptide 6 for -D-Trp5-Cys6-Asp7-Arg8-Phe9-Gly10- matched very well with the NMR derived pharmacophore of ASIP(117-122) -Arg117-Phe118-Phe119-Arg120-Ser121-Ala122-.
Table 7.
Binding and cAMP activities of Peptide 6 at human melanoma cells (A375)
| Compound | Structure | IC50 | Binding Efficiency |
EC50t | Act% |
|---|---|---|---|---|---|
| Peptide 6 | c[(CH2)3-Gly-His-D-Phe-Arg-D-Trp-Cys(S-)]- Asp-Arg-Phe-Gly-NH2 | 92 ± 65 | 98 ± 41 | 22 ± 2.5 | NA |
| MT-II | Ac-Nle-c[Asp-His-D-Phe-Arg-Trp-Lys]-NH2 | 6.7 ± 2.9 | 100 ± 10 | 110 ± 0.6 | 100 |
| SHU9119 | Ac-Nle-c[Asp-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | 100 ± 96 | 80 ± 34 | 3.3 ± 2.6 | NA |
IC50 = concentration of peptide at 50% specific binding (N=4). Binding Efficiency = maximal % of [125I]-NDP-α-MSH displacement observed at 10 μM. EC50 = Effective concentration of peptide that was able to generate 50% maximal intracellular cAMP accumulation (N=4). Act% = % of cAMP produced at 10 μM ligand concentration, in relation to MT-II. NA = 0% cAMP accumulation observed at 10 μM. The peptides were tested at a range of concentration from 10−10 to 10−5 M.
In summary, structure-activity relationships (SAR) of cyclic γ-MSH analogues were examined to evaluate the multiple factors that contribute to melanocortin 1 receptor selectivity, and are illustrated by the Figures 2-9 and Tables 2-6. Figures 8 and 9 illustrate the new binding site of gamma MSH, and the D-Trp5 configuration which contributes to selective antagonist activities. Table 3 compares the binding affinities of the D-Trp5 analogues having different constraints and extension of the C termini, and thus it establishes a trend towards hMC1R selectivity. Finally, the absence of Nle in the gamma-MSH cyclized analogues leads to weak binding compared to the cyclized peptide with Nle (e.g. MTII) and this is consistent with our earlier findings.47 Also, NMR analysis reveals a new binding site which is closely related to that of the endogenous antagonist ASIP (117-122) fragment. Table 7 illustrates the nano-molar binding affinity of this peptide towards melanoma cells (A375) which may be very useful for therapeutics.
Conclusions
SAR and conformational studies of novel cyclized γ-MSH analogues have identified a highly selective hMC1R antagonist (analogue 6). NMR studies of this constrained peptide reveals a new binding site, -D-Trp5-Cys6-Asp7-Arg8-Phe9-Gly10- of peptide 6, which is different from the more general -His-D-Phe-Arg-Trp- pharmacophore of melanocortin receptor agonists. The backbone conformation of peptide 6 matches well with the endogenous antatagonist conformation of the hMC1R selective ASIP (117-122) fragment. In this new binding site, D-Trp5 and Phe9 of Peptide 6 plays a critical role for hMC1R antagonist activity and the residue D-Trp5 is also crucial for binding to the 6th transmembrane domain (6TM) of hMC1R56. The newly developed selective melanotropin peptides will facilitate the elucidation of the physiological roles of the hMC1R in pigmentation and immune response related disorders. Finally, the nano-molar binding affinity of this peptide towards melanoma cells indicate that it can be used as a molecular probe for the diagnosis and treatment of skin cancer. Furthermore, the backbone conformation of Peptide 6 can be used for future melanoma inhibitor discovery.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by grants from the U.S. Public Health Service, National Institutes of Health, DK17420 and DA06284.
Abbreviations
Abbreviations used for amino acids and designation of peptides follow the rules of the IUPAC-IUB Commission of Biochemical Nomenclature in J. Biol. Chem. 1972, 247, 977-983. The following additional abbreviations are used:
- AAA
amino acid analysis
- Boc
tert-butyloxycarbonyl
- Fmoc
fluorenylmethoxycarbonyl
- Fmo
fluorenylmethyl
- Bzl
benzyl
- tBu
tert-butyl
- CH3CN
acetonitrile
- DCM
dichloromethane
- DIEA
diisopropylethylamine
- DMF
N,N-dimethylformamide
- DIC
diisopropyl carbodiimide
- HOBt
N-hydroxybenzotriazole
- BOP reagent
(benzotriazolyloxy) tris-(dimethylamino)-phosphonium hexa-fluorophosphate
- Nal(2′)-
2′-naphthylalanine
- TFA
trifluoroacetic acid
- TIPS
triisopropylsilyl
- SPPS
solid-phase peptide synthesis
- RP-HPLC
reversed phase high performance liquid chromatography
- hMC1R
human melanocortin-1 receptor
- α-MSH
Ac-Ser-Tyr-Ser-Met-Glu-His-Phe-Arg-Trp-Gly-Lys-Pro-Val-NH2
- NDP-α-MSH
Ac-Ser-Tyr-Ser-Nle-Glu-His-D-Phe-Arg-Trp-Gly-Lys-Pro-Val-NH2
- γ-MSH
H-Tyr-Val-Met-Gly-His-Phe-Arg-Trp-Asp-Arg-Phe-Gly-OH
- ASIP(117-122)
Agouti Signal Protein fragment
Footnotes
SUPPORTING INFORMATION AVAILABLE: High resolution mass spectra of peptide analogues and extensive 2D NMR of peptide 6 which provide data needed for conformational analysis are provided. The material is available free of charge via the internet at http://pubs.acs.org/.
References
- (1).Denning-Kendall PA, Sumpter JP, Lowry PJ. Peptide derived from proopiomelanocortin in the pituitary gland of the dogfish, Squalus acanthias. J. Endocrinology. 1982;93:381–390. doi: 10.1677/joe.0.0930381. [DOI] [PubMed] [Google Scholar]
- (2).Tatjana H, Janis K, Akiyoshi K, Maja L, Aneta R, Johan E, Hiroshi K, Earl LT, Robert F, Helgi SB. Functional characterization of two melanocortin (MC) receptors in lamprey showing orthology to the MC1 and MC4 receptor subtypes. BMC Evolutionary Biology. 2007;7:101. doi: 10.1186/1471-2148-7-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Krieger DT, Ganong WF, editors. ACTH and Related Peptides: Structure, Regulation, and Action. Ann. N.Y. Acad. Sci. 1977;297:664. [PubMed] [Google Scholar]
- (4).Vandry H, Eberle AN, editors. The Melanotropic Peptides. Ann. N.Y. Acad. Sci. 1993;680:687. [Google Scholar]
- (5).Montjoy KG, Robins LS, Mortruel MT, Cone RD. The Cloning of a Family of Genes that Encode the Melanotropin Receptors. Science. 1992;257:1248–1251. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- (6).Chhajlani V, Wikberg JES. Molecular cloning and expression of the human melanocyte stimulating hormone receptor cDNA. FEBS Letters. 1992;309(3):417–420. doi: 10.1016/0014-5793(92)80820-7. [DOI] [PubMed] [Google Scholar]
- (7).Gantz I, Konda Y, Tashiro T, Shimoto Y, Miwa H, Munzert G, Watson SJ, DelValle J, Yamada TT. Molecular cloning of a novel melanocortin receptor. J. Biol. Chem. 1993;268:8246–8250. [PubMed] [Google Scholar]
- (8).Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, DelValle J, Yamada T. Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J. Biol. Chem. 1993;268:15174–15179. [PubMed] [Google Scholar]
- (9).Gantz I, Tashiro T, Barcroft C, Konda Y, Shimoto Y, Miwa H, Glover T, Munzert G, Yamada T. Localization of the genes encoding the melanocortin-2 (adrenocorticotropic hormone) and melanocortin-3 receptors to chromosomes 18p11.2 and 20q13.2-q13.3 by fluorescence in situ hybridization. Genomics. J. Biol. Chem. 1993;18:166–167. doi: 10.1006/geno.1993.1448. [DOI] [PubMed] [Google Scholar]
- (10).Gantz I, Shimoto Y, Konda Y, Miwa H, Dickinson CJ, Yamada T. Molecular cloning, expression, and characterization of a fifth melanocortin receptor. Biochem. Biophys. Res Comm. 1994;200(3):1214–1220. doi: 10.1006/bbrc.1994.1580. [DOI] [PubMed] [Google Scholar]
- (11).Cone RD, editor. The Melanocortin Receptors. Humana Press; 2000. pp. 1–551. [Google Scholar]
- (12).The Melanocortin System Ann. N.Y. Acad. Sci. 2003;994:1–367. [Google Scholar]
- (13).Hadley ME, editor. The Melanotropin Peptides CRC Press; Boca Raton, FL: 1989. 1,2 & 3. [Google Scholar]; Eberle AN, Karger B. The Melanotropines: Chemistry, Physiology and Mechanisms of Action. 1988 [Google Scholar]
- (14).Roselli-Rehfuss L, Montjoy KG, Robbins LS, Mortrud MT, Low MJ, Tatro ML, Entwistle ML, Simerly RB, Cone RD. Identification of a Receptor for α-Melanotropin and Other Proopiomelanocortin Peptides in the Hypothalamus and Limbic Systems. Proc. Natl. Acad. Sci. U.S.A. 1993;90:8856–8860. doi: 10.1073/pnas.90.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Xia Y, Wikberg J, Chhajlani V. Express(ion of melanocortin 1 receptors in periaqueductal gray matter. Neuroreport. 1995;6:2193–2196. doi: 10.1097/00001756-199511000-00022. [DOI] [PubMed] [Google Scholar]
- (16).Liem E, Joiner T, Tsueda K, Sessler D. Increased sensitivity to thermal pain and reduced subcutaneous lidocaine efficacy in redheads. Anesthesiology. 2005;102:509–514. doi: 10.1097/00000542-200503000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Mogil JS, Wilson SM, Chesler EJ, Rankin AL, Nemmani KVS, Lariviere WR, Groce MK, Wallace AR, Kaplan L, Staud R, Ness TJ, Clover TL, Stakova M, Mayorov A, Hruby VJ, Grisel JE, Fillingim RB. The melanocortin-1 receptor gene mediates female-specific mechanisms of analgesia in mice and humans. Proc. Natl. Acad. Sci. U.S.A. 2003;100:4867–4872. doi: 10.1073/pnas.0730053100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Hruby VJ, Al-Obeidi F, Kazmierski W. Emerging approaches in the molecular design of receptor-selective peptide ligands: conformational, topographical and dynamic considerations. Biochem. J. 1990;268:249–262. doi: 10.1042/bj2680249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hruby VJ. Designing peptide receptor agonists and antagonists. Nature Reviews Drug Discovery. 2002;1(11):847–858. doi: 10.1038/nrd939. [DOI] [PubMed] [Google Scholar]
- (20).Hruby VJ, Cai M, Grieco P, Han G, Kavarana M, Trivedi D. Exploring the stereostructural requirements of peptide ligands for the melanocortin receptors. Ann. N.Y. Acad. Sci. 2003;994:12–20. doi: 10.1111/j.1749-6632.2003.tb03157.x. [DOI] [PubMed] [Google Scholar]
- (21).Hruby VJ, Smith CW, Bower SA, Hadley ME. Melanophore Stimulating Hormone: Release inhibition by ring structures of neurohypophysial hormones. Science. 1972;176:1331–1332. doi: 10.1126/science.176.4041.1331. [DOI] [PubMed] [Google Scholar]
- (22).Hruby VJ, Wilkes BC, Hadley ME, Al-Obeidi F, Sawyer TK, Staples DJ, de Vaux AE, Dym O, de Lauro Castrucci A-M, Hintz MF, Riehm JR, Rao RR. α-Melanotropin: The Minimum Active Sequence in the Frog Skin Bioassay. J. Med. Chem. 1987;30:2126–2130. doi: 10.1021/jm00394a033. [DOI] [PubMed] [Google Scholar]
- (23).de Lauro Castrucci A-M, Hadley ME, Sawyer TK, Wilkes BC, Al-Obeidi F, Staples DJ, de Vaux AE, Dym O, Hintz MF, Riehm JP, Rao RR, Hruby VJ. α-Melanotropin: The Minimal Active Sequence in the Lizard Skin Bioassay. Gen. Comp. Endocrinol. 1989;73:157–163. doi: 10.1016/0016-6480(89)90066-x. [DOI] [PubMed] [Google Scholar]
- (24).Grieco P, Balse PM, Weinberg D, MacNeil T, Hruby VJ. D-Amino Acid Scan of γ-Melanocyte-Stimulating Hormone: Importance of Trp8 on Human MC3 Receptor Selectivity. J. Med. Chem. 2000;43:4998–5002. doi: 10.1021/jm000211e. [DOI] [PubMed] [Google Scholar]
- (25).Grieco P, Balse-Srinivasan P, Han G, Weinberg D, MacNeil T, Van der Ploeg LHT, Hruby VJ. Synthesis and Biological Evaluation on hMC3, hMC4 and hMC5 Receptors of γ-MSH Analogs Substituted with L-Alanine. J. Peptide Res. 2002;59:203–210. doi: 10.1034/j.1399-3011.2002.01966.x. [DOI] [PubMed] [Google Scholar]
- (26).Cai M, Nyberg J, Hruby VJ. Melanotropins as Drugs for the Treatment of Obesity and Other Feeding Disorders: Potential and Problems. Curr. Topics Med. Chem. 2009;9:554–563. doi: 10.2174/156802609788897817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hruby VJ, Cai M, Nyberg J, Muthu D. Approaches to the Rational Design of Selective Melanocortin Receptor Antagonists. Expert Opinion on Drug Discovery. 2011;6:543–557. doi: 10.1517/17460441.2011.565743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Doedens L, Opperer F, Cai M, Beck JG, Dedek M, Palmer E, Hruby VJ, Kessler Horst. Multiple N-methylation of MT-II backbone amide bonds leads to Melanocortin receptor subtype hMC1R selectivity: Pharmacological and conformational studies. J. Am. Chem. Soc. 2010;132:8115–8128. doi: 10.1021/ja101428m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Joseph CM, Pilgrim JJ, Darren AT, Biaoxin C, Ira Gantz., Gregory SB, Philip ED, Glenn LM. Structures of the Agouti Signaling Protein. J. Mol. Biol. 2005;346(4):1059–1070. doi: 10.1016/j.jmb.2004.12.030. [DOI] [PubMed] [Google Scholar]
- (30).Lebl M, Hruby VJ. Synthesis of cyclic peptides by solid-phase methodology. Tetrahedron Lett. 1984;25:2067–2068. [Google Scholar]
- (31).Krchnak V, Vagner J, Lebl M. Noninvasive monitoring of solid-phase peptide synthesis by acid-base indicator. Int. J. Pept. Protein Res. 1988;32:415–416. doi: 10.1111/j.1399-3011.1988.tb01276.x. [DOI] [PubMed] [Google Scholar]
- (32).Polinsky A, Cooney MG, Toy-Palmer A, Osapay G, Goodman M. Synthesis and conformational properties of the lanthionine-bridged opioid peptide [D-AlaL2, AlaL5]enkephalin as determined by NMR and computer simulations. J. Med. Chem. 1992;35:4185–4194. doi: 10.1021/jm00100a026. [DOI] [PubMed] [Google Scholar]
- (33).Mayer JP, Heil JR, Zhang J, Munson MC. An alternative solid-phase approach to C1-oxytocin. Tetrahedron Lett. 1995;36:7387–7390. [Google Scholar]
- (34).Mosberg HI, Omnaas JR. Dithioether-containing cyclic peptides. J. Am. Chem. Soc. 1985;107:2986–2987. [Google Scholar]
- (35).Kataoka T, Beusen DD, Clark JD, Yodo M, Marshall GR. The utility of side-chain cyclization in determining the receptor-bound conformation of peptides: cyclic tripeptides and angiotensin II. Biopolymers. 1992;32:1519–1533. doi: 10.1002/bip.360321110. [DOI] [PubMed] [Google Scholar]
- (36).Hruby VJ, Bonner GG. Design of novel synthetic peptides including cyclic conformationally and topographically constrained analogs. Methods in Molecular Biology. 1994;35:201–240. doi: 10.1385/0-89603-273-6:201. [DOI] [PubMed] [Google Scholar]
- (37).Jung G. Lantibiotics-ribosomally synthesized biologically active polypeptides containing sulfide bridges and α-didehydroamino acids. Angew. Chem. Int. Ed. Engl. 1991;30:1051–1068. [Google Scholar]
- (38).Jack RW, Sahl HG. Unique peptide modifications involved in the biosynthesis of lantibiotics. Trends in Biotechnology. 1995;13:269–278. doi: 10.1016/S0167-7799(00)88962-3. [DOI] [PubMed] [Google Scholar]
- (39).Sahl HG, Jack RW, Bierbaum G. Biosynthesis and biological activities of lantibiotics with unique post-translational modifications. Eur. J. Biochem. 1995;230:827–853. doi: 10.1111/j.1432-1033.1995.tb20627.x. [DOI] [PubMed] [Google Scholar]
- (40).Kaiser E, Colescott RL, Bossinger CD, Cook PI. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Analyt. Biochem. 1970;34:595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- (41).Al-Obeidi F, Hadley ME, Pettitt BM, Hruby VJ. Design of a new class of superpotent α-melanotropins based on quenched dynamic simulations. J. Am. Chem. Soc. 1989;111:3413–3416. [Google Scholar]
- (42).Al-Obeidi F, Castrucci AML, Hadley ME, Hruby VJ. Potent and prolonged acting cyclic lactam analogues of alpha-melanotropin: design based on molecular dynamics. J. Med. Chem. 1989;32:2555–2561. doi: 10.1021/jm00132a010. [DOI] [PubMed] [Google Scholar]
- (43).Hruby VJ, Lu D, Sharma SD, Castrucci AML, Kesterson R, Al-Obeidi F, Hadley ME, Cone RD. Cyclic lactam α-melanotropin analogues of Ac-Nle4-cyclo[Asp5,D-Phe7,Lys10] α-melanocyte-stimulating hormone-(4-10)-NH2 with bulky aromatic amino acids at position 7 show high antagonist potency and selectivity at specific melanocortin receptors. J. Med. Chem. 1995;38:3454–3461. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]
- (44).Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. 4-Norleucine, 7-D-phenylalanine-alpha-melanocyte-stimulating hormone: a highly potent alpha-melanotropin with ultralong biological activity. Proc. Nat. Acad. Sci. U.S.A. 1980;77:5754–5758. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Grieco P, Balse PM, Weinberg D, MacNeil T, Hruby VJ. D-Amino acid scan of gamma-melanocyte-stimulating hormone: importance of Trp(8) on human MC3 receptor selectivity. J. Med. Chem. 2000;43:4998–5002. doi: 10.1021/jm000211e. [DOI] [PubMed] [Google Scholar]
- (46).Cai M, Mayorov AV, Cabello C, Stankova M, Trivedi D, Hruby VJ. Novel 3D Pharmacophore of α-MSH/γ-MSH Hybrids Leads to Selective Human MC1R and MC3R Analogues. J. Med. Chem. 2005;48:1839–1848. doi: 10.1021/jm049579s. [DOI] [PubMed] [Google Scholar]
- (47).Cai M, Cai C, Mayorov AV, Xiong C, Cabello CM, Soloshonok VA, Swift JR, Trivedi D, Hruby VJ. Biological and conformational study of β-substituted prolines in MT-II template: Steric effects leading to human MC5 receptor selectivity. J. Pept. Res. 2004;63(2):116–131. doi: 10.1111/j.1399-3011.2003.00105.x. [DOI] [PubMed] [Google Scholar]
- (48).Wüthrich K, Billeter M, Braun W. Polypeptide secondary structure determination by nuclear magnetic resonance observation of short proton-proton distances. J. Mol. Biol. 1984;180:715–740. doi: 10.1016/0022-2836(84)90034-2. [DOI] [PubMed] [Google Scholar]
- (49).Williamson MP, Waltho J. Peptide structure from NMR. Chem. Soc. Rev. 1984;21:227–236. [Google Scholar]
- (50).Cierpicki T, Otlewski J. Amide proton temperature coefficients as hydrogen bond indicators in proteins. J. Biomol. NMR. 2001;21:249–261. doi: 10.1023/a:1012911329730. [DOI] [PubMed] [Google Scholar]
- (51).Wishart DS, Sykes BD, Richards FM. The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry. 1992;31:1647–1651. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- (52).Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963;7:95–99. doi: 10.1016/s0022-2836(63)80023-6. [DOI] [PubMed] [Google Scholar]
- (53).Ying J, Koever KE, Gu X, Han G, Trivedi DB, Kavarana MJ, Hruby VJ. Solution structures of cyclic melanocortin agonists and antagonists by NMR. Biopolymers. 2003;71:696–716. doi: 10.1002/bip.10596. [DOI] [PubMed] [Google Scholar]
- (54).Grieco P, Lavecchia A, Cai M, Trivedi D, Weinberg TM, Van der D, Ploeg LHT, Hruby VJ. Structure-activity studies of the melanocortin peptides: Discovery of potent and selective antagonists at MC3 and MC4 receptors. J. Med. Chem. 2002;45:5287–5294. doi: 10.1021/jm0202526. [DOI] [PubMed] [Google Scholar]
- (55).Patel MP, Cribb Fabersunne CS, Yang Y, Kaelin CB, Barsh GS, Millhauser Glenn L. Loop-swapped chimeras of the Agouti-Related Protein and the Agouti Signaling Protein identify contacts required for Melanocortin 1 Receptor selectivity and antagonism. J. Mol. Biol. 2010;404:45–55. doi: 10.1016/j.jmb.2010.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Chen M, Cai M, McPherson D, Hruby VJ, Harmon CM, Yang Y. Contribution of the transmembrane domain 6 of melanocortin-4 receptor to peptide [Pro5,DNal(2′)8]-γ-MSH selectivity. Biochem. Pharmaco. 2009;77:114–124. doi: 10.1016/j.bcp.2008.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Gudbjartsson DF, Sulem P, Stacey SN, Goldstein AM, Rafnar T, Sigurgeirsson B, Benediktsdottir KR, Thorisdottir K, Ragnarsson R, Sveinsdottir SG, et al. ASIP and TYR pigmentation variants associate with cutaneous melanoma and basal cell carcinoma. Nature Genetics. 2008;40(7):886–891. doi: 10.1038/ng.161. [DOI] [PubMed] [Google Scholar]
- (58).Mayorov AV, Cai M, Palmer ES, Liu Z, Cain JP, Vagner J, Trivedi D, Hruby VJ. Solid-Phase Peptide Head-to-Side Chain Cyclodimerization: Discovery of C2-Symmetric Cyclic Lactam Hybrid α-Melanocyte-Stimulating Hormone (MSH)/Agouti-Signaling Protein (ASIP) Analogues with Potent Activities at the Human Melanocortin Receptors. Peptides. 2010;31(10):1894–1905. doi: 10.1016/j.peptides.2010.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.