

Meta-analysis of cytokinin-regulated gene expression across multiple transcriptome studies identifies the transcriptional changes occurring in response to cytokinin.
Abstract
Cytokinins are N6-substituted adenine derivatives that play diverse roles in plant growth and development. We sought to define a robust set of genes regulated by cytokinin as well as to query the response of genes not represented on microarrays. To this end, we performed a meta-analysis of microarray data from a variety of cytokinin-treated samples and used RNA-seq to examine cytokinin-regulated gene expression in Arabidopsis (Arabidopsis thaliana). Microarray meta-analysis using 13 microarray experiments combined with empirically defined filtering criteria identified a set of 226 genes differentially regulated by cytokinin, a subset of which has previously been validated by other methods. RNA-seq validated about 73% of the up-regulated genes identified by this meta-analysis. In silico promoter analysis indicated an overrepresentation of type-B Arabidopsis response regulator binding elements, consistent with the role of type-B Arabidopsis response regulators as primary mediators of cytokinin-responsive gene expression. RNA-seq analysis identified 73 cytokinin-regulated genes that were not represented on the ATH1 microarray. Representative genes were verified using quantitative reverse transcription-polymerase chain reaction and NanoString analysis. Analysis of the genes identified reveals a substantial effect of cytokinin on genes encoding proteins involved in secondary metabolism, particularly those acting in flavonoid and phenylpropanoid biosynthesis, as well as in the regulation of redox state of the cell, particularly a set of glutaredoxin genes. Novel splicing events were found in members of some gene families that are known to play a role in cytokinin signaling or metabolism. The genes identified in this analysis represent a robust set of cytokinin-responsive genes that are useful in the analysis of cytokinin function in plants.
The phytohormone cytokinin was discovered based on its ability to stimulate cell division in cultured plant cells (Skoog and Miller, 1957). In intact plants, cytokinins play diverse roles in nearly every aspect of plant cell growth and development, including meristem function, leaf senescence, sink/source relationships, vascular development, and biotic and abiotic responses (Taniguchi et al., 2007; Séguéla et al., 2008; Argueso et al., 2009; Nishiyama et al., 2011; Argueso et al., 2012). Cytokinins regulate these processes through interactions with other plant hormones, which depend on cell type, developmental stage, and exogenous inputs (Werner and Schmülling, 2009; Wang et al., 2011b; Vanstraelen and Benková, 2012). The availability of mutants affecting distinct steps in cytokinin signaling has facilitated a general analysis of how cytokinins regulate gene expression (Kieber, 2002).
Cytokinin signal transduction involves a multistep phosphorelay system that begins with autophosphorylation of a membrane-bound receptor (ARABIDOPSIS HISTIDINE KINASE2 [AHK2], AHK3, and CYTOKININ RESPONSE1 [CRE1]/AHK4) upon cytokinin binding and subsequent transfer of the phosphate group to His phosphotransfer proteins (AHPs) and then finally to the response regulators (ARRs). There are two types of response regulators in Arabidopsis (Arabidopsis thaliana). Type-B ARRs are activated by phosphorylation and mediate cytokinin-regulated gene expression, including the up-regulation of the type-A ARRs, which act as negative feedback elements in cytokinin signaling (Brandstatter and Kieber, 1998; D’Agostino et al., 2000; Sakai et al., 2000; Imamura et al., 2003; Tajima et al., 2004; To et al., 2004; Taniguchi et al., 2007; Argyros et al., 2008). A subclade of APETALA2/Ethylene Response Factor (AP2/ERF) transcription factors, the Cytokinin Response Factors (CRFs), some of which are up-regulated in response to cytokinin, also play a role in the cytokinin-regulated gene expression (Rashotte et al., 2006).
Microarrays have been widely used in the genome-wide analysis of hormone responses in Arabidopsis, including the response to altered cytokinin function (Rashotte et al., 2003; Kiba et al., 2004; Brenner et al., 2005; Lee et al., 2007; Taniguchi et al., 2007; Yokoyama et al., 2007; Argyros et al., 2008; Dello Ioio et al., 2008; Goda et al., 2008; Müller and Sheen, 2008; Argueso et al., 2010; Bishopp et al., 2011; Köllmer et al., 2011; Brenner and Schmülling, 2012). These data sets have identified many genes regulated by cytokinin, and several have been validated by other methods (Supplemental Table S1). Nevertheless, there is only modest overlap in the sets of genes identified in these studies, which likely results from multiple factors, including lab/growth-specific effects, treatment-specific effects, and the identification of false positives/negatives. A complementary approach to query transcriptome changes is RNA-seq (Mortazavi et al., 2008; Chen et al., 2010; Filichkin et al., 2010; Lu et al., 2010). Despite biases related to transcript size and sequence composition, RNA-seq data represent mostly random samplings from expressed sequences and thus, in addition to determining transcript abundance, can provide new information about splicing patterns, alternative splicing, and previously unannotated genes, even in well-annotated, well-characterized genomes such as the model plant Arabidopsis. An important advantage of RNA-seq data over microarrays is that prior knowledge of the expressed transcriptome is not required. This is particularly important in the case of Arabidopsis and cytokinin signaling, because the majority of hormone-related array studies were performed using the older model ATH1 Affymetrix array, which lacks specific probe sets for more than 11,000 annotated genes. However, the statistical methods needed to analyze RNA-seq data are less well developed compared with microarray data analysis.
Meta-analysis of microarrays has been used extensively in animal systems to define robust sets of regulated genes (Nazri and Lio, 2012; Plank et al., 2012). Thus, a meta-analysis of the microarray data associated with the cytokinin responses, taking advantage of the variety of conditions employed for these analyses, would be a powerful tool to define a robust set of cytokinin-regulated genes. A recent study presented a preliminary meta-analysis of cytokinin-treated microarrays in Arabidopsis, though this study was limited to comparable growth conditions, treatment, and time points, and only a small subset of the genes identified were presented (Brenner et al., 2012).
The aim of this work was to identify a robust set of genes regulated by cytokinin, defining genes that are induced in multiple conditions using meta-analysis of the publicly available, as well as previously unpublished, microarray data sets. Filters were applied using empirically defined criteria with appropriate statistical limits. Most of the genes from this set were verified by an independent RNA-seq experiment using very stringent statistical criteria. Furthermore, the RNA-seq analysis identified a set of cytokinin-regulated genes that were not present on microarrays and also provided evidence for alternative splicing events among cytokinin signaling/metabolism genes. The gene sets identified were analyzed for enrichment of Gene Ontology (GO) terms as well as using MapMan to identify processes and pathways affected by cytokinin.
RESULTS
Hierarchical Clustering of Expression Profiles of Experiments Used for Microarray Meta-Analysis
As the basis for our meta-analysis, we employed 13 experiments querying cytokinin-regulated gene expression changes, all of which were performed using the ATH1 Affymetrix array platform. These experiments included data from seven published studies as well as six previously unpublished data sets from our laboratories (Table I). The raw data (.CEL) files from these experiments were obtained from either ArrayExpress (http://www.ebi.ac.uk/arrayexpress/) or NASCArrays databases (http://affymetrix.arabidopsis.info/narrays/experimentbrowse.pl) or were generated in our laboratories. These microarray experiments included distinct treatment regimes, growth conditions, and/or developmental stages and were generated in multiple laboratories, resulting in potential differences in global gene expression and cytokinin responses. We incorporated all of these experiments in our analysis to identify the robustly cytokinin-regulated genes (i.e. those that are differentially regulated in multiple conditions).
Table I. Microarray experiments used for meta-analysis.
| Experiment No. | Plants Used in the Experiments |
Treatment |
No. of Microarrays |
Principal Investigator | Publication | ArrayExpress/NASCArrays | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tissue | Age | Day Length | Chemical | Amount | Time | Delivery | Control | Treated | ||||
| d | µm | min | ||||||||||
| 1A | Seedling | 10 | Continuous | Zeatin | 1 | 120 | Immersed | 1 | 1 | Kieber | This work | E-MTAB-1601 |
| 1B | Seedling | 10 | Continuous | BA | 1 | 120 | Immersed | 1 | 1 | Kieber | This work | E-MTAB-1601 |
| 2 | Seedling | 21 | 16/8 | Zeatin | 20 | 180 | Immersed | 3 | 3 | Mizuno | Kiba et al. (2004) | NASCARRAY S-181 |
| 3 | Shoots | 14 | Continuous | BA | 10 | 60 | Immersed | 2 | 2 | Schaller | Argyros et al. (2008) | E-MEXP-1573 |
| 4 | Shoots | 14 | Continuous | BA | 10 | 60 | Immersed | 3 | 3 | Schaller | This work | E-MTAB-1596 |
| 5 | Shoots | 14 | N/A | Zeatin | 20 | 60 | Sprayed | 3 | 3 | Sakakibara | Yokoyama et al. (2007) | NASCARRAY S-405 |
| 6 | Roots | 14 | N/A | Zeatin | 20 | 60 | Sprayed | 2 | 2 | Sakakibara | Yokoyama et al. (2007) | NASCARRAY S-406 |
| 7A | Seedling | 7 | 16/8 | BA | 5 | 30 | Immersed | 2 | 2 | Kim | Lee et al. (2007) | E-ATMX-3 |
| 7B | Seedling | 7 | 16/8 | BA | 5 | 120 | Immersed | 2 | 2 | Kim | Lee et al. (2007) | E-ATMX-3 |
| 8A | Seedling | 10 | Dark | BA | 1 | 45 | Immersed | 2 | 2 | Kieber | This work | E-MTAB-1597 |
| 8B | Seedling | 10 | Dark | BA | 1 | 120 | Immersed | 2 | 2 | Kieber | This work | E-MTAB-1597 |
| 9A | Seedling | 7 | N/A | Zeatin | 1 | 30 | Immersed | 2 | 2 | Shimada | Goda et al. (2008) | NASCARRAY S-173 |
| 9B | Seedling | 7 | N/A | Zeatin | 1 | 180 | Immersed | 2 | 2 | Shimada | Goda et al. (2008) | NASCARRAY S-173 |
To gain insight into the effect of the varying conditions/treatments on transcript expression profiles, we performed a hierarchical clustering analysis of all the transcripts in control and cytokinin-treated samples across different experiments as described in Table I. Using GeneSpring software, the expression data were gene-wise normalized (Cheng et al., 2009), and a hierarchical clustering of signal intensities of gene expression patterns was carried out using Euclidean distance correlation for two entities (the genes and the experimental conditions). This clustering revealed the relatedness of the various data sets (Fig. 1). Duplicate or triplicate microarrays clustered as nearest neighbors in all cases. The microarrays that examined root tissues clustered together, as did the experiments using etiolated seedlings. However, experiments involving shoots and whole seedlings did not clade separately from each other, perhaps reflecting the preponderance of RNA from the shoot when RNA is prepared from a whole seedling. The method of application (spraying versus immersion) did have an effect on gene expression, as these experiments did tend to cluster together. Overall, this analysis revealed substantial differences in overall gene expression profiles based on the experimental designs and tissues used for these microarrays, which suggests that genes regulated by cytokinin across these diverse conditions will reflect robustly regulated genes.
Figure 1.

Hierarchical clustering of expression profiles of experiments used for microarray meta-analysis. Clustering of all the transcripts in control (C) and cytokinin-treated samples (T). The numbers below the image refer to the various experimental regimes described in Table I. The expression data were gene-wise normalized, and hierarchical clustering of gene expression patterns was carried out using GeneSpring software with Euclidean distance correlation for the two series of experiments, entities and variables. The color scale at the bottom represents the relative signal intensities.
Identification of a Set of Cytokinin-Responsive Genes Using Microarray Meta-Analysis
A Fisher’s combined probability test (Brown, 1975; Kippola and Santorico, 2010) was used to analyze the microarray data and identified 4,485 differentially regulated genes (P ≤ 0.01; Supplemental Table S2). To identify genes that are robustly regulated by cytokinin, a variety of empirically defined filters were then applied to this data set. For the analysis of microarray data, 1.5- to 2-fold changes are often used as minimum cutoffs when identifying regulated genes. We explored various cutoffs values using a set of genes that were previously demonstrated to be regulated by cytokinin using additional methods beyond microarray analysis (e.g. quantitative reverse transcription [RT]-PCR and northern blotting; Supplemental Table S1), including genes involved in cytokinin signal transduction (type-A ARRs and AHK4), metabolism (CYTOKININ OXIDASEs CKX3, CKX4, and CKX5) and transcription factors. The latter were particularly useful because transcription factors are generally expressed at low levels (Holland, 2002; Vaquerizas et al., 2009), and thus their expression is difficult to accurately assess using microarrays. The relative expression of these genes in control and cytokinin-treated samples as determined by each microarray was plotted with different midpoint values (Fig. 2A). Most of these transcription factors fell below a threshold of 1.8-fold induction in this analysis, indicating that a cutoff of greater than or equal to 1.8-fold excluded many known cytokinin-regulated genes (Fig. 2A). By contrast, a midpoint of 1.5-fold induction captured most of the known cytokinin-regulated transcription factors (Fig. 2A).
Figure 2.

Selection and validation of filters used for microarray meta-analysis of cytokinin-treated microarrays. A, Selection of appropriate fold change cutoff to use for the meta-analysis. A subset of the genes that has previously been confirmed to be induced by cytokinin (Supplemental Table S1) was used for this analysis. The relative expression of each gene in cytokinin versus control samples in each microarray experiment (Table I) was plotted with a midpoint value on the color scale of 1.3-, 1.5-, 1.8-, or 2-fold change as indicated. B, Examination of the number of genes that are up-regulated at least 1.5-fold in at least the number of microarray experiments noted on the x axis (blue line, left y axis), plotted along with the percentage of those genes thus identified that test as cytokinin regulated using a Fisher’s combined probability test with a P value of 0.01 or less (red line, right y axis). C, Number of microarrays in which a set of 15 genes previously demonstrated to be induced by cytokinin (Supplemental Table S1) show at least a 1.5-fold induction in the microarray experiments shown in Table I. The dashed line indicates the six or more microarrays chosen as the cutoff for inclusion in the list of robustly regulated genes.
We next examined the relationship between the number of array experiments in which a gene was up-regulated greater than or equal to 1.5-fold and its identification based on the Fisher’s combined probability test (P ≤ 0.01; Fig. 2B). There is a sharp decline in the percentage of genes that passed the threshold of P ≤ 0.01 for the Fisher’s test for genes that appear in less than six out of 13 experiments (Fig. 2B). In addition, we examined a subset of genes confirmed to be regulated by cytokinin, including several transcription factors, and almost all (15 out of 16) were found to be induced by greater than or equal to 1.5-fold in at least six microarray experiments (Fig. 2C). We thus queried for all genes that displayed a greater than or equal to 1.5-fold change in response to cytokinin in at least six experiments (Table I) and that had a P value of 0.01 or less in the Fisher’s test. This analysis identified 226 genes (Tables II and III), including 39 out of 55 genes previously validated by other methods in addition to microarray (Supplemental Table S1). We refer to this list of 226 genes as the “golden list” of cytokinin-regulated genes, containing 158 up-regulated genes (Table II) and 68 down-regulated genes (Table III).
Table II. Up-regulated genes identified by microarray meta-analysis.
Genes in boldface indicate candidates validated by methods other than microarrays.
| Microarrays/13 | Up-Regulated Genes |
|---|---|
| 13 | AT1G10470 (ARR4); AT1G16530 (ASL9); AT1G19050 (ARR7); AT1G74890 (ARR15); AT2G40670 (ARR16); AT3G48100 (ARR5); AT3G57040 (ARR9); AT4G29740 (CKX4); AT5G62920 (ARR6); AT4G03610 (Metallohydrolase) |
| 12 | AT1G13420 (ST4B); AT1G67110 (CYP735A2); AT2G38760 (ANNAT3); AT2G40230 (Acyl-transferase); AT2G41310 (ARR8); AT2G46310 (CRF5); AT1G12740 (CYP87A2); AT1G13740 (AFP2); AT1G75440 (UBC16); AT2G17820 (ATHK1); AT2G25160 (CYP82F1); AT3G50300 (Acyl-transferase); AT5G05860 (UGT76C2); AT5G47980 (Acyl-transferase) |
| 11 | AT1G59940 (ARR3); AT2G01830 (WOL); AT2G30540 (Thioredoxin); AT2G38750 (ANNAT4); AT1G58170 (Dirigent-like protein); AT1G64590 (Rossmann-fold); AT1G72140 (Unknown); AT3G15720 (Pectin lyase like); AT3G29575 (AFP3); AT3G45700 (Major Facilitator); AT3G57010 (Strictosidine synthase) |
| 10 | AT1G69040 (ACR4); AT1G69530 (ATEXPA1); AT2G22770 (NAI1); AT2G29490 (ATGSTU1); AT4G23750 (CRF2); AT1G31320 (LBD4); AT1G61800 (GPT2); AT2G01890 (PAP8); AT2G25150 (Acyl-transferase); AT2G35980 (YLS9); AT2G43200 (Methyltransferases); AT3G29250 (Rossmann-fold); AT3G45070 (Hydrolases); AT3G45710 (Major Facilitator); AT4G02850 (PhzC/PhzF); AT4G15680 (Glutaredoxin); AT4G19030 (NLM1); AT4G29690 (Alkaline phosphatase like); AT5G19260 (FAF); AT5G60890 (MYB34) |
| 9 | AT1G03850 (GRXS13); AT1G75450 (CKX5); AT3G62930 (Glutaredoxin); AT4G11190 (Dirigent-like protein); AT4G26150 (CGA1); AT1G11670 (MATE efflux); AT1G14960 (Polyketide cyclase); AT1G17190 (ATGSTU26); AT1G26770 (ATEXPA10); AT1G68360 (C2H2); AT2G03090 (ATEXPA15); AT2G36870 (XTH32); AT4G22570 (APT3); AT4G27970 (SLAH2); AT4G29700 (Alkaline phosphatase like); AT4G37400 (CYP81F3); AT5G04770 (CAT6); AT5G11420 (Unknown); AT5G40390 (SIP1); AT5G47990 (CYP705A5); AT5G48000 (CYP708A2) |
| 8 | AT1G04240 (SHY2); AT1G10480 (ZFP5); AT4G11210 (Dirigent-like protein); AT1G18400 (BEE1); AT1G28100 (Unknown); AT1G64390 (AtGH9C2); AT1G66800 (Unknown); AT1G73300 (scpl2); AT2G22930 (UDP-Glycosyltransferase); AT2G34610 (Unknown); AT2G46660 (CYP78A6); AT3G50700 (AtlDD2); AT4G15660 (Glutaredoxin); AT4G25410 (bHLH); AT4G35150 (O-methyltransferase); AT4G39770 (HAD); AT5G19110 (Aspartyl protease); AT5G26220 (Unknown); AT5G26260 (TRAF-like); AT5G42590 (CYP71A16) |
| 7 | AT5G50915 (bHLH); AT1G04250 (AXR3); AT1G15100 (RHA2A); AT1G21520 (Unknown); AT1G54300 (Unknown); AT1G70560 (TAA1); AT1G78120 (TPR12); AT1G78580 (ATTPS1); AT2G17500 (Auxin efflux); AT2G27840 (HDT4); AT2G32990 (AtGH9B8); AT2G39705 (RTFL8); AT2G39920 (HAD); AT2G43550 (DEFL); AT3G10960 (AZG1); AT3G45680 (Unknown); AT3G54720 (AMP1); AT3G61640 (AGP20); AT4G34590 (GBF6); AT4G34950 (Major Facilitator); AT5G04120 (Unknown); AT5G14070 (ROXY2); AT5G28640 (AN3); AT5G46230 (Unknown); AT5G47950 (Acyl-transferase); AT5G48010 (THAS1); AT5G64620 (C/VIF2) |
| 6 | AT1G15550 (GA3OX1); AT2G21650 (MEE3); AT3G61630 (CRF6); AT4G39070 (B-box zinc finger); AT1G03110 (TRM82); AT1G03360 (ATRRP4); AT1G47400 (Unknown); AT1G49450 (WD40); AT1G65730 (YSL7); AT1G67030 (ZFP6); AT1G75580 (SAUR-like); AT1G76410 (ATL8); AT2G04090 (MATE efflux); AT2G20080 (Unknown); AT2G47990 (SWA1); AT3G15990 (SULTR3;4); AT3G21670 (Unknown); AT3G26960 (Unknown); AT3G44320 (NIT3); AT3G55010 (PUR5); AT3G59670 (Unknown); AT4G10500 (Oxygenase); AT4G16000 (Unknown); AT4G21120 (AAT1); AT4G26600 (Methyltransferase); AT4G29720 (ATPAO5); AT4G35720 (Unknown); AT4G38840 (SAUR-like); AT5G13180 (ANAC083); AT5G38020 (SAM like); AT5G42580 (CYP705A12); AT5G56320 (ATEXPA14); AT5G56970 (Unknown); AT5G62420 (Oxidoreductase); AT5G63450 (CYP94B1) |
Table III. Down-regulated genes identified by microarray meta-analysis.
Genes in boldface indicate candidates validated by methods other than microarrays.
| Microarrays/13 | Down-Regulated Genes |
|---|---|
| 11 | AT2G35270 (GIK); AT3G13760 (C1 domain); AT5G43520 (C1 domain) |
| 10 | AT1G72360 (ERF73); AT4G33420 (Peroxidase); AT5G15830 (bZIP3) |
| 9 | AT2G28160 (bHLH029); AT1G77330 (ACC oxidase); AT1G78000 (SULTR1;2); AT5G43180 (Unknown); AT5G50760 (SAUR-like) |
| 8 | AT1G23390 (F-box protein); AT1G80440 (Galactose oxidase); AT5G61590 (AP2 family); AT1G49470 (Unknown); AT1G61590 (Protein Kinase); AT2G40970 (MYBC1); AT3G01190 (Peroxidase); AT3G11340 (UGT76B1); AT3G48920 (AtMYB45); AT4G14130 (XTR7); AT5G01740 (NTF2); AT5G10580 (Unknown); AT5G22270 (Unknown); AT5G45070 (AtPP2-A8); AT5G66985 (Unknown) |
| 7 | AT1G80920 (J8); AT1G74940 (Unknown); AT5G57785 (Unknown); AT1G61840 (C1 domain); AT1G68880 (AtbZIP); AT1G71030 (MYBL2); AT2G28120 (Unknown); AT2G44500 (O-fucosyltransferase); AT3G04530 (PPCK2); AT3G29410 (Terpenoid cyclase); AT3G62270 (Transporter); AT4G23700 (ATCHX17); AT5G09440 (EXL4); AT5G40590 (C1 domain); AT5G54040 (C1 domain) |
| 6 | AT3G48360 (BT2); AT1G06160 (ORA59); AT1G70290 (TPS8); AT2G02710 (PLPB); AT2G23170 (GH3.3); AT2G44130 (Galactose oxidase); AT3G27170 (CLC-B); AT3G62550 (Hydrolase like); AT4G03510 (RMA1); AT5G01210 (Acyl-transferase); AT5G37540 (Aspartyl protease); AT1G09090 (ATRBOHB); AT1G11450 (Nodulin transporter); AT1G13670 (Unknown); AT2G36950 (Heavy metal transporter); AT2G39400 (Hydrolase); AT3G04070 (anac047); AT3G14280 (Unknown); AT3G25930 (Hydrolase like); AT4G30170 (Peroxidase); AT4G31320 (SAUR-like); AT4G39675 (Unknown); AT5G03380 (Heavy metal transport); AT5G05790 (Homeodomain); AT5G60520 (LEA); AT5G64260 (EXL2); AT5G65210 (TGA1) |
The up-regulated genes on the golden list include nine out of the 10 type-A response regulators. The sole exception, ARR17, did not have a significant meta-P value, which may reflect its low level of expression and hence poor detection on microarray experiments. Multiple cytokinin metabolism genes are also part of the golden list, including CKX4 and CKX5, which encode cytokinin oxidases/dehydrogenases (Werner et al., 2001), and a UDP-glucosyl transferase gene (UGT76C2) encoding a cytokinin O-glycosyltransferase (Hou et al., 2004). Ten cytochrome P450 genes are present in the golden list as up-regulated genes, including CYP735A2, which encodes a cytokinin transhydroxylase (Takei et al., 2004), all of which are up-regulated in response to cytokinin. The cytokinin-regulated CRF genes (Rashotte et al., 2006) are also part of this list, as are multiple additional AP2/ERF transcription factors. Four thioredoxin genes, which likely play a role in biotic and/or abiotic stress, are present on the golden list as up-regulated in response to cytokinin, as was a glutaredoxin gene. Other interesting genes induced by cytokinin on the golden list include two annexins, three dirigent-like proteins, an abscisic acid-dependent aquaporin (NOD26-LIKE MAJOR INTRINSIC PROTEIN1), five acyl transferases, and several genes involved in auxin signaling, including two SMALL AUXIN UP-REGULATED (SAUR)-like genes, an auxin efflux carrier, and AUXIN RESISTANT3 (AXR3). Furthermore, there were multiple genes involved in cell wall function, including expansins, pectin lyase-like, and xyloglucan endotransglucosylase/hydrolase32. Among the 68 down-regulated genes on the golden list (Table III), there is an uncharacterized UDP-glucosyl transferase gene, UGT76B1, that is down-regulated in most of the microarray experiments, suggesting a potential role in cytokinin homeostasis. Several genes encoding transporters are down-regulated, as are three peroxidase genes. The golden list also includes numerous transcription factors (21 up-regulated and 12 down-regulated; Tables II and III), at least some of which likely play a role in mediated cytokinin-regulated gene expression. The known and putative functions of the genes in the golden list are shown in Supplemental Table S3.
Validation of Genes Identified by the Meta-Analysis
We validated the regulation by cytokinin and characterized the response kinetics of a representative group of genes encoding transcription factors from the golden list using the NanoString nCounter system. The NanoString system allows for assessment of transcript levels with similar sensitivity to what is found with quantitative RT-PCR but does not require enzymatic reactions (e.g. reverse transcription) or amplification of targets and thus eliminates the potential biases introduced by these steps (Geiss et al., 2008; Heyl et al., 2008; Malkov et al., 2009). We chose genes encoding transcription factors for this first analysis, as they tend to be expressed at relatively low levels and thus represent a fairly stringent test of the golden list. Non-cytokinin-regulated genes were included as controls, and the cytokinin-treated seedlings were compared with the dimethyl sulfoxide (DMSO)-treated seedlings at matched treatment times. All nine of these up-regulated genes from the golden list retested as being induced by cytokinin, though the kinetics of the response to cytokinin varied (Fig. 3A). AXR3 (AT1G04250) is only induced at later time points (>120 min). ZINC FINGER PROTEIN5 (ZFP5; AT1G10480) and ZFP6 (AT1G67030) showed a rapid but transient response to cytokinin, reaching almost basal level of expression in 120 min. We also examined seven genes encoding transcription factors from the golden list that were down-regulated in response to cytokinin (Fig. 3B). The transcript levels for all seven were lower than their corresponding controls.
Figure 3.

Validation of golden list candidates by nCounter gene expression analysis and quantitative RT-PCR. A, NanoString nCounter gene expression analysis of up-regulated representative genes from the golden list. B, NanoString nCounter gene expression analysis of down-regulated representative genes from the golden list. For A and B, 10-d-old light-grown seedlings were treated with 5 μm BA or a DMSO vehicle control for 60 or 120 min as indicated, and the normalized counts for indicated transcripts were determined. Error bars indicate the mean ± se from three biological replicates. C, Relative transcript level of the up-regulated representative genes from the golden list as determined by quantitative RT-PCR. D, Relative transcript level of the down-regulated representative genes from the golden list as determined by quantitative RT-PCR. Ten-day-old light-grown seedlings treated with 5 μm BA or a DMSO vehicle control for 120 min were used for quantitative RT-PCR. The level of transcript relative to the DMSO control is shown as a mean of three biological replicates ± se of the mean.
In addition to the NanoString analysis, we validated additional genes from the golden list using quantitative RT-PCR. For this, we examined the expression of 11 up-regulated genes (Fig. 3C) and three down-regulated genes (Fig. 3D) in response to treatment with cytokinin. With the sole exception of the GIANT KILLER (GIK; AT2G35270) gene, this quantitative RT-PCR analysis confirmed the regulation of these genes in response to cytokinin. Overall, these analyses validated the quality of the golden list and support the notion that these represent a set of genes that is robustly regulated by cytokinin.
A Type-B Response Regulator Binding Motif Is Enriched in the Promoters of the Up-Regulated Genes
Previous studies have shown that type-B ARRs mediate the majority of cytokinin-regulated gene expression (Argyros et al., 2008). We thus examined the putative regulatory regions of the genes comprising the golden list to determine if they were enriched for type-B binding sequences. The core DNA sequence responsible for in vitro binding of ARR1, ARR2, ARR10, and ARR11 has been identified as AGAT (T/C; Sakai et al., 2000; Hosoda et al., 2002; Imamura et al., 2003). Further analysis revealed that the ARR1 consensus binding site includes an additional two bases on either side of the core sequence (AAGAT [T/C] TT; Taniguchi et al., 2007). While the AGAT (T/C) core motif is likely functional in planta (Ross et al., 2004), its frequent occurrence precludes its use as a diagnostic for the cytokinin responsiveness of a given promoter. This core sequence is not significantly enriched in the putative regulatory regions (defined for this purpose as 1 kb upstream of the translational start site) of the genes comprising the golden list (Fig. 4). By contrast, the extended version of the ARR1 binding site is substantially overrepresented in the putative regulatory regions of the cytokinin-induced genes present in the golden list (Fig. 4) relative to the putative regulatory regions of the cytokinin nonresponsive genes (i.e. genes that displayed fold change of <1 in response to cytokinin in all 13 microarray experiments), consistent with prior studies (Brenner et al., 2012). As expected, there was no enrichment of the TATA box element (TTATTT) in the putative regulatory regions of the cytokinin-responsive genes and also no enrichment of the auxin response element (Aux-RE; TGTCTC) that we used as a negative control (Fig. 4). Another putative cytokinin response element (TATTAG) has been identified in cucumber (Cucumis sativus; Kuroda et al., 2000; Fusada et al., 2005), but this element is not overrepresented in the putative regulatory regions of the genes in the golden list. The enrichment of the type-B binding sites in the up-regulated genes suggests a direct regulation of these genes by type-B ARRs in response to cytokinin, consistent with the role of the type-B ARRs in cytokinin-regulated transcription (Mason et al., 2004, 2005; Argyros et al., 2008). Interestingly, there is no enrichment of the type-B element motifs in the putative regulatory regions of the cytokinin-down-regulated genes on this list, which may reflect an indirect effect of the type-B ARRs on these genes or the presence of an alternative type-B binding site not represented by the extended ARR1 binding site.
Figure 4.

The frequency of various transcription factor motifs in the promoters of cytokinin-regulated versus unregulated genes. The frequency of various transcription factor binding motifs as indicated (a type-B core motif, AGAT [T/C]; the extended motif determined for ARR1, AAGAT [T/C]TT; a type-B motif defined from cucumber, TATTAG; a TATA box element, TTATTT; and an Aux-RE element, TGTCTC) were quantified in the promoter regions (defined as 1,000 bp upstream of the translational start site) of the validated up-regulated genes and the nonvalidated up- and down-regulated genes identified by microarray meta-analysis (Tables II and III) using the Motif Mapper of the TOUCAN 2 workbench. The number of motifs present in the promoters of the regulated genes was divided by the number of motifs present in an equal number of promoters from random genes whose level was found to be unchanged by cytokinin in all microarray experiments used in the meta-analysis. Aux-RE was chosen as a negative control.
Differential Expression Analysis of RNA-Seq Data
To further explore cytokinin-regulated gene expression, we used RNA-seq to characterize the response of the transcriptome to cytokinin. We analyzed three biological replicates of 10-d-old seedlings treated for 2 h with 5 μm cytokinin or a DMSO vehicle control. The sequencing reads were aligned to the Arabidopsis genome using TopHAT, and differential expression analysis was performed with DESeq with a false discovery rate of 5% as described in “Materials and Methods.” This analysis revealed 573 genes (P ≤ 0.05) that were differentially regulated by cytokinin (423 up-regulated and 150 down-regulated), including many genes previously demonstrated to be up- or down-regulated under similar conditions, as well as new genes not previously identified as cytokinin regulated (Supplemental Table S4).
As expected, the type-A response regulators were among the most highly up-regulated genes, with log2 fold changes (as calculated by DESeq) ranging from 1.68 to 5.5. Nine out of 10 type-A ARRs showed differential regulation in this experiment, with the sole exception again being ARR17. Overall, the fold induction of the type-A ARRs in RNA-seq was similar to that found in a microarray experiment that employed similar experimental conditions (Rashotte et al., 2003; Lee et al., 2007). Other known cytokinin-responsive genes appeared among the top 50 differentially up-regulated genes sorted by ascending P values, including cytokinin metabolism (CKX4 and CKX5) and signaling genes (AHK4), annexins (ANNAT3 and ANNAT4), GLUTATHIONE S-TRANSFERASE19 (GSTU19; AT2G29490), CYP735A2 as well as well-characterized cytokinin-induced transcription factors such as CRF2, CRF5, NAI1, Cytokinin responsive GATA factor1 (CGA1), and ASYMMETRIC LEAVES2 LIKE9 (ASL9), and basic helix-loop-helix (bHLH) transcription factors (Supplemental Table S4).
Most prior studies exploring the response of the Arabidopsis transcriptome to cytokinin have used the ATH1 microarray, which was developed using the TIGRv5 annotations of the Arabidopsis genome (Redman et al., 2004). Many additional genes (approximately 11,000) have been annotated in the genome since the release of TIGRv5, and these are not represented on the ATH1 microarray. RNA-seq allows an exploration of the effects of cytokinin on these recently annotated genes. The RNA-seq analysis of cytokinin-treated seedlings revealed 73 new genes that were differentially regulated in the presence of cytokinin (60 up-regulated and 13 down-regulated; Table IV). The up-regulated genes encode proteins such as eight transcription factors (including five bHLH family members), three defensin-like proteins, two F-box proteins, a cytochrome P450, and CKX7 (Table IV). The down-regulated genes include one bHLH family member and a peroxidase gene. We used quantitative RT-PCR to independently examine the expression of the top (by fold change) 18 up-regulated and three down-regulated genes among the list of genes uniquely identified by RNA-seq (Fig. 5). With the sole exception of an uncharacterized peroxidase (AT1G77100), all of the other genes tested were confirmed to be regulated in response to cytokinin.
Table IV. Novel cytokinin responsive genes (not on ATH microarray) identified by RNA-seq.
| Arabidopsis Genome Initiative | Fold Change | P value | Description |
|---|---|---|---|
| AT3G29252 | 13.8 | 9.77E-03 | Dehydrogenase/reductase |
| AT3G44326 | 13.4 | 1.83E-02 | F-box family protein |
| AT2G43470 | 8.6 | 3.42E-07 | Unknown protein |
| AT5G44417 | 8.5 | 9.12E-05 | Similar to CPRD2 |
| AT5G51780 | 7.0 | 2.81E-04 | Basic helix-loop-helix |
| AT1G70860 | 5.8 | 5.85E-03 | Lipid transport superfamily |
| AT2G20080 | 5.6 | 9.27E-03 | Unknown protein |
| AT1G74458 | 5.3 | 3.99E-04 | Unknown protein |
| AT5G26290 | 5.2 | 6.77E-17 | TRAF-like family |
| AT3G14540 | 4.9 | 1.49E-05 | Protein prenyltransferases |
| AT5G07475 | 4.7 | 2.70E-02 | Cupredoxin superfamily |
| AT4G25400 | 4.5 | 4.99E-05 | Basic helix-loop-helix |
| AT2G31220 | 4.2 | 4.85E-02 | Basic helix-loop-helix |
| AT2G36295 | 4.1 | 1.16E-02 | Unknown protein |
| AT4G37620 | 4.1 | 3.54E-02 | RNase H domain |
| AT3G49330 | 3.9 | 2.69E-06 | Plant invertase protein |
| AT1G62190 | 3.5 | 5.03E-02 | Kua-ubiquitin protein |
| AT5G44005 | 3.5 | 4.38E-05 | Unknown protein |
| AT4G22214 | 3.4 | 2.90E-02 | Defensin-like |
| AT4G20240 | 3.4 | 4.38E-05 | Cytochrome P450-71A27 |
| AT2G26370 | 3.4 | 1.79E-02 | MD-2-related |
| AT3G13404 | 3.4 | 2.67E-04 | Unknown protein |
| AT1G61050 | 3.4 | 4.91E-03 | Glycosyltransferase |
| AT5G02090 | 3.4 | 2.83E-02 | Unknown protein |
| AT5G42930 | 3.3 | 4.42E-02 | α-/β-Hydrolases |
| AT3G13403 | 3.2 | 3.98E-02 | Defensin-like |
| AT4G28840 | 3.2 | 6.42E-04 | Unknown protein |
| AT4G22230 | 3.1 | 4.65E-03 | Arabidopsis defensin-like |
| AT2G34350 | 3.1 | 4.27E-03 | Major Facilitator |
| AT3G01513 | 3.0 | 9.58E-03 | Unknown protein |
| AT1G36622 | 2.9 | 2.76E-03 | Unknown protein |
| AT5G65030 | 2.8 | 3.15E-04 | Unknown protein |
| AT4G15417 | 2.7 | 8.48E-04 | RNAse II-like1 |
| AT1G68870 | 2.6 | 9.12E-05 | SOB five-like2 |
| AT3G11720 | 2.6 | 1.63E-07 | Polyketide cyclase |
| AT1G14630 | 2.6 | 5.00E-03 | Unknown protein |
| AT4G29930 | 2.6 | 3.78E-06 | Basic helix-loop-helix |
| AT2G01818 | 2.6 | 1.37E-02 | PLATZ transcription factor |
| AT3G15340 | 2.5 | 5.79E-04 | PPI2 |
| AT4G36820 | 2.4 | 6.52E-03 | Unknown protein |
| AT4G37409 | 2.4 | 1.61E-03 | Unknown protein |
| AT3G25597 | 2.4 | 4.08E-03 | Unknown protein |
| AT4G16008 | 2.3 | 1.65E-02 | Unknown protein |
| AT1G54217 | 2.3 | 1.08E-02 | Ribosomal L18ae |
| AT1G53541 | 2.1 | 4.87E-02 | Unknown protein |
| AT3G50840 | 2.1 | 4.23E-03 | NPH3 |
| AT1G12420 | 2.1 | 3.23E-04 | ACT domain8 |
| AT4G24015 | 2.1 | 1.21E-02 | RING/U-box |
| AT4G15670 | 2.0 | 1.12E-03 | Thioredoxin |
| AT1G21520 | 2.0 | 2.10E-02 | Unknown protein |
| AT1G62975 | 2.0 | 8.79E-03 | Basic helix-loop-helix |
| AT1G68862 | 2.0 | 4.73E-02 | Unknown protein |
| AT3G52910 | 2.0 | 2.01E-02 | GRF4 |
| AT5G21482 | 1.9 | 1.08E-04 | Cytokinin oxidase7 |
| AT4G29905 | 1.9 | 2.05E-05 | Unknown protein |
| AT5G44490 | 1.9 | 3.50E-02 | FBD, F-box protein |
| AT4G30850 | 1.8 | 1.16E-03 | Transmembrane |
| AT4G21215 | 1.7 | 3.91E-02 | Unknown protein |
| AT5G51540 | 1.7 | 1.95E-02 | Metalloproteases |
| AT2G43110 | 1.6 | 2.31E-02 | Unknown protein |
| AT1G31835 | 1.6 | 4.16E-02 | Unknown protein |
| AT3G55340 | 1.6 | 4.21E-02 | Phragmoplastin |
| AT1G25440 | 0.6 | 2.99E-02 | B-box type zinc finger |
| AT3G49970 | 0.6 | 3.13E-02 | NPH3 |
| AT5G50150 | 0.6 | 1.80E-03 | Unknown protein |
| AT4G13395 | 0.5 | 1.34E-02 | Rotundifoluia like |
| AT3G22142 | 0.5 | 3.98E-05 | Bifunctional inhibitor |
| AT3G21330 | 0.5 | 2.36E-02 | Basic helix-loop-helix |
| AT4G13540 | 0.4 | 2.41E-03 | Unknown protein |
| AT2G41231 | 0.4 | 3.40E-03 | Unknown protein |
| AT5G35525 | 0.4 | 2.40E-03 | PLAC8 family protein |
| AT2G26695 | 0.4 | 2.58E-03 | Ran BP2/NZF |
| AT1G15125 | 0.4 | 1.02E-06 | Methyltransferases |
| AT5G57685 | 0.3 | 1.02E-06 | Gln dumper3 |
| AT1G77100 | 0.1 | 1.97E-02 | Peroxidase protein |
Figure 5.

Validation of the cytokinin regulation of a subset of genes identified by RNA-seq that is not present on the ATH1 microarrays. A, Relative transcript level of the up-regulated indicated genes. B, Relative transcript level of the down-regulated indicated genes. Ten-day-old light-grown seedlings were treated with 5 µm BA or a DMSO vehicle control for 120 min and were determined by quantitative RT-PCR. The level of transcript relative to the DMSO control is shown as a mean of three biological replicates ± se of the mean.
We identified several noncoding genes as being differentially expressed in response to treatment with benzyladenine (BA; Fig. 6A), including two microRNA genes that were up-regulated, MIR163 and MIR414. The top candidate for a target of miRNA163 identified by the AthaMap prediction tool (Bülow et al., 2012) is an S-adenosyl-l-Met-dependent methyltransferases (AT1G15125). Consistent with the up-regulation of its cognate microRNA, AT1G15125 was down-regulated in response to cytokinin (Fig. 6B). No potential targets of MIR414 were predicted using this prediction tool. The other nonprotein-coding genes (AT3G27884, AT1G31935, AT5G03668, and AT5G01215) have not been characterized.
Figure 6.

Cytokinin-regulated expression of nonprotein-coding genes as identified by RNA-seq. A, List of genes regulated by cytokinin with the fold change of the transcript level for each gene in cytokinin-treated versus control samples indicated. B, Expression of MIR163 and its potential target (AT1G66725, S-adenosyl-l-Met-dependent methyltransferase) in cytokinin-treated and control samples as determined by RNA-seq. Expression is expressed as reads per million (RPM).
Assigning Biological Functions to the Novel Cytokinin-Regulated Genes: GO Term and MapMan Analysis of Cytokinin-Regulated Genes
To query physiological processes potentially targeted by the transcript changes that occur in response to cytokinin, we analyzed the GO term enrichment of the cytokinin-regulated genes identified by RNA-seq using AmiGO (Carbon et al., 2009) and DAVID (Supplemental Fig. S1; Supplemental Table S5; Huang da et al., 2009). This analysis revealed not only the expected enrichment of genes involved in cytokinin signaling and metabolism, but also enrichment of genes involved in secondary metabolism (anthocyanin, flavonoid, and suberin biosynthetic pathways), nutrient assimilation (nitrate transport and iron binding), and reductase activities. An analysis of the golden list resulted in an enrichment of similar GO term categories and is also consistent with the GO term enrichment from a previous meta-analysis of cytokinin-regulated genes (Brenner et al., 2012).
The cytokinin-regulated genes present in the GO-enriched group cytokinin signaling and metabolism included almost all the type-A ARRs, the CRFs, and AHP1 (AT3G21510), as well as a cluster of cytokinin oxidases (CKX3, CKX4, CKX5, and CKX7) and a group of five N-glucosyl transferases, only one of which, AT5G05860, has been shown to act on cytokinins. The largest group of cytokinin-regulated genes (approximately 70) was in a GO term group consisting of transcription factors, including some previously identified as cytokinin responsive (e.g. NAI1, AT2G22770; BR Enhanced Expression2 [BEE2], AT4G36540; etc.), as well as many not previously described as being regulated by cytokinin (e.g. ZFP6, AT1G67030; NGATHA3, AT1G01030; etc.). These likely mediate the secondary transcriptional responses to cytokinin that occur in response to extended cytokinin treatments.
Disease resistance genes were also found to have a high GO term enrichment in the cytokinin-regulated gene list. The cytokinin-regulated genes from this group included defense-associated dirigent-like proteins, defense-associated oxygenase (DOWNY MILDEW RESISTANT6), methyltransferase (AT1G77530), and chalcone isomerase like (AT5G05270). This is consistent with recent studies linking cytokinin to defense responses (Choi et al., 2010; Argueso et al., 2012). Other enriched groups of genes include GDSL family of Ser esterases/lipases, carboxypeptidases, thioredoxins, phototropic-responsive Nonphototrophic hypocotyl3 (NPH3) family proteins, ubiquitin-conjugating enzyme family members, several cytochrome P450s, and peroxidases. A number of kinase genes were identified that could link cytokinin to other signaling pathways in the plant, including many Ser/Thr kinases such as WITH NO LYSINE9, CBL-interacting kinases, Nek6, BR1-LIKE3, WAG1, CLAVATA1, several Leu-rich receptor-like protein kinases, a MAPKKK, and a senescence-induced kinase (AT2G19190).
There was an enrichment of genes related to auxin function in the set of cytokinin-regulated genes. These included a number of auxin responsive genes, including GH3 family and SAUR-like auxin responsive genes, and multiple ATP-binding cassette transporters, some of which may play a role in auxin transport. The induction of these genes involved in auxin function by cytokinin is consistent with the previously described cross talk between these two plant hormones (Moubayidin et al., 2009).
As a complementary approach to the GO term enrichment analysis, we explored the putative functions of the genes identified by microarray meta-analysis and RNA-seq experiment using MapMan (Thimm et al., 2004), which allows the visualization of cytokinin-induced changes in different metabolic processes (Fig. 7; Supplemental Fig. S3). Each gene in MapMan is initially organized in bins rather than as pathways, which allows genes to be assigned into a pathway even when their function is only tentatively known. Figure 7 shows the overall mapping of the cytokinin-regulated genes identified by microarray meta-analysis and RNA-seq, and Supplemental Figure S3 highlights relevant transcriptional changes related to overall metabolism, cellular responses, and regulation. As with the GO analysis, the cytokinin-regulated genes identified by the meta-analysis and RNA-seq results in the enrichment of similar functional categories and pathways in MapMan. As expected, there were a large number of genes assigned to the hormone metabolism bin (Fig. 7; Table V). A large number of genes were linked to biotic and abiotic stress, consistent with a role of cytokinin in the response to pathogens (Choi et al., 2010; Argueso et al., 2012) and abiotic stress (Argueso et al., 2009). An additional bin identified from both the golden list and RNA-seq is comprised of genes involved in secondary metabolism (Fig. 7), and in most cases, these genes are up-regulated in response to cytokinin. This includes genes involved in the biosynthesis of terpenes, flavonoids, and phenylpropanoids. This suggests that cytokinin may up-regulate the synthesis of these compounds. Consistent with this, cytokinin increases the synthesis of anthocyanin, a flavonoid compound synthesized via the phenylpropanoid pathway (Deikman and Hammer, 1995). Another striking category in this analysis was a group of genes regulating the redox state of the cell; more specifically, cytokinin caused the up-regulation of multiple genes encoding glutaredoxins. Glutaredoxins are oxidoreductases that use glutathione to catalyze the reduction of disulphide bridges and protein-GSH adducts (S-glutathionylated proteins). Nine of the 30 glutaredoxin genes in the Arabidopsis genome were identified as being up-regulated by cytokinin. The functions of glutaredoxins in plants are only beginning to be elucidated (Rouhier et al., 2008), but include roles in the protection against photooxidative stress (Laporte et al., 2012), iron-sulfur cluster assembly, flower development (Wang et al., 2009), and pathogen response (Wang et al., 2009). The cytokinin regulation of multiple glutaredoxin genes suggests a link to these processes.
Figure 7.

Functional analysis of cytokinin-regulated genes by MapMan. Overview of the distribution of genes in different functional groups as identified by MapMan analysis from the golden list (A) and RNA-seq (B). Gray/red boxes correspond to up-regulated genes and black/blue boxes to down-regulated genes. The numbers refer to the bin designations as defined in MapMan. [See online article for color version of this figure.]
Table V. Known and predicted functions of the microarray meta-analysis identified cytokinin induced genes using the MapMan bioinformatics tool.
References for genes with previously defined functions are listed in the footnotes.
| Broad-Function Category | Functions | Genes from the Golden List as Predicted by MapMan Analysis to be Associated with Known Functional Category |
|---|---|---|
| Stress (biotic/abiotic) | Pathogen, defense, drought, freezing tolerance, immunity | AT4G11190 (Dirigent-like)1, AT4G11210 (Dirigent-like)1, AT1G09090 (RBOHB)2, AT5G45070 (AtPP2-A8), AT2G43550 (DEFL), AT1G80920 (J8), AT2G43200 (Methyltransferases), AT1G14960 Polyketide cyclase), AT3G25930 (Hydrolase) |
| Redox | Oxidative burst, senescence, stress, photooxidative stress | AT1G03850 (GRXS13)3, AT5G14070 (ROXY2)4, AT4G15660 (Glutaredoxin), AT4G15680 (Glutaredoxin), AT2G30540 (Glutaredoxin), AT3G62930 (Glutaredoxin) |
| Protein degradation/ modification | Ubiquitination, posttranslational modification | AT3G48360 (BT2)5, AT3G54720 (AMP1)6, AT4G03510 (RMA1)7, AT2G44130 (Galactose oxidase)8, AT1G61590 (Protein kinase), AT1G75440 (UBC16), AT1G76410 (ATL8), AT1G15100 (RHA2A), AT1G80440 (Galactose oxidase), AT1G23390 (F-box protein), AT5G37540 (Aspartyl protease) |
| Signaling | Nutrient, receptor kinases, light | AT3G48360 (BT2)5, AT5G60890 (MYB34)9, AT5G64260 (EXL2), AT5G09440 (EXL4), AT5G01740 (NTF2), AT2G02710 (PLPB) |
| Cell wall | Modification, degradation, biosynthesis | AT1G69530 (EXPA1)10,11, AT3G61640 (AGP20), AT2G32990 (GH9B8), AT3G15720 (Pectin lyase like), AT2G36870 (XTH32), AT2G03090 (EXPA15), AT4G14130 (XTR7), AT5G56320 (EXPA14), AT1G26770 (EXPA10) |
| Development | Cell proliferation and differentiation, nodulation, seed development | AT1G09090 (RBOHB)2, AT5G14070 (ROXY2)4, AT2G35980 (YLS9)12, AT5G60520 (LEA), AT5G13180 (ANAC083), AT3G04070 (ANAC047), AT2G39705 (RTFL8), AT1G74940 (Unknown), AT4G34950 (Major Facilitator), AT2G28120 (Unknown) |
| Hormone action/ interaction | Biosynthesis and metabolism of auxin, cytokinin, brassinosteroid, salicylic acid, ethylene | AT1G75450 (CKX5)13, AT4G29740 (CKX4)13, AT2G01830 (WOL)14, AT1G15550 (GA3OX1)15, AT5G05860 (UGT76C2)16, AT5G56970 (CKX3)17, AT2G17820 (AHK1)18, AT1G75580 (SAUR-like), AT5G50760 (SAUR-like), AT4G31320 (SAUR-like), AT4G38840 (SAUR-like), AT2G23170 (GH3.3), AT5G38020 (SAM-like), AT1G77330 (ACC oxidase), AT1G06160 (ORA59), AT5G61590 (AP2 family), AT3G62550 (hydrolase-like) |
| Secondary metabolism | Flavanoids, lignin, phenylpropanoids, anthocyanin, suberin, alkaloids | AT5G48010 (THAS1)19, AT5G60890 (MYB34)9, AT1G70560 (TAA1)20, AT2G40230 (Acyltransferase), AT3G29410 (Terpenoid cyclase), AT5G47950 (Acyltransferase), AT3G50300 (Acyltransferase), AT5G01210 (Acyltransferase), AT5G47980 (Acyltransferase), AT3G57010 (Strictosidine synthase), AT3G44320 (NIT3), AT2G22930 (UDP-glucosyltransferase) |
| Gene expression/ regulation | Transcription factors, miRNA, Type A ARRs, histone modifiers | AT2G46310 (CRF5)21, AT1G74890 (ARR15)22, AT1G59940 (ARR3)23, AT1G19050 (ARR7)23, AT3G48100 (ARR5)23, AT3G57040 (ARR9)23, AT5G62920 (ARR6)23, AT2G41310 (ARR8)23, AT2G28160 (bHLH029)24, AT2G22770 (NAI1)25, AT4G26150 (CGA1)26,27, AT1G10480 (ZFP5)28,29,30, AT1G16530 (ASL9)31, AT1G04240 (SHY2)13, AT1G72360 (ERF73)32, AT2G40970 (MYBC1)33, AT1G71030 (MYBL2)34, AT5G65210 (TGA1)35, AT2G21650 (MEE3), AT4G39070 (B-box zinc finger), AT5G50915 (bHLH), AT4G23750 (CRF2), AT2G40670 (ARR16), AT1G10470 (ARR4), AT4G25410 (bHLH), AT1G18400, AT1G04250 (BEE1), AT2G27840 (HDT4), AT1G67030 (ZFP6), AT1G68360 (C2H2), AT3G48920 (bHLH), AT5G05790 (Homeodomain), AT3G04070 (ANAC047), AT1G68880 (ZIP), AT5G15830 (ZIP3), AT1G31320 (LBD4), AT3G50700 (IDDT2) |
1Ralph et al. (2007). 2Müller et al. (2009). 3Laporte et al. (2012). 4Wang et al. (2009). 5Mandadi et al. (2009). 6Griffiths et al. (2011). 7Lee et al. (2009). 8Curtis et al. (2013). 9Celenza et al. (2005). 10Wei et al. (2011). 11Zhang et al. (2011). 12Zheng et al. (2005). 13Bartrina et al. (2011). 14Riefler et al. (2006). 15Zhang et al. (2010). 16Wang et al. (2011a). 17Holst et al. (2011). 18Urao et al. (1999). 19de Silva et al. (2011). 20Yamada et al. (2009). 21Liang et al. (2010). 22Müller and Sheen (2008). 23To et al. (2004). 24Yuan, et al. (2005). 25Matsushima et al. (2003). 26Hudson et al. (2011). 27Chiang et al. (2012). 28Zhou et al. (2011). 29Zhou et al. (2012). 30An et al. (2012). 31Naito et al. (2007). 32Licausi et al. (2010). 33Zhai et al. (2010). 34Ye et al. (2012). 35Shearer et al. (2012).
New Gene Models and Alternative Splicing in Genes Involved in Cytokinin Signaling and Metabolism
In addition to providing information regarding overall transcript levels, RNA-seq read alignments can reveal alternative splicing, alternative transcription start and termination sites, and the relative prevalence of transcript variants. According to The Arabidopsis Information Resource 10 (TAIR10) gene annotations, nearly 20% of annotated genes produce multiple transcripts via one or more of these mechanisms. For most of these, little is known regarding relative expression levels of individual transcript isoforms or which isoforms are the most abundant. Moreover, it is possible that many of these genes may produce novel, previously undiscovered variants. We investigated these questions using Integrated Genome Browser (IGB), a visualization tool that allows in-depth inspection of individual loci (Nicol et al., 2009).
Using IGB, we inspected read alignments and junction files for 62 genes, focusing on gene families whose members are known to play a role in cytokinin signaling or metabolism (Supplemental Table S6). These included cytokinin receptors (AHKs), phosphotransfer proteins (AHPs), type-A and type-B ARRs, CRFs, cytokinin oxidases (CKXs), isopentyltransferases (IPTs), and the Lonely Guy (LOG) gene family.
Of the genes examined, 14 (23%) were already annotated in the TAIR10 genome annotations as producing more than one transcript variant. Of the 14 genes annotated as producing multiple variants, 11 had sufficient coverage to allow identification of the most abundant isoform (Table V). In the majority of cases, more than 90% of spliced reads overlapping differentially transcribed regions supported expression of one isoform, recapitulating a trend observed previously in ESTs (English et al., 2010). We also asked whether the cytokinin treatment triggered changes in the relative amounts of alternative isoforms; we found no significant difference in isoform ratios in the treated versus control samples in any of the alternatively spliced genes, suggesting that alternatively spliced isoforms are produced at constitutive levels independent of cytokinin signaling.
The RNA-seq data also revealed previously unannotated introns and allowed improvement of gene structure annotations. Among the 62 genes examined, 45 new splicing events affecting 28 genes were detected (Supplemental Table S7). However, among these 45 new splicing events, 25 were supported by only one spliced read, suggesting these were either low abundance events or artifacts of library construction or alignment. Of the novel events supported by more than one read, 13 events (in 11 genes) were supported by at least three reads from at least two libraries, suggesting that these events are less likely to be artifacts of library preparation or alignment (Table VI). We selected eight of these well-supported events in eight genes for validation by PCR (Table VII).
Table VI. Relative expression levels of transcript isoforms.
Genes with multiple TAIR10 models are listed along with the number of isoforms. Main isoform indicates the most abundantly expressed isoform(s) as determined by visual inspection of RNA-seq read alignments. Relative expression levels were determined by counting reads that were unique to each variant. V1, V2, etc. refer to the variant number shown after the decimal in an Arabidopsis Genome Initiative code.
| Gene type | Symbol | Arabidopsis Genome Initiative | No. of Models | Main Isoform |
|---|---|---|---|---|
| Cytokinin receptor | AHK4 | AT2G01830 | 3 | V2=V1>V3 |
| Phosphotransferprotein HPt | AHP2 | AT3G29350 | 2 | V1 |
| Phosphotransferprotein HPt | AHP4 | AT3G16360 | 2 | V2 |
| Pseudo-HPt | AHP6 | AT3G04580 | 2 | V1 |
| Type-A ARR | ARR16 | AT2G40670 | 2 | V1 |
| Type-B ARR | ARR1 | AT3G16857 | 2 | V1=V2 |
| Type-B ARR | ARR19 | AT1G49190 | 2 | Insufficient data |
| Cytokinin Response Factor | CRF2 | AT4G23750 | 2 | V1 |
| IPT | AtIPT9 | AT5G20040 | 3 | V1 |
| Cytokinin oxidase | CKX4 | AT4G29740 | 2 | V2 |
| Cytokinin oxidase | CKX5 | AT1G75450 | 2 | V1 |
| Lonely Guy | AtLOG2 | AT2G35990 | 3 | Insufficient data |
| Lonely Guy | AtLOG3 | AT2G37210 | 2 | Insufficient data |
| Lonely Guy | AtLOG8 | AT5G11950 | 2 | V1 |
Table VII. Novel splicing events and supporting reads.
Events with at least three supporting reads in at least two samples are listed. Supporting reads indicate the number of spliced reads supporting either the new event or its annotated TAIR10 alternative. Reads per gene is the number of spliced and unspliced reads mapping to the gene from all samples combined.
| Symbol | Arabidopsis Genome Initiative | Event | Reads Supporting New Event | Reads Supporting TAIR Model | Reads per Gene |
|---|---|---|---|---|---|
| AHP1 | AT3G21510 | N11 | 3 | 411 | 7,096 |
| AHP5 | AT1G03430 | N1 | 3 | 137 | 2,599 |
| N2 | 3 | 137 | |||
| ARR4 | AT1G10470 | N41,2 | 23 | NA | 25,540 |
| ARR7 | AT1G19050 | N21 | 4 | 1,090 | 15,831 |
| ARR16 | AT2G40670 | N1 | 5 | 106 | 2,361 |
| ARR17 | AT3G56380 | N11,2 | 7 | NA | 171 |
| ARR1 | AT3G16857 | N11 | 12 | 147 | 8,019 |
| N2 | 1 | 206 | |||
| AtIPT2 | AT2G27760 | N11,2 | 34 | 34 | 2,009 |
| AtLOG1 | AT2G28305 | N4 | 4 | 409 | 7,750 |
| AtLOG6 | AT5G03270 | N11,2 | 3 | 0 | 84 |
| AtLOG8 | AT5G11950 | N21,2 | 6 | 323 | 6,183 |
1Events tested by PCR. 2Events validated by PCR.
RT-PCR analysis confirmed new splicing events in five of the eight genes (Supplemental Fig. S4). For four genes (ARR4, LOG6, LOG8, and IPT2), the validated events included examples of alternative splicing, and new gene models were created that represent the new alternatively spliced isoforms. In the case of ARR17, the validated event added a new exon and intron to the 5′ end of the gene and was compatible with the existing model. Rather than create a new gene model for ARR17, we updated the boundaries of its existing gene model AT3G46380.1. In LOG6, the RNA-seq data contradicted the 3′ boundary of the final intron. Because the expression level for LOG6 was relatively low, we also examined spliced reads from another RNA-seq data set (Gulledge et al., 2012). Altogether, the RNA-seq data contradicted the annotated gene model (AT5G03270.1), consistent with a previous report investigating the LOG6 gene structure (Tokunaga et al., 2012). To capture LOG6 alternative splicing, we created five new gene models, including one new model (AT5G03270.2) encoding what appears to be a full-length protein compared with the other variants (Supplemental Fig. S4).
In most cases, the conceptual translations for the RT-PCR-verified, novel alternatively spliced isoforms were shorter than the annotated gene models’ translation. Alternatively spliced isoforms for IPT2 (AT2G27760.2), LOG6 (AT5G03270.3, AT5G03270.4, AT5G03270.5, and AT5G03270.6) and LOG8 (AT5G11950.3) introduced frame shifts and a premature termination codon. The alternatively spliced isoform for ARR4 (AT1G10470.2) contained a new 5′ intron that removed the annotated start codon, resulting in a conceptual translation that removed 14 amino acids on the N terminus of the gene’s conceptual translation. According to the RNA-seq data, most novel isoforms were lower in abundance than their annotated counterparts, with the exceptions of IPT2 and LOG6. In IPT2, both variants were equally abundant. For LOG6 (involved in cytokinin activation), the annotated form was not detected, and the alternatively spliced isoforms appeared equally abundant. Taken together, these results highlight how RNA-seq experiments, in addition to providing new information about differential expression, allow revision and improvement of gene models. However, tools and mechanisms to permit sharing of new models are needed to maximize the impact and relevance of RNA-seq data sets. Pending development of new data sharing tools, these new gene models will be made available via the publicly accessible IGBQuickLoad site (www.IGBQuickLoad.org).
DISCUSSION
Various microarray studies have shown that many processes influenced by cytokinin are reflected in changes of the abundance of transcripts encoding proteins of diverse function under specific conditions (Rashotte et al., 2003; Kiba et al., 2004; Brenner et al., 2005; Lee et al., 2007; Argyros et al., 2008; Goda et al., 2008). Our meta-analysis shows that there are many genes identified as cytokinin regulated by only one or a few studies (Fig. 2B), which likely reflects a combination of false positives/negatives and/or context-dependent regulation. The meta-analysis involving microarrays from multiple labs analyzing plants grown under differing conditions and treated with cytokinin in different ways defines a set of robustly cytokinin-regulated genes, a so-called “golden list” that serves as a valuable resource for subsequent studies of cytokinin function and as a foundation for future network analyses in Arabidopsis.
To construct the golden list of cytokinin-regulated genes, we considered only those genes whose transcripts were differentially regulated by at least 1.5-fold in at least 40% of the microarray experiments (i.e. six out of 13). This percentage is higher than used in previous microarray meta-analysis (Brenner et al., 2012), where only 25% of the microarrays were used without any further filters. While the prior approach is useful to identify as many genes as possible (though only the top 25 were reported), we sought to identify genes that can be assigned as cytokinin regulated with high confidence. Our choice of filters used for the meta-analysis in this work is supported by the observation that with a 1.5-fold change (up-regulation), 73% of the identified genes in six out of 13 microarrays are shown to be up-regulated by an independent experiment (RNA-seq; Fig. 8B). Several published studies further support the quality of the golden list. Taniguchi et al. (2007) identified 23 genes that are direct targets of ARR1, and 20 of these are present on the golden list. The top 25 genes identified as up-regulated by cytokinin that are presented in the Brenner et al. (2012) meta-analysis are all found on the golden list. The golden list also contains 39 genes previously validated to be cytokinin responsive by methods other than microarrays (Supplemental Table S1), accounting for 71% of the cytokinin-regulated genes described in the literature. The filtering criteria determined to be optimal for the set of microarrays used here may not be optimal for the meta-analysis of other sets of microarrays, but it does provide a starting point and a reasonable rubric for such studies.
Figure 8.

Percentage of genes present in multiple microarrays validated by RNA-seq at different fold change cutoffs. A to D, The number of genes that are up-regulated at least 1.3-fold (A), 1.5-fold (B), 1.8-fold (C), or 2-fold (D), in at least the number of microarray experiments noted on the x axis (squares, left y axis), plotted along with the percentage of those genes thus identified that test as cytokinin regulated from the RNA-seq analysis using P ≤ 0.05 (circles, right y axis). [See online article for color version of this figure.]
The generation of a list of regulated genes from transcriptome analysis is always a tradeoff between the level of stringency and inclusiveness, trying to achieve an optimal balance between the number of false positives and negatives. The existence of multiple experiments querying cytokinin-regulated gene expression provides an opportunity to identify cytokinin-regulated genes with a higher degree of confidence, independent of experimental variation due to differing growth and treatment conditions. Our meta-analysis focuses on genes regulated by cytokinin across a range of growth conditions, treatment regimens, and, in some cases, distinct tissue types. It is clear that this analysis will exclude genes induced in limited conditions, such as only in etiolated seedlings, or only at specific time points or under specific growth conditions. The hierarchical clustering of the microarray data shows distinct patterns of gene expression in etiolated seedling and roots relative to shoots and whole seedlings. A prior transcript profiling study has shown that roots and shoots display largely similar responses to cytokinin (Brenner and Schmülling, 2012), consistent with the notion that many genes will be induced by cytokinin in multiple tissues and supporting the inclusion of microarray data from both root and shoot tissues in our meta-analysis.
There was an overall 54% overlap in the genes identified as cytokinin regulated by the meta-analysis and the RNA-seq. However, when only up-regulated genes were considered, this overlap rose to 73% and dropped to 14% for the down-regulated genes (Supplemental Fig. S2). This low percentage of overlap with down-regulated genes may reflect the limitations of RNA-seq and/or of microarrays to identify down-regulated transcripts, the difficulty in robustly identifying transcript levels for genes whose expression level is decreased further by application of a stimulus, or an inherent variability in these genes (Fig. 3). In any case, the lower level of reproducibility for down-regulated genes compared with up-regulated genes has also been found when examining Arabidopsis responses to the phytohormone ethylene (Hall et al., 2012), and thus the difference we observe here is not unusual.
In addition to known early-responsive cytokinin signaling and metabolism genes (type-A ARRs, CKXs, etc.), there were many groups of genes that were overrepresented in both the gene list obtained by microarray meta-analysis and by RNA-seq. A particularly interesting group of genes are the transcription factors, as they likely play a role in mediating the diverse responses to cytokinin. For example, prior research has implicated the cytokinin-regulated CRF and CGA1 transcription factor families in regulating the stimulatory effect of cytokinin on chloroplast division (Rashotte et al., 2003; Okazaki et al., 2009; Chiang et al., 2012). Genetic analysis of the novel cytokinin-regulated transcription factors uncovered here may lend similar insight into how cytokinin regulates its diversity of downstream responses. Several other regulated genes can be associated with demonstrated functions of cytokinin, such as cross talk with other hormones, including auxin (e.g. SAUR like, IAA3 and AXR3), or its functions in cell wall (e.g. AT5G64620; CELL WALL/VACUOLAR INHIBITOR OF FRUCTOSIDASE2 [C/VIF2], AT3G15720; and pectin-lyase). Another set of interesting genes identified encodes TNF-receptor associated factor (TRAF)-like proteins (AT5G26290, AT3G20370, AT5G26260, AT5G52330, and AT1G582700), identified by both microarray meta-analysis and RNA-seq. The TRAF-like genes are a large gene family in Arabidopsis encoding potential E3 ligase proteins (Huang et al., 2004; Gingerich et al., 2007; McBerry et al., 2012), only a very few of which have been functionally characterized. These genes may play a role in the regulation of protein turnover in response to cytokinin.
RNA-seq allowed the examination of regulation by cytokinin of nonprotein-coding genes. We found both the up-regulation of miRNA163 and the down-regulation of its potential target, S-adenosyl-l-Met-dependent methyltransferase (AT1G15125), in response to cytokinin. This family of enzymes has diverse roles (Loenen, 2006) in biotic and abiotic stress and in secondary metabolism. RNA-seq also allows the refinement of existing gene models, the definition of novel gene models, and quantification of alternate splicing variants. We used the data from the RNA-seq, coupled with the IGB tool, to analyze the gene models for various genes involved in cytokinin function. This analysis has substantially refined these gene models, which is an important foundation for subsequent analysis of these genes.
The defining of a list of genes that are robustly regulated by cytokinin will help elucidate the mechanisms by which this phytohormone achieves its pleiotropic effects. It provides markers useful for subsequent research and will enable the delineation of a transcriptional network linking cytokinin to other signaling networks in the coordination of growth and development, as well as to biotic and abiotic stress responses.
MATERIALS AND METHODS
Plant Materials, Growth Conditions, and Cytokinin Treatment
Arabidopsis (Arabidopsis thaliana; accession Columbia-0 [Col-0]) was used in all experiments. Seeds were sown onto six separate plates containing Murashige and Skoog (MS) media (Murashige and Skoog, 1962) and grown under constant light (150 μmol m–2 s–1) at 22°C. After 10 d of growth, seedlings from each plate were collected and added to flasks containing liquid MS media supplemented with 5 µm BA dissolved in DMSO or an equal aliquot of DMSO only. Seedlings from each plate were added to separate flasks. The six flasks were then incubated on a rotating shaker in constant light for 120 min. Samples were collected by and flash frozen in liquid nitrogen. Seedlings from the same plate and the same flask served as biological replicates and were pooled for RNA isolation, complementary DNA (cDNA) library synthesis, and sequencing.
Microarray Meta-Analysis
Microarrays used in this study are described in Table I. Raw data files (.CEL) files were downloaded from NASCArrays (http://affymetrix.arabidopsis.info/narrays/experimentbrowse.pl) or ArrayExpress (http://www.ebi.ac.uk/arrayexpress/). The raw files were investigated using GeneSpring version 12.1-GX (Agilent Technologies) with default values. Because some microarrays were represented with only a single replicate, no P-value filter was applied. The fold change and P value for each gene were further processed using Microsoft Excel. The P values obtained from this analysis were analyzed using Fisher’s method. This test combines P value probabilities from each test into one test statistic (X2) using the formula
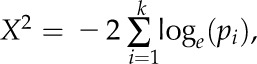 |
where pi is the P value for the ith hypothesis test. When all the null hypotheses are true and pi (or its corresponding test statistic) is independent, X2 has a χ2 distribution with 2k degrees of freedom, where k is the number of tests being combined. This fact can be used to determine the P value for X2. In this experiment, k = 13 (13 microarrays used). For the data sets for which no P value was available (one replicate, experiments number 1 and 2), the P value was assumed to be 1 for all the genes. This test provides a meta-P value; a cutoff of meta-P ≤ 0.01 was used to filter the differential regulated genes. The gene list was further filtered using a fold change cutoff of 1.5 or greater and further filtered for genes displaying a fold change of 1.5 or greater in six or more microarray experiments.
Hierarchical Clustering of the Microarray Data
GeneSpring was used to gene-wise normalize the expression data, and hierarchical clustering of signal intensities with respect to gene expression patterns was carried out with Euclidean distance correlation for the two series of experiments, all genes (referred to in GeneSpring as entities) and variables (referred to as conditions). The graphic result was downloaded from the analysis.
In Silico Promoter Analysis
The genomic sequences 1,000 bp upstream of the translational start sites of the genes identified as cytokinin regulated from the meta-analysis (Tables II and III) were downloaded from the TAIR Web site (http://www.arabidopsis.org/tools/bulk/sequences/index.jsp). This set of sequences was tested for conserved promoter elements, including the core type-B binding element (AGAT [T/C]; Sakai et al., 2000; Hosoda et al., 2002; Imamura et al., 2003), the extended type-B element (AAGAT[T/C]TT; Taniguchi et al., 1998), a cucumber (Cucumis sativus) putative cytokinin response element (TATTAG; Kuroda et al., 2000; Fusada et al., 2005), a TATA box (TTATTT), and Aux-RE (TGTCTC) as a negative control using the Motif Mapper of the TOUCAN 2 workbench (Aerts et al., 2005). Random lists derived from the same number of promoters were generated from genes that were unchanged by cytokinin treatment (i.e. no differential regulation in any microarray). The motifs were counted, and the frequency of their occurrence in the promoters of cytokinin-regulated genes was divided by their frequency in promoters of an equal number of nonregulated genes.
RNA-Seq
RNA Isolation
Total RNA was isolated using RNAeasy Mini RNA kit (Qiagen) according to the manufacturer’s instructions. Concentration, integrity, and extent of contamination by ribosomal RNA were monitored using the ND-1000 spectrophotometer (Thermo Fisher Scientific) and the Bioanalyzer 2100 (Agilent Technologies).
cDNA Libraries
Barcoded cDNA libraries were prepared for multiplex sequencing on the Illumina HiSeq2000 platform using the TruSeq sample preparation kit according to manufacturer’s instructions. Six libraries were prepared from each of three controls and three treatment groups. Starting amount for each sample was 5 µg of RNA. Libraries were named BA1, BA2, and BA3 and Control 1, Control 2, and Control 3 according to the treatment. Libraries BA1, BA2, and BA3 received barcoded TruSeq adapters 9, 10, and 11 and libraries Control 1, Control 2, and Control 3 received adapter barcodes 1, 3, and 8. To obtain sufficient material for sequencing, the libraries were amplified by PCR for 15 cycles following the recommendations of the TruSeq sample preparation protocol. Final elution of each library was in 25 µL of total volume. Library concentrations and quality were assessed using the Qubit 2.0 Fluorometer (Life Technologies) and the Bioanalyzer 2100. BA1 and Control 1, BA2 and Control 2, and BA3 and Control 3 were combined for clustering and sequencing in three separate flow cell lanes. Sequencing was performed on the HiSeq2000 instrument, 50 cycles per flow cell, at the Lineberger Cancer Research High-Throughput Sequencing Center at University of North Carolina, Chapel Hill. Initial sequencing yields for libraries BA1 and Control 1 were low, so a second round of sequencing was performed on these two libraries, thus providing technical replicates for BA1 and Control 1. To distinguish technical replicates, we designate data from these different runs as BA1_1, BA1_2, Control 1_1, and Control 1_2.
Sequence Processing
Sequence data in FastQ format were downloaded from the sequencing facility Web site and processed as follows using a combination of publicly available tools and custom programs. Quality was assessed using FastQC; all samples, including the first low-yielding runs of Control 1 and BA1 libraries, had high overall quality. Sequences were aligned onto the latest Arabidopsis Col-0 genome assembly (released June 2009) using TopHat version 2.0.5 (Langmead et al., 2009) and BowTie version 2.0.5 (Langmead et al., 2009) with maximum and minimum intron size parameters set to 2,000 and 30 bases, reflecting the unusual compactness of Arabidopsis genes compared with mammalian genomes.
The accepted_hits.bam file produced by TopHat was used in subsequent analysis steps. This binary alignment file (Li et al., 2009) contained both spliced and unspliced read alignments. SAMtools version 0.1.18 (Li et al., 2009) and the “NH” (number of hits) Sequence Alignment Map flag were used to separate alignments according to the number of times a read mapped onto the reference genome. Reads mapping to a unique location in the genome were used for differential expression and splicing analysis. The wiggles program (from the TopHat distribution) was used to created coverage graphs in bedGraph format for each binary alignment file. Browser extensible format and bedGraph files were sorted and indexed using the tabix utility (Li, 2011) and deployed on an IGB QuickLoad site to facilitate visualization in IGB (Nicol et al., 2009). The number of single-mapping reads that overlap each annotated gene in this Arabidopsis TAIR10 gene model annotations release was counted and supplied as inputs to DESeq for statistical analysis of differential gene expression. Differential expression analysis of RNA-seq read alignment counts was performed using Bioconductor 2.11 (Gentleman et al., 2004) and Bioconductor library DESeq version 1.2.1 (Anders and Huber, 2010). Probe set annotations used to identify genes not represented on the ATH1 microarray (Gene Expression Omnibus platform accession GPL138) were from the TAIR probe set annotation file available from ftp://ftp.arabidopsis.org/Microarrays/Affymetrix/affy_ATH1_array_elements-2010-12-20.txt. The false discovery rate was controlled at 5% using the method of Benjamini and Hochberg (1995). For each gene, probe sets that uniquely match each gene (unique.ps) and probe sets that also cross hybridized with related genes (promiscuous.ps) were identified using annotations from the TAIR file.
For splicing analysis, a custom program (FindJunctions) was developed to predict exon-exon junctions from spliced alignments produced by TopHat. FindJunctions was run with options –u (unique) and –n 5 (five flanking bases) so that junction features were predicted from uniquely mapping reads with gapped alignments in which at least five bases of the read aligned to both sides of the gap representing a possible intron. Each junction feature thus represents a distinct splicing event and defines the boundaries of a putative intron inferred from the data. Strand of origin for junctions was inferred from the corresponding genomic sequence using canonical splice site (GT-intron-AG) consensus sequences, where possible. The program (developed in Java) is freely available as part of the open-source GenoViz project (http://sourceforge.net/projects/genoviz/). Novel splicing variants for individual genes were detected by visual analysis of junction features displayed in IGB alongside TAIR10 structural annotations. Numbers of spliced reads supporting annotated or novel introns were tabulated and compared to identify the most abundantly expressed isoform. For genes in which alternative splicing was detected, single-mapping read alignments were used to extend, where possible, the 5′ and 3′ ends of genes. IGB was used to identify the longest possible reading frame in cases where new splicing events disrupted the annotated open reading frame.
RNA Extraction Quantitative RT-PCR and NanoString nCounter Gene Expression Analysis
Seedlings were grown on MS media (Murashige and Skoog, 1962) for 10 d in continuous light, treated with either DMSO or BA for 120 min, and subjected to total RNA extraction using an RNeasy Plus kit (Qiagen). cDNA was prepared from the total RNA using SuperScript III reverse transcriptase (Invitrogen) as described by the manufacturer. Quantitative RT-PCR was performed using SYBR Premix Ex Taq polymerase (TaKaRa) in a DNA Engine OPTICON 2 (MJ Research). Primers used are listed in Supplemental Tables S8 and S9. Three biological samples each were analyzed with three technical replicates per sample. The relative expression for candidate genes (normalized to β-tubulin as a reference gene and to the wild type grown on DMSO as a control sample) and ses were determined using REST 2009 software (Qiagen). Error bars represent se of the mean of the three biological replicates, and Student’s t test was performed as a test for significance.
The NanoString nCounter gene expression analysis was performed as described previously (Geiss et al., 2008) by the University of North Carolina Genomics and Bioinformatics Core Facilities using 100 ng of total RNA extracted from the treated and control seedling at different time points as stated. The NanoString probes targeting ANGUSTIFOLIA3 (AN3; AT5G28640), SHORT HYPOCOTYL2 (SHY2; AT1G04240), ZINC FINGER PROTEIN5 (bZIP5; AT1G10480), bZIP6 (AT1G67030), MYB34 (AT5G60890), ERF73 (AT1G72360), bZIP3 (AT5G15830), C2H2 (AT1G68360), BEE1 (AT1G18400), MYBC1 (AT2G40970), MYB45 (AT3G48920), Homeodomain (AT5G05790), AXR3 (AT1G04250), TGACG SEQUENCE SPECIFIC BINDING PROTEIN1 (TGA1; AT5G65210), LOB DOMAIN-CONTAINING PROTEIN4 (LBD4; AT1G31320), and bHLH029 (AT2G28160) and the control probes for PROTEIN PHOSPHATASE2A (PP2A; AT1G13320) and UBIQUITIN10 (UBQ10; AT4G05320) were designed and synthesized by NanoString Technologies (http://www.nanostring.com). The mRNA counts for candidate genes were normalized to the reference genes PP2A and UBQ10 as described in the nCounter gene expression assay manual.
Validation of Splicing Variants by RT-PCR
Wild-type Arabidopsis (accession Col-0) seedlings were sown onto plates containing MS media (Murashige and Skoog, 1962) under constant light of 150 μmol m–2 s–1 at 22°C. After 10 d of growth, seedlings from each of five plates were divided between two flasks containing liquid MS and either 20 μm BA dissolved via DMSO or the comparable quantity of DMSO. Seedlings were treated for 120 min, with 90 min of the treatment on a shaker set to 150 rpm. Seedlings from the same plate and treatment were pooled for each sample and collected with RNALater (Invitrogen). RNA was extracted using a Qiagen RNA mini kit following manufacturer’s instructions. cDNA for PCR was synthesized by SMARTMMLV reverse transcriptase using 1 μg of RNA per 20 μL reaction.
Splicing events were tested based using the primers shown in Supplemental Table S10 using a DNAEngine Peltier Thermal Cycler set to either 30 or 35 cycles of 94°C denaturation, 58°C or 60°C annealing, and 72°C elongation using 0.2 μL or more of cDNA in a 25-μL reaction. For most primers, a series of PCR reactions over a gradient of 50°C to 70°C annealing was done to optimize annealing temperatures and check for nonspecific products.
GO and Pathways Analysis
GO term enrichment was performed using the GO Web site (http://amigo.geneontology.org/cgi-bin/amigo/term_enrichment; Carbon et al., 2009) and the DAVID tool (http://david.abcc.ncifcrf.gov/home.jsp; Huang da et al., 2009). The graphic result was downloaded from the Web site.
MapMan Analysis
MapMan (Thimm et al., 2004) was downloaded following author’s instructions (http://mapman.gabipd.org/web/guest/mapman), and maps were produced to visualize processes affected by the cytokinin treatment. Averaged fold changes of 13 experiments used for microarray meta-analysis was used for the genes from the golden list, and log2-transformed fold change for RNA-seq (Supplemental Table S4) served as input to MapMan. Pathway images were saved as .JPG files. Genes were assigned to individual reactions using BINCodes (Thimm et al., 2004).
Sequence data are available from GenBank under BioProject accession no. PRJNA194426.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure S1. GO term enrichment of cytokinin-regulated genes.
Supplemental Figure S2. Comparison of genes from RNA-seq and the golden list.
Supplemental Figure S3. MapMan illustrations of cytokinin-regulated genes.
Supplemental Figure S4. New gene models.
Supplemental Table S1. Validated (by method[s] other than microarray) cytokinin-responsive genes.
Supplemental Table S2. Genes identified as cytokinin responsive in the microarray meta-analysis by Fisher’s combined probability test.
Supplemental Table S3. Golden list genes and functions.
Supplemental Table S4. Cytokinin-responsive genes identified by RNA-seq.
Supplemental Table S5. Genes in functional groups significantly enriched from GO term analysis (DAVID).
Supplemental Table S6. Genes examined using IGB.
Supplemental Table S7. Novel splicing events and supporting reads.
Supplemental Table S8. Primers used for RT-PCR validation of cytokinin-regulated genes identified in the golden list.
Supplemental Table S9. Primers used for RNA-seq candidates validation.
Supplemental Table S10. Primers used to test novel splicing events.
Acknowledgments
We thank Hemant Kelkar (University of North Carolina Center for Bioinformatics) for his support with the bioinformatics tools used in this work. We also thank Kevin Thompson (University of North Carolina at Charlotte) for early analysis of the RNA-seq and microarray data and Selena Tran (University of North Carolina at Chapel Hill) for help in organizing data.
Glossary
- GO
Gene Ontology
- RT
reverse transcription
- DMSO
dimethyl sulfoxide
- BA
benzyladenine
- IGB
Integrated Genome Browser
- Col-0
Columbia-0
- MS
Murashige and Skoog
- TAIR
The Arabidopsis Information Resource
- TAIR10
The Arabidopsis Information Resource 10
- cDNA
complementary DNA
- cDNA
complementary DNA
References
- Aerts S, Van Loo P, Thijs G, Mayer H, de Martin R, Moreau Y, De Moor B. (2005) TOUCAN 2: the all-inclusive open source workbench for regulatory sequence analysis. Nucleic Acids Res 33: W393–W396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An L, Zhou Z, Sun L, Yan A, Xi W, Yu N, Cai W, Chen X, Yu H, Schiefelbein J, et al. (2012) A zinc finger protein gene ZFP5 integrates phytohormone signaling to control root hair development in Arabidopsis. Plant J 72: 474–490 [DOI] [PubMed] [Google Scholar]
- Anders S, Huber W. (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argueso CT, Ferreira FJ, Epple P, To JP, Hutchison CE, Schaller GE, Dangl JL, Kieber JJ. (2012) Two-component elements mediate interactions between cytokinin and salicylic acid in plant immunity. PLoS Genet 8: e1002448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argueso CT, Ferreira FJ, Kieber JJ. (2009) Environmental perception avenues: the interaction of cytokinin and environmental response pathways. Plant Cell Environ 32: 1147–1160 [DOI] [PubMed] [Google Scholar]
- Argueso CT, Raines T, Kieber JJ. (2010) Cytokinin signaling and transcriptional networks. Curr Opin Plant Biol 13: 533–539 [DOI] [PubMed] [Google Scholar]
- Argyros RD, Mathews DE, Chiang YH, Palmer CM, Thibault DM, Etheridge N, Argyros DA, Mason MG, Kieber JJ, Schaller GE. (2008) Type B response regulators of Arabidopsis play key roles in cytokinin signaling and plant development. Plant Cell 20: 2102–2116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartrina I, Otto E, Strnad M, Werner T, Schmülling T. (2011) Cytokinin regulates the activity of reproductive meristems, flower organ size, ovule formation, and thus seed yield in Arabidopsis thaliana. Plant Cell 23: 69–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. JR Stat Soc 57: 289–300 [Google Scholar]
- Bishopp A, Lehesranta S, Vatén A, Help H, El-Showk S, Scheres B, Helariutta K, Mähönen AP, Sakakibara H, Helariutta Y. (2011) Phloem-transported cytokinin regulates polar auxin transport and maintains vascular pattern in the root meristem. Curr Biol 21: 927–932 [DOI] [PubMed] [Google Scholar]
- Brandstatter I, Kieber JJ. (1998) Two genes with similarity to bacterial response regulators are rapidly and specifically induced by cytokinin in Arabidopsis. Plant Cell 10: 1009–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner WG, Ramireddy E, Heyl A, Schmülling T. (2012) Gene regulation by cytokinin in Arabidopsis. Front Plant Sci 3: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner WG, Romanov GA, Köllmer I, Bürkle L, Schmülling T. (2005) Immediate-early and delayed cytokinin response genes of Arabidopsis thaliana identified by genome-wide expression profiling reveal novel cytokinin-sensitive processes and suggest cytokinin action through transcriptional cascades. Plant J 44: 314–333 [DOI] [PubMed] [Google Scholar]
- Brenner WG, Schmülling T. (2012) Transcript profiling of cytokinin action in Arabidopsis roots and shoots discovers largely similar but also organ-specific responses. BMC Plant Biol 12: 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M. (1975) A method for combining non-independent, one-sided tests of significance. Biometrics 31: 987–992 [Google Scholar]
- Bülow L, Bolívar JC, Ruhe J, Brill Y, Hehl R. (2012) ‘MicroRNA Targets’, a new AthaMap web-tool for genome-wide identification of miRNA targets in Arabidopsis thaliana. BioData Min 5: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S. (2009) AmiGO: online access to ontology and annotation data. Bioinformatics 25: 288–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celenza JL, Quiel JA, Smolen GA, Merrikh H, Silvestro AR, Normanly J, Bender J. (2005) The Arabidopsis ATR1 Myb transcription factor controls indolic glucosinolate homeostasis. Plant Physiol 137: 253–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Farmer AD, Langley RJ, Mudge J, Crow JA, May GD, Huntley J, Smith AG, Retzel EF. (2010) Meiosis-specific gene discovery in plants: RNA-seq applied to isolated Arabidopsis male meiocytes. BMC Plant Biol 10: 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Shen K, Song C, Luo J, Tseng GC. (2009) Ratio adjustment and calibration scheme for gene-wise normalization to enhance microarray inter-study prediction. Bioinformatics 25: 1655–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang YH, Zubo YO, Tapken W, Kim HJ, Lavanway AM, Howard L, Pilon M, Kieber JJ, Schaller GE. (2012) Functional characterization of the GATA transcription factors GNC and CGA1 reveals their key role in chloroplast development, growth, and division in Arabidopsis. Plant Physiol 160: 332–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Huh SU, Kojima M, Sakakibara H, Paek KH, Hwang I. (2010) The cytokinin-activated transcription factor ARR2 promotes plant immunity via TGA3/NPR1-dependent salicylic acid signaling in Arabidopsis. Dev Cell 19: 284–295 [DOI] [PubMed] [Google Scholar]
- Curtis RH, Pankaj, Powers SJ, Napier J, Matthes MC. (2013) The Arabidopsis F-box/Kelch-repeat protein At2g44130 is upregulated in giant cells and promotes nematode susceptibility. Mol Plant Microbe Interact 26: 36–43 [DOI] [PubMed] [Google Scholar]
- D’Agostino IB, Deruère J, Kieber JJ. (2000) Characterization of the response of the Arabidopsis response regulator gene family to cytokinin. Plant Physiol 124: 1706–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deikman J, Hammer PE. (1995) Induction of anthocyanin accumulation by cytokinins in Arabidopsis thaliana. Plant Physiol 108: 47–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dello Ioio R, Linhares FS, Sabatini S. (2008) Emerging role of cytokinin as a regulator of cellular differentiation. Curr Opin Plant Biol 11: 23–27 [DOI] [PubMed] [Google Scholar]
- de Silva K, Laska B, Brown C, Sederoff HW, Khodakovskaya M. (2011) Arabidopsis thaliana calcium-dependent lipid-binding protein (AtCLB): a novel repressor of abiotic stress response. J Exp Bot 62: 2679–2689 [DOI] [PubMed] [Google Scholar]
- English AC, Patel KS, Loraine AE. (2010) Prevalence of alternative splicing choices in Arabidopsis thaliana. BMC Plant Biol 10: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filichkin SA, Priest HD, Givan SA, Shen R, Bryant DW, Fox SE, Wong WK, Mockler TC. (2010) Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res 20: 45–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusada N, Masuda T, Kuroda H, Shimada H, Ohta H, Takamiya K. (2005) Identification of a novel cis-element exhibiting cytokinin-dependent protein binding in vitro in the 5′-region of NADPH-protochlorophyllide oxidoreductase gene in cucumber. Plant Mol Biol 59: 631–645 [DOI] [PubMed] [Google Scholar]
- Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, et al. (2008) Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 26: 317–325 [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingerich DJ, Hanada K, Shiu SH, Vierstra RD. (2007) Large-scale, lineage-specific expansion of a bric-a-brac/tramtrack/broad complex ubiquitin-ligase gene family in rice. Plant Cell 19: 2329–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda H, Sasaki E, Akiyama K, Maruyama-Nakashita A, Nakabayashi K, Li W, Ogawa M, Yamauchi Y, Preston J, Aoki K, et al. (2008) The AtGenExpress hormone and chemical treatment data set: experimental design, data evaluation, model data analysis and data access. Plant J 55: 526–542 [DOI] [PubMed] [Google Scholar]
- Griffiths J, Barrero JM, Taylor J, Helliwell CA, Gubler F. (2011) ALTERED MERISTEM PROGRAM 1 is involved in development of seed dormancy in Arabidopsis. PLoS ONE 6: e20408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AA, Roberts AD, Vora H, Patel K, Loraine AE. (2012) Mining Arabidopsis thaliana RNA-seq data with Integrated Genome Browser reveals stress-induced alternative splicing of the putative splicing regulator SR45a. Am J Bot 99: 219–231 [DOI] [PubMed] [Google Scholar]
- Hall BP, Shakeel SN, Amir M, Ul Haq N, Qu X, Schaller GE. (2012) Histidine kinase activity of the ethylene receptor ETR1 facilitates the ethylene response in Arabidopsis. Plant Physiol 159: 682–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyl A, Ramireddy E, Brenner WG, Riefler M, Allemeersch J, Schmülling T. (2008) The transcriptional repressor ARR1-SRDX suppresses pleiotropic cytokinin activities in Arabidopsis. Plant Physiol 147: 1380–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland MJ. (2002) Transcript abundance in yeast varies over six orders of magnitude. J Biol Chem 277: 14363–14366 [DOI] [PubMed] [Google Scholar]
- Holst K, Schmülling T, Werner T. (2011) Enhanced cytokinin degradation in leaf primordia of transgenic Arabidopsis plants reduces leaf size and shoot organ primordia formation. J Plant Physiol 168: 1328–1334 [DOI] [PubMed] [Google Scholar]
- Hosoda K, Imamura A, Katoh E, Hatta T, Tachiki M, Yamada H, Mizuno T, Yamazaki T. (2002) Molecular structure of the GARP family of plant Myb-related DNA binding motifs of the Arabidopsis response regulators. Plant Cell 14: 2015–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou B, Lim EK, Higgins GS, Bowles DJ. (2004) N-Glucosylation of cytokinins by glycosyltransferases of Arabidopsis thaliana. J Biol Chem 279: 47822–47832 [DOI] [PubMed] [Google Scholar]
- Huang CJ, Chen CY, Chen HH, Tsai SF, Choo KB. (2004) TDPOZ, a family of bipartite animal and plant proteins that contain the TRAF (TD) and POZ/BTB domains. Gene 324: 117–127 [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson D, Guevara D, Yaish MW, Hannam C, Long N, Clarke JD, Bi YM, Rothstein SJ. (2011) GNC and CGA1 modulate chlorophyll biosynthesis and glutamate synthase (GLU1/Fd-GOGAT) expression in Arabidopsis. PLoS ONE 6: e26765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura A, Kiba T, Tajima Y, Yamashino T, Mizuno T. (2003) In vivo and in vitro characterization of the ARR11 response regulator implicated in the His-to-Asp phosphorelay signal transduction in Arabidopsis thaliana. Plant Cell Physiol 44: 122–131 [DOI] [PubMed] [Google Scholar]
- Kiba T, Aoki K, Sakakibara H, Mizuno T. (2004) Arabidopsis response regulator, ARR22, ectopic expression of which results in phenotypes similar to the wol cytokinin-receptor mutant. Plant Cell Physiol 45: 1063–1077 [DOI] [PubMed] [Google Scholar]
- Kieber JJ. (2002) Cytokinins. The Arabidopsis Book 1: e0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kippola TA, Santorico SA. (2010) Methods for combining multiple genome-wide linkage studies. Methods Mol Biol 620: 541–560 [DOI] [PubMed] [Google Scholar]
- Köllmer I, Werner T, Schmülling T. (2011) Ectopic expression of different cytokinin-regulated transcription factor genes of Arabidopsis thaliana alters plant growth and development. J Plant Physiol 168: 1320–1327 [DOI] [PubMed] [Google Scholar]
- Kuroda H, Masuda T, Fusada N, Ohta H, Takamiya K. (2000) Expression of NADPH-Protochlorophyllide oxidoreductase gene in fully green leaves of cucumber. Plant Cell Physiol 41: 226–229 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte D, Olate E, Salinas P, Salazar M, Jordana X, Holuigue L. (2012) Glutaredoxin GRXS13 plays a key role in protection against photooxidative stress in Arabidopsis. J Exp Bot 63: 503–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DJ, Park JY, Ku SJ, Ha YM, Kim S, Kim MD, Oh MH, Kim J. (2007) Genome-wide expression profiling of ARABIDOPSIS RESPONSE REGULATOR 7 (ARR7) overexpression in cytokinin response. Mol Genet Genomics 277: 115–137 [DOI] [PubMed] [Google Scholar]
- Lee HK, Cho SK, Son O, Xu Z, Hwang I, Kim WT. (2009) Drought stress-induced Rma1H1, a RING membrane-anchor E3 ubiquitin ligase homolog, regulates aquaporin levels via ubiquitination in transgenic Arabidopsis plants. Plant Cell 21: 622–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2011) Tabix: fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics 27: 718–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang YS, Ermawati N, Cha JY, Jung MH, Su’udi M, Kim MG, Ha SH, Park CG, Son D. (2010) Overexpression of an AP2/ERF-type transcription factor CRF5 confers pathogen resistance to Arabidopsis plants. J Korean Soc Appl Biol Chem 53: 142–148 [Google Scholar]
- Licausi F, van Dongen JT, Giuntoli B, Novi G, Santaniello A, Geigenberger P, Perata P. (2010) HRE1 and HRE2, two hypoxia-inducible ethylene response factors, affect anaerobic responses in Arabidopsis thaliana. Plant J 62: 302–315 [DOI] [PubMed] [Google Scholar]
- Loenen WA. (2006) S-Adenosylmethionine: jack of all trades and master of everything? Biochem Soc Trans 34: 330–333 [DOI] [PubMed] [Google Scholar]
- Lu T, Lu G, Fan D, Zhu C, Li W, Zhao Q, Feng Q, Zhao Y, Guo Y, Li W, et al. (2010) Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq. Genome Res 20: 1238–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkov VA, Serikawa KA, Balantac N, Watters J, Geiss G, Mashadi-Hossein A, Fare T. (2009) Multiplexed measurements of gene signatures in different analytes using the Nanostring nCounter Assay System. BMC Res Notes 2: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandadi KK, Misra A, Ren S, McKnight TD. (2009) BT2, a BTB protein, mediates multiple responses to nutrients, stresses, and hormones in Arabidopsis. Plant Physiol 150: 1930–1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason MG, Li J, Mathews DE, Kieber JJ, Schaller GE. (2004) Type-B response regulators display overlapping expression patterns in Arabidopsis. Plant Physiol 135: 927–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason MG, Mathews DE, Argyros DA, Maxwell BB, Kieber JJ, Alonso JM, Ecker JR, Schaller GE. (2005) Multiple type-B response regulators mediate cytokinin signal transduction in Arabidopsis. Plant Cell 17: 3007–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima R, Kondo M, Nishimura M, Hara-Nishimura I. (2003) A novel ER-derived compartment, the ER body, selectively accumulates a beta-glucosidase with an ER-retention signal in Arabidopsis. Plant J 33: 493–502 [DOI] [PubMed] [Google Scholar]
- McBerry C, Gonzalez RM, Shryock N, Dias A, Aliberti J. (2012) SOCS2-induced proteasome-dependent TRAF6 degradation: a common anti-inflammatory pathway for control of innate immune responses. PLoS ONE 7: e38384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. (2008) Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat Methods 5: 621–628 [DOI] [PubMed] [Google Scholar]
- Moubayidin L, Di Mambro R, Sabatini S. (2009) Cytokinin-auxin crosstalk. Trends Plant Sci 14: 557–562 [DOI] [PubMed] [Google Scholar]
- Müller B, Sheen J. (2008) Cytokinin and auxin interaction in root stem-cell specification during early embryogenesis. Nature 453: 1094–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller K, Carstens AC, Linkies A, Torres MA, Leubner-Metzger G. (2009) The NADPH-oxidase AtrbohB plays a role in Arabidopsis seed after ripening. New Phytol 184: 885–897 [DOI] [PubMed] [Google Scholar]
- Murashige T, Skoog F. (1962) A revised medium for the rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15: 473–497 [Google Scholar]
- Naito T, Yamashino T, Kiba T, Koizumi N, Kojima M, Sakakibara H, Mizuno T. (2007) A link between cytokinin and ASL9 (ASYMMETRIC LEAVES 2 LIKE 9) that belongs to the AS2/LOB (LATERAL ORGAN BOUNDARIES) family genes in Arabidopsis thaliana. Biosci Biotechnol Biochem 71: 1269–1278 [DOI] [PubMed] [Google Scholar]
- Nazri A, Lio P. (2012) Investigating meta-approaches for reconstructing gene networks in a mammalian cellular context. PLoS ONE 7: e28713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol JW, Helt GA, Blanchard SG, Jr, Raja A, Loraine AE. (2009) The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics 25: 2730–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama R, Watanabe Y, Fujita Y, Le DT, Kojima M, Werner T, Vankova R, Yamaguchi-Shinozaki K, Shinozaki K, Kakimoto T, et al. (2011) Analysis of cytokinin mutants and regulation of cytokinin metabolic genes reveals important regulatory roles of cytokinins in drought, salt and abscisic acid responses, and abscisic acid biosynthesis. Plant Cell 23: 2169–2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki K, Kabeya Y, Suzuki K, Mori T, Ichikawa T, Matsui M, Nakanishi H, Miyagishima SY. (2009) The PLASTID DIVISION1 and 2 components of the chloroplast division machinery determine the rate of chloroplast division in land plant cell differentiation. Plant Cell 21: 1769–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plank M, Wuttke D, van Dam S, Clarke SA, de Magalhães JP. (2012) A meta-analysis of caloric restriction gene expression profiles to infer common signatures and regulatory mechanisms. Mol Biosyst 8: 1339–1349 [DOI] [PubMed] [Google Scholar]
- Ralph SG, Jancsik S, Bohlmann J. (2007) Dirigent proteins in conifer defense II: extended gene discovery, phylogeny, and constitutive and stress-induced gene expression in spruce (Picea spp.). Phytochemistry 68: 1975–1991 [DOI] [PubMed] [Google Scholar]
- Rashotte AM, Carson SD, To JP, Kieber JJ. (2003) Expression profiling of cytokinin action in Arabidopsis. Plant Physiol 132: 1998–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashotte AM, Mason MG, Hutchison CE, Ferreira FJ, Schaller GE, Kieber JJ. (2006) A subset of Arabidopsis AP2 transcription factors mediates cytokinin responses in concert with a two-component pathway. Proc Natl Acad Sci USA 103: 11081–11085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman JC, Haas BJ, Tanimoto G, Town CD. (2004) Development and evaluation of an Arabidopsis whole genome Affymetrix probe array. Plant J 38: 545–561 [DOI] [PubMed] [Google Scholar]
- Riefler M, Novak O, Strnad M, Schmülling T. (2006) Arabidopsis cytokinin receptor mutants reveal functions in shoot growth, leaf senescence, seed size, germination, root development, and cytokinin metabolism. Plant Cell 18: 40–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross EJ, Stone JM, Elowsky CG, Arredondo-Peter R, Klucas RV, Sarath G. (2004) Activation of the Oryza sativa non-symbiotic haemoglobin-2 promoter by the cytokinin-regulated transcription factor, ARR1. J Exp Bot 55: 1721–1731 [DOI] [PubMed] [Google Scholar]
- Rouhier N, Lemaire SD, Jacquot JP. (2008) The role of glutathione in photosynthetic organisms: emerging functions for glutaredoxins and glutathionylation. Annu Rev Plant Biol 59: 143–166 [DOI] [PubMed] [Google Scholar]
- Sakai H, Aoyama T, Oka A. (2000) Arabidopsis ARR1 and ARR2 response regulators operate as transcriptional activators. Plant J 24: 703–711 [DOI] [PubMed] [Google Scholar]
- Séguéla M, Briat JF, Vert G, Curie C. (2008) Cytokinins negatively regulate the root iron uptake machinery in Arabidopsis through a growth-dependent pathway. Plant J 55: 289–300 [DOI] [PubMed] [Google Scholar]
- Shearer HL, Cheng YT, Wang L, Liu J, Boyle P, Després C, Zhang Y, Li X, Fobert PR. (2012) Arabidopsis clade I TGA transcription factors regulate plant defenses in an NPR1-independent fashion. Mol Plant Microbe Interact 25: 1459–1468 [DOI] [PubMed] [Google Scholar]
- Skoog F, Miller CO. (1957) Chemical regulation of growth and organ formation in plant tissues cultured in vitro. Symp Soc Exp Biol 11: 118–130 [PubMed] [Google Scholar]
- Tajima Y, Imamura A, Kiba T, Amano Y, Yamashino T, Mizuno T. (2004) Comparative studies on the type-B response regulators revealing their distinctive properties in the His-to-Asp phosphorelay signal transduction of Arabidopsis thaliana. Plant Cell Physiol 45: 28–39 [DOI] [PubMed] [Google Scholar]
- Takei K, Yamaya T, Sakakibara H. (2004) Arabidopsis CYP735A1 and CYP735A2 encode cytokinin hydroxylases that catalyze the biosynthesis of trans-Zeatin. J Biol Chem 279: 41866–41872 [DOI] [PubMed] [Google Scholar]
- Taniguchi M, Kiba T, Sakakibara H, Ueguchi C, Mizuno T, Sugiyama T. (1998) Expression of Arabidopsis response regulator homologs is induced by cytokinins and nitrate. FEBS Lett 429: 259–262 [DOI] [PubMed] [Google Scholar]
- Taniguchi M, Sasaki N, Tsuge T, Aoyama T, Oka A. (2007) ARR1 directly activates cytokinin response genes that encode proteins with diverse regulatory functions. Plant Cell Physiol 48: 263–277 [DOI] [PubMed] [Google Scholar]
- Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M. (2004) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37: 914–939 [DOI] [PubMed] [Google Scholar]
- To JP, Haberer G, Ferreira FJ, Deruère J, Mason MG, Schaller GE, Alonso JM, Ecker JR, Kieber JJ. (2004) Type-A Arabidopsis response regulators are partially redundant negative regulators of cytokinin signaling. Plant Cell 16: 658–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga H, Kojima M, Kuroha T, Ishida T, Sugimoto K, Kiba T, Sakakibara H. (2012) Arabidopsis lonely guy (LOG) multiple mutants reveal a central role of the LOG-dependent pathway in cytokinin activation. Plant J 69: 355–365 [DOI] [PubMed] [Google Scholar]
- Urao T, Yakubov B, Satoh R, Yamaguchi-Shinozaki K, Seki M, Hirayama T, Shinozaki K. (1999) A transmembrane hybrid-type histidine kinase in Arabidopsis functions as an osmosensor. Plant Cell 11: 1743–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanstraelen M, Benková E. (2012) Hormonal interactions in the regulation of plant development. Annu Rev Cell Dev Biol 28: 463–487 [DOI] [PubMed] [Google Scholar]
- Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. (2009) A census of human transcription factors: function, expression and evolution. Nat Rev Genet 10: 252–263 [DOI] [PubMed] [Google Scholar]
- Wang J, Ma XM, Kojima M, Sakakibara H, Hou BK. (2011a) N-Glucosyltransferase UGT76C2 is involved in cytokinin homeostasis and cytokinin response in Arabidopsis thaliana. Plant Cell Physiol 52: 2200–2213 [DOI] [PubMed] [Google Scholar]
- Wang Y, Li L, Ye T, Zhao S, Liu Z, Feng YQ, Wu Y. (2011b) Cytokinin antagonizes ABA suppression to seed germination of Arabidopsis by downregulating ABI5 expression. Plant J 68: 249–261 [DOI] [PubMed] [Google Scholar]
- Wang Z, Xing S, Birkenbihl RP, Zachgo S. (2009) Conserved functions of Arabidopsis and rice CC-type glutaredoxins in flower development and pathogen response. Mol Plant 2: 323–335 [DOI] [PubMed] [Google Scholar]
- Wei PC, Zhang XQ, Zhao P, Wang XC. (2011) Regulation of stomatal opening by the guard cell expansin AtEXPA1. Plant Signal Behav 6: 740–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner T, Motyka V, Strnad M, Schmülling T. (2001) Regulation of plant growth by cytokinin. Proc Natl Acad Sci USA 98: 10487–10492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner T, Schmülling T. (2009) Cytokinin action in plant development. Curr Opin Plant Biol 12: 527–538 [DOI] [PubMed] [Google Scholar]
- Yamada M, Greenham K, Prigge MJ, Jensen PJ, Estelle M. (2009) The TRANSPORT INHIBITOR RESPONSE2 gene is required for auxin synthesis and diverse aspects of plant development. Plant Physiol 151: 168–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye H, Li L, Guo H, Yin Y. (2012) MYBL2 is a substrate of GSK3-like kinase BIN2 and acts as a corepressor of BES1 in brassinosteroid signaling pathway in Arabidopsis. Proc Natl Acad Sci USA 109: 20142–20147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Yamashino T, Amano Y, Tajima Y, Imamura A, Sakakibara H, Mizuno T. (2007) Type-B ARR transcription factors, ARR10 and ARR12, are implicated in cytokinin-mediated regulation of protoxylem differentiation in roots of Arabidopsis thaliana. Plant Cell Physiol 48: 84–96 [DOI] [PubMed] [Google Scholar]
- Yuan YX, Zhang J, Wang DW, Ling HQ. (2005) AtbHLH29 of Arabidopsis thaliana is a functional ortholog of tomato FER involved in controlling iron acquisition in strategy I plants. Cell Res 15: 613–621 [DOI] [PubMed] [Google Scholar]
- Zhai H, Bai X, Zhu Y, Li Y, Cai H, Ji W, Ji Z, Liu X, Liu X, Li J. (2010) A single-repeat R3-MYB transcription factor MYBC1 negatively regulates freezing tolerance in Arabidopsis. Biochem Biophys Res Commun 394: 1018–1023 [DOI] [PubMed] [Google Scholar]
- Zhang XQ, Wei PC, Xiong YM, Yang Y, Chen J, Wang XC. (2011) Overexpression of the Arabidopsis α-expansin gene AtEXPA1 accelerates stomatal opening by decreasing the volumetric elastic modulus. Plant Cell Rep 30: 27–36 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu Z, Wang L, Zheng S, Xie J, Bi Y. (2010) Sucrose-induced hypocotyl elongation of Arabidopsis seedlings in darkness depends on the presence of gibberellins. J Plant Physiol 167: 1130–1136 [DOI] [PubMed] [Google Scholar]
- Zheng MS, Takahashi H, Miyazaki A, Yamaguchi K, Kusano T. (2005) Identification of the cis-acting elements in Arabidopsis thaliana NHL10 promoter responsible for leaf senescence, the hypersensitive response against Cucumber mosaic virus infection, and spermine treatment. Plant Sci 168: 415–422 [Google Scholar]
- Zhou Z, An L, Sun L, Gan Y. (2012) ZFP5 encodes a functionally equivalent GIS protein to control trichome initiation. Plant Signal Behav 7: 28–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, An L, Sun L, Zhu S, Xi W, Broun P, Yu H, Gan Y. (2011) Zinc finger protein5 is required for the control of trichome initiation by acting upstream of zinc finger protein8 in Arabidopsis. Plant Physiol 157: 673–682 [DOI] [PMC free article] [PubMed] [Google Scholar]