

Abstract
The ATP4A encodes α subunit of H+, K+-ATPase that contains catalytic sites of the enzyme forming pores through cell membrane which allows the ion transport. H+, K+-ATPase is a membrane bound P-type ATPase enzyme which is found on the surface of parietal cells and uses the energy derived from each cycle of ATP hydrolysis that can help in exchanging ions (H+, K+ and Cl−) across the cell membrane secreting acid into the gastric lumen. The 3-D model of α-subunit of H+, K+-ATPase was generated by homology modeling. It was evaluated and validated on the basis of free energies and amino acid residues. The inhibitor binding amino acid active pockets were identified in the 3-D model by molecular docking. The two drugs Omeprazole and Rabeprazole were found more potent interactions with generated model of α-subunit of H+, K+-ATPase on the basis of their affinity between drug–protein interactions. We have generated ATP4A gene regulatory networks for interactions with other proteins which involved in regulation that can help in fine-tuning of proton pump and ion channels. These findings provide a new dimension for discovery and development of proton pump inhibitors and gene regulation of the ATPase. It can be helpful in better understanding of human physiology and also using synthetic biology strategy for reprogramming of parietal cells for control of gastric ulcers.
Electronic supplementary material
The online version of this article (doi:10.1007/s11693-012-9103-1) contains supplementary material, which is available to authorized users.
Keywords: H+, K+-ATPase; Homology modeling; Docking; Gastric ulcer; ATP4A gene network
Introduction
The gastric hydrogen potassium ATPase or H+, K+-ATPase is a proton pump of the stomach which is responsible for secretion of acid. The H+, K+-ATPase is found in parietal cells which are highly specialized epithelial cells located in inner cell lining of the stomach called the gastric mucosa (Munson et al. 2007). It is a member of P-type ATPase (ion pumps) that catalyzes active ion transport by coupling autophosphorylation and dephosphorylation to ion movement across lipid bilayers. A proton pump is an integral membrane protein that is capable of moving proton across the cell membrane, mitochondria and other organelles. It consists of α and β-subunits. The α-subunit is a catalytic domain which contains ATP binding site for ion recognition. Whereas, the β-subunit is essential for functional expression of H+, K+-ATPase which is also involved in the maturation of intracellular transport and stabilization of functional holoenzymes (Asano et al. 2004). It catalyzes an electro neutral exchange of cytoplasmic H+ and external K+ coupled with ATP hydrolysis (Slayman and Zuckier 1989).
Gastric acid is a digestive fluid, formed in the stomach with pH in between of 1.5 and 3.5 which is composed of hydrochloric acid (0.5 %), and large quantities of potassium chloride and sodium chloride. It plays an important role in digestion of proteins by activating digestive enzymes that can breakdown long chain of amino acid into smaller chains or amino acids. Increasing the production of gastric acid causes major health problems in human which includes dyspepsia, peptic ulcer, cancer and gastro esophageal reflux and it can inhibit the gastric acid secretion via blocking its activity. The process of activation of acid secretion requires a change in location of the ATPase from cytoplasmic tubules into the microvilli of the secretory canaliculus of the parietal cells. Gastric acid production would be controlled by targeting H+, K+ ATPase of the stomach. The proton pump has become an attractive drug target for therapeutic application in the regulation of acid suppression (Sachs et al. 1989). Proton-pump inhibitors (PPIs) are a group of drugs that can use in suppression of gastric acid production. However, there are numbers of adverse effect which have been recorded such as headache, nausea, diarrhea, abdominal pain, fatigue, dizziness, rashes, itching and depression by using long-term or high dose of drugs for control of acid production (Rossi 2006). Consequently; we require potent and specific drug for binding with proton pump for regulatable production of gastric acid in the stomach.
A number of reports available on inhibitors which binds transmembrane helices of the α-subunit of H+, K+-ATPase but the function of the β-subunit are still undetermined (Glynn and Karlish 1990; Toyoshima et al. 1993; Carafoli 1992). The effectiveness of inhibitors controlled to the introduction of omeprazole as therapy for acid-related disease in 1989 followed by several other PPIs which includes lansoprazole, pantoprazole and rabeprazole (Sachs et al. 1989). The inhibitors bind and inactivate the activity of H+/K+ ATPase enzyme for treatment of gastric ulcer. The drug lansoprazole has been reported as an acid secretion inhibitor in pigs from gastric ulceration (Melnichouk et al. 1999) while Omeprazole has been shown to block gastric acid secretion by specific inhibition of the H+, K+-ATPase (Tuukkanen and Väänänen 1986). In other study, omeprazole displays an inhibitor of acid secretion in vivo and in vitro test models including partially purified H+, K+-ATPase (Wallmark et al. 1983).
For the regulation of gastric acid production we require three dimensional (3-D) structure of H+, K+-ATPase and inhibitors. The 3-D structure of the α-subunit of H+, K+-ATPase is not yet determined although it plays an important role in the exchange of ions through the membrane and ATP hydrolysis. Recent development of bioinformatics tools and software’s could help for generation of 3-D model using known crystal structure. Homology modeling is a method of choice between sequence of target protein and single known structure (Kroemer et al. 1996). Molecular dynamics simulations on homology models of the E2P and E1 states have been performed to investigate the mechanism of K+ movement. The mechanism of transport against the established electrochemical gradients includes intermediate conformations in which the transferred ions are trapped (occluded) within the membrane domain of the pump. The pump cycle involves switching between the E1 and E2P states (Law et al. 2008) which regulate the acid production in stomach.
Knowledge of the amino acids in the H+, K+-ATPase which affect the mode of inhibition by SCH28080 and inhibitor affinity would provide insight into the regions of the membrane domain influencing the inhibitor selectivity and the luminal route to the ion transport site (Vagin et al. 2001). The catalytic α-subunit of H+, K+-ATPase has transmembrane segments with a cluster of intramembranal carboxylic amino acids located in the middle of transmembrane segments TM4, TM5, TM6, and TM8. Comparison to the known structure of the SERCA pump, mutagenesis, and molecular modeling has been identified and these are constituents of the ion binding domain (Shin et al. 2009). Consequently; there is a need to identify a cluster of gene network for designing of effective inhibitors.
Knowledge about gene encoding protein and enzyme interaction with each other in the gene network for regulation of gene expression would be helpful for the designing of drugs and inhibitors. The gene networks coordination is a complex interaction among biomolecules which includes DNA, RNA and proteins; and these elements contribute in biological process. The Systems Biology uses for depth understanding of biological system level phenomena that needs to explore the relationship between network structure and the dynamics of genes, proteins and other biomolecules (Samal and Jain 2008). The gene regulatory network for α subunit of the H+, K+-ATPase is still unknown. There are numbers of enzyme interacted to each other in gene networks that help in drug discovery and development. It could further help to repress and activate the gene regulatory networks for control of cellular activity (Singh et al. 2012a, b). However, proteins/enzymes serve as to activate or repress the other genes which are known as transcription factors that play a dynamic role in regulatory network. It binds to operator sites of promoter for regulation of gene expression by switching off and on system. The aim of present study was to generate 3-D model of α-subunit of H+, K+-ATPase of Homo sapiens which could use for screening of potent drugs for better treatment of gastric ulcers. We generated a novel gene regulatory network for fine-tuning of proton pump and ion channels.
Materials and methods
Collection and alignment sequences
The complete protein sequences of the H+, K+-ATPase of H. sapiens was retrieved (http://www.ncbi.nlm.nih.gov/) and the homology was obtained using Basic Local Alignment Search Tool (BLAST: http://www.ncbi.nim.nih.gov/blast) (Altschul et al. 1997). BlastP program was performed for searching structural similarity of the sequence with any template in Protein Data Bank. The alignment was performed using Clustal X (Thompson et al. 1997) with the target protein sequence (PDB: 3B8E).
Generation and validation of 3-D model
The resolution of X-ray crystal structure (3B8E) was 3.5 Å which belongs to pig renal Na+, K+-ATPase (Morth et al. 2007) and was retrieved from Protein Data Bank (http://www.rcsb.org/pdb/). It was used as a template structure for generation of the 3-D model of H+, K+-ATPase of H. sapiens using Modeller9v7 (Sali and Blundell 1993) and visualization by PYMOL (DeLano 2002). The evaluation of generated 3-D model was performed on the basis of free energy of model and template. PROCHECK was used to validate the 3-D model of H+, K+-ATPase (Laskowski et al. 1993). It generates Ramachandran plot and the amino acid residues in allowed, disallowed region and overall G-factor were considered.
Identification and analysis of potent drugs using molecular docking
The drugs dataset were obtained from NCBI-Pubchem compound library (http://pubchem.ncbi.nlm.nih.gov/) in SDF format that was converted into the 3-D structure using OpenBabel (Guha et al. 2006). The qualitative and quantitative characterization such as physiochemical property was taken from the same database (supplementary Table 1). The 3-D model of H+, K+-ATPase and drugs were used for molecular docking by AutoDock4.0 (Morris et al. 1998). There are docking parameters used as follows: 100 docking trials, population size 150, random starting position and conformational translation step ranges of 1.5 Ǻ, rotation step ranges 35, elitism of 1, mutation rate 0.02, cross over rate of 0.8, local search rate of 0.06 and 25 million energy evaluations. Distance dependent function of dielectric constant was used for calculating the energetic maps and all other parameters were used by their default value. The selection of suitable drugs was selected based on the minimum docked energy.
Protein–protein interactions
We used STRING database for generation of gene regulatory network of ATP4A of H. sapiens based on known and predicted protein interactions (http://string-db.org/). The protein–protein interactions include direct (physical) and indirect (functional) associations; these are derived from four sources: (1) genomic context, (2) high-throughput experiments, (3) conserved co-expression and (4) literature knowledge. In the STRING quantitatively integrates interaction data from these sources for a large number of organisms, and translates genetic information between these organisms. The database currently covers 5214234 proteins from 1133 organisms during protein–protein interaction in gene regulatory network.
Protein model identification number
The 3-D model of the α-subunit H+, K+-ATPase of H. sapiens was submitted to protein model database (http://mi.caspur.it/PMDB/) as PM0075582. It can be easily accessible for further experiments.
Results
Structural and functional analysis of H+/K+-ATPase
The primary transporter responsible for acidity of the stomach is H+, K+-ATPase, which is located on the apical surface of the cell. As shown in Fig. 1 the schematic representation of transport of H+ and Cl− ions into stomach by H+, K+-ATPase hydrolysis of ATP inhibited by Omeprazole drug for controlling the ulcer. We showed that the systematic approach for down-regulation of acid production in human lumen. ATPase hydrolyzed the ATP molecule into ADP as well as inorganic phosphate molecules and releases more hydrogen and chloride ions which combined together in the form of hydrochloric acid (HCl). Parietal cells secrete a proton (H+) into the lumen of the stomach; exchange the potassium ion (K+) into the parietal cells. The H+, K+-ATPase pumps against H+ concentration gradient of over 1 million: HCl is produced when the chloride ions (Cl−) passively flow out through the Cl− channels into the lumen for balancing the electroneutrality. The source of Cl− ions exchanges with Cl−/HCO3− and blood CO2 and H2O produce HCO3− and H+ through the enzyme carbonic anhydrase. The HCO3− is secreted into the interstitial fluid in exchange for Cl− ions into the parietal cell. Consequently, an alkaline tide can induce alkalosis in a person with extreme H+ secretion. Therefore, excess amount of HCl (acid) created gastric ulcer. Omeprazole drug is shown (Fig. 1) inhibition of ATPase activity for controlling the acid formation that can be used in treatment of ulcers.
Fig. 1.

Schematic representation of transport H+ and Cl− ions into stomach by H+ K+-ATPase hydrolysis of ATP inhibited by Omeprazole drug for control of ulcer
The homology of protein sequence of H+, K+-ATPase showed 63 % identity with X-ray crystal structure of pig renal Na+, K+-ATPase (3B8E) (Fig. 2). Total five 3-D of H+, K+-ATPase was generated using known crystal 3-D structure model (3B8E) and their free energies were evaluated. The free energy of H+, K+-ATPase model was nearly similar with 3-D structure of template. The 3-D model of H+, K+-ATPase of H. sapiens was shown (Fig. 3) and was observed being rich in α-helixes and the most favored Ramachandran plot (RP) (Supplementary Fig. S1). The favored amino acid residues in RP of 3-D model of H+, K+-ATPase was 84.4 % and disallowed region was 1.0 %. The overall G-factor was 2.8 and all these properties of 3-D model of H+, K+-ATPase shows a good quality model.
Fig. 2.

Alignment of H+ K+-ATPase sequences with template crystal structure 3B8E by Clustal X. Asterisk (*) indicates the identical amino acids
Fig. 3.
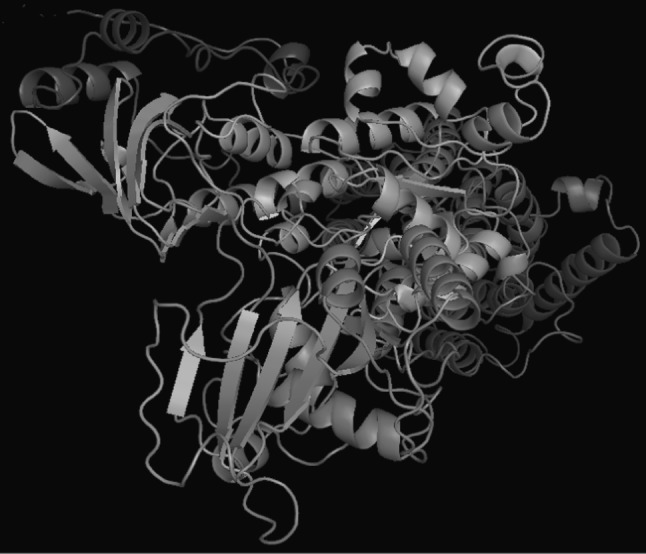
The 3-D model of H+, K+ ATPase of H. sapiens showing the α + β sheets
Atomic level interaction of potent proton pump inhibitors
The 3-D model of H+, K+-ATPase of H. sapiens was used for screening of suitable anti-ulcer drugs based on binding affinity. Lipinski’s rule of five (Ghose et al. 1999) was applied to narrow down search limiting the number of inhibitors by molecular docking. Physicochemical properties of drugs were given in Supplementary Table 1. The Omeprazole and Rabeprazole showed the highest binding affinity with active amino acid pockets in H+, K+-ATPase. The interaction of drugs with H+, K+-ATPase on the basis of several type of energies involved in molecular docking such as docking energy, inter molecular energy, torsional energy, RMS and internal energy which were given in Table 1. In this study, we performed ten dockings experiment with the generated 3-D model of H+, K+-ATPase and the lowest free energy of docked complex with hydrogen bonds was considered. The docking energy of Omeprazole and Rabeprazole were −19.00 and −17.04 kcal mol−1, respectively. However other drugs like, Perprazole, Losec and Omepral were shown comparatively less affinity with active amino acid pocket of 3-D model. The amino acids residues present in the 3-D model of H+, K+-ATPase during drug interaction using molecular docking strategies were shown (Supplementary Table 2).
Table 1.
The interaction energy of H+, K+-ATPase and drug obtained from molecular docking
| S. no. | Drugs | Binding energy (kcal mol−1) | Docked energy (kcal mol−1) | Inter molecular energy (kcal mol−1) | Torsional energy (kcal mol−1) | Internal energy (kcal mol−1) | RMSD (Ǻ) |
|---|---|---|---|---|---|---|---|
| 1. | Omeprazole | −19.80 | −19.00 | −20.5 | 0.62 | 1.5 | 58.13 |
| 2. | Perprazole | −16.89 | −5.89 | −19.07 | 2.18 | 13.18 | 62.96 |
| 3. | Rabeprazole | −16.65 | −17.04 | −18.21 | 1.56 | 1.17 | 52.6 |
| 4. | Losec | −12.76 | −6.99 | 13.69 | 0.93 | 6.7 | 59.06 |
| 5. | Omepral | −0.71 | −1.64 | −1.64 | 0.93 | 0.0 | 86.53 |
The active pocket of amino acid residues like Asp740, Ala739, Asn290, Asn371, Lys370, Leu736, and Asp758 in 3-D model of H+, K+-ATPase was found during Omeprazole interaction (Fig. 4). When drug binds with the amino acids of H+, K+-ATPase then only one hydrogen bond (HB) was formed between the UNK1:H–Asp758:OD1 atoms having 1.905 Ǻ distance (Supplementary Fig. 2). The Asp387, Asp728, Thr389, Ser750 and Ile747 amino acid residues in H+, K+ ATPase were observed in interaction of Rabeprazole drug (Fig. 5). As shown in Supplementary Fig. 3 stick-ball model of amino acid residue with the Rabeprazole drug during docking.
Fig. 4.

Interaction of high affinity potent Omeprazole with H+, K+ ATPase showing the hydrogen bonds
Fig. 5.

Interaction of high affinity potent Rabeprazole drug with amino acids in 3-D of H+, K+ ATPase
Analysis of ATP4A gene regulatory network
The cellular mechanism of ATP4A of H. Sapiens at gene network level is not well understood, consequently; we showed the interaction of ATP4A (present in centre of network) in cellular gene regulatory network and the interaction with other proteins/enzymes. The H+/K+ ATPase (α subunit with 1,035 aa) catalyzes the hydrolysis of ATP coupled with the exchange of H+ and K+ ions across the plasma membrane. It is responsible for acid production in the stomach. It seems like that, several other proteins are regulated the expression of ATP4A in cellular system. In the ATP4A gene regulatory network, we found the closely related proteins such as ATP4B, PPA1, LHPP, PPA2, FXYD5, APLP1, COX7A1 and CTCF with more than 0.702 score (Supplementary Table 3). We obtained a newly constructed gene regulatory network of H. Sapiens which would be useful for analysis of gene regulation of ATP4A that can help in fine tuning of proton pump and ion transport. The protein–protein interaction of ATP4A in H. Sapiens gene regulatory network was shown in Fig. 6. On the basis of these interactions, it is categorized into the three different clusters like, clusters I, II and III consequently, strong interaction of ATP4A was observed in between cluster I and cluster III proteins/enzymes.
Fig. 6.

Protein–protein interactions of ATPase (ATP4A) of H. sapiens with other cellular proteins and enzymes
Discussion
The glands of the stomach secrete hydrochloric (HCl) acid and protein digesting enzymes. The acid of stomach also serves to kill bacteria entering the organ. The H+, K+-ATPase is large family of related proteins that transport ions in all the biological membranes in each species. It transports one hydrogen ion (H+) from the cytoplasm of the parietal cell exchanges of potassium ion (K+) from the gastric lumen. The H+/K+ ATPase is able to transport ions against a concentration gradient using energy derived from the hydrolysis of ATP to ADP and phosphate helps to drive ion transport by conformational changes. In the present study, H+, K+-ATPase of H. sapiens was used for construction of 3-D model using 3-D X-ray crystal structure. The α-subunit of human H+, K+-ATPase is comprised of approximately 1,035 amino acid residues and arranged in 10 transmembrane segments (Jaunin et al. 1992; Shin et al. 2009).The 3-D model of aerolysin and hemolysin of Aeromonashydrophila have been generated through homology modeling (Singh et al. 2009; Singh et al. 2010). The H+, K+-ATPase in the E (2)-P predicted interaction of two known classes of specific inhibitors by modeling (Munson et al. 2005).
In this study, we obtained H+, K+-ATPase model with enriched α-helix and β-sheets which provides clue for proper functioning. In an earlier study, homology models have been generated of gastric H+/K+-ATPase reflecting the E1 and E2 conformations adopted by P-type ATPases in their catalytic cycle (Chang et al. 2005). The total 28 cysteine amino acids present in α-subunit of the gastric H+, K+-ATPase, in which 10 were found in the predicted transmembrane segments and their connecting loop, and 9 were presented in the β-subunit, and 6 were found in the form of disulfide linkage (Besancon et al. 1997).
The 3-D model of H+, K+-ATPase of H. sapiens was used for screening of suitable and potent anti-ulcer drugs based on their binding affinity. Similarly, inhibitor forms a disulfide bond with cysteine813 that is accessible from the luminal surface. This allows allocation of the binding site to a luminal vestibule adjacent to Cys813 enclosed by part of TM4 and the loop between TM5 and TM6. The K+ competitive imidazo-1, 2 alpha-pyridines also bind to the luminal surface of the E (2)-P conformation and their binding excludes the proton pump inhibitor reaction (Munson et al. 2005). The 2-methyl-8-(phenylmethoxy) imidazo [1, 2-a] pyridine-3-acetonitrile (SCH 28080) is a reversible inhibitor specific for the gastric proton pump. The binding sites of inhibitor on the 3-D structure of the gastric proton pump α-subunit have been demonstrated (Asano et al. 2004).
In the present study, Asp758 is strongly bound with Omeprazole via hydrogen bond (1.905 Å). The Omeprazole and Pantoprazole reacted with Cys813 in the intercellular M5–M6 region of the H+, K+-ATPase. The model suggests a mechanism for inhibitors interacting with enzyme by comparing the 3-D-structure of P-type ATPase (Yan et al. 2004; Kim et al. 2005). Lansoprazole has bound to cysteines residues of two domains and cysteine 321 toward the extracytoplasmic end of the third transmembrane segments. Pantoprazole bound only either cysteine 813 or 822 in the fifth and sixth transmembrane regions. The inhibition of Rabeprazole correlated with binding to active region of protein, but this compound continued to bind after full inhibition when binds with cysteines 321 and 892 amino acids (Besancon et al. 1997).
In this study, amino acid residues such as Asp740, Ala739, Asn290, Asn371, Lys370, Leu736 and Asp758 present in active pocket of 3-D model of H+, K+-ATPase and it was found during interaction with Omeprazole. Similarly, identification of the amino acids in the membrane domain found with SCH28080 in the crystal structure of the Ca-ATPase. They found total five conserved carboxylic residues which include Glu343, Glu795, Glu820, Asp824 and Glu936 whereas unique Lys791 was investigated in the H, K-ATPase (Vagin et al. 2001). The integrity of the SCH28080 binding site depends on the presence of Lys791, Glu936, and Glu795 in H+, K+-ATPase.
Both the proton and sodium pumps consist of a catalytic α-subunit and a glycosylated β-subunit that is essential for normal pump maturation and trafficking. Individual N-glycans linked to the β-subunits of the Na+, K+-ATPase and H+, K+-ATPase. These are important for stable membrane integration in the respective of α-subunits, folding, stability, subunit assembly; and enzymatic activity of the pumps. Importance of N-glycans for the maturation and quality control of the H+, K+-ATPase is greater than Na+, K+-ATPase. The roles of individual N-glycans of the β-subunits in the post-ER trafficking, membrane targeting and plasma membrane retention of the Na+, K+-ATPase and H+, K+-ATPase are different (Vagin et al. 2007). The acid transport by the gastric H+, K+-ATPase is covalently inhibited by several substitute pyridyl methylsulfinyl benzimidazoles and omeprazole (Lambrecht et al. 1998). Therefore, we require atomic level knowledge and understanding for targeting the combinatorial genetic network by designing of more potent and specific inhibitors.
In the ATP4A gene regulatory network, we found the several interactive proteins such as ATP4B, PPA1, LHPP, PPA2, FXYD5, APLP1, COX7A1 and CTCF. We showed combinatorial influence of these newly constructed genetic networks of H. Sapiens would be useful in gene regulation of ATP4A for controlling of acidity. In this study, protein–protein interactions of ATP4A in H. Sapiens gene regulatory network has been acknowledged. The ATP4A encodes H+/K+ ATPase α subunit with ~1,000-amino acids which contains the catalytic sites of the enzyme and forms the pore in cell membrane that allows the transport of ions. Whereas, ATP4B encodes the β subunit of H+/K+ ATPase, which is an ~300-amino acid protein with a 36-amino acid N-terminal cytoplasmic domain. The H+/K+ ATPase β subunit stabilizes the H+/K+ ATPase α subunit and is required for function of the enzyme. It appears to contain signals which direct the heterodimer to membrane destinations within the cell, although some of these signals are subordinate to signals found in H+/K+ ATPase α subunit (Shin et al. 2009).
The FXYD domain has been identified which containing ion transport regulator 5 that involved in down-regulation of E-cadherin; and also reduced the cell adhesion and promotes metastasis. The FXYD5 is a member of the FXYD family of single span type I membrane proteins. There are five members of this group have been shown to interact with Na+, K+-ATPase and to modulate its properties (Lubarski et al. 2005). It is structurally different from other family members and has been suggested to play a role in regulating E-cadherin and promoting metastasis (Ino et al. 2002). In our study, we found the interaction of CCCTC-binding factor (zinc finger protein) also known as chromatin binding factor which binds to DNA sequence at specific sites. It involves in transcriptional regulation by binding to chromatin insulators and preventing interaction between promoter and nearby enhancers and silencers. Identification of a cryptic upstream orchestrator of interferon-gamma (IFNG) transcription is embedded within human IL26 gene. It is compromised of a single CCCTC-binding factor (CTCF) retained in all mammals, even surviving near-complete evolutionary deletion of the homologous gene encoding IL-26 in rodents (Sekimata et al. 2009). NDRG is a family member-2 involves in dendritic cell and neuron differentiation which plays an anti-tumor activity. The DNAH8 is a dynein, axonemal, heavy chain 8 produced force towards the minus ends of microtubules. The Dynein has ATPase activity; the force-producing power stroke is thought to occur on release of ADP. It is involved in sperm motility that implicated in sperm flagellar assembly. The N-myc downstream-regulated gene 2 (NDRG2) is involved in tumor cell differentiation and apoptosis, but its function in tumor angiogenesis (Ma et al. 2012).
Gene regulatory network of multicopper oxidase and B subunit of DNA gyrase of A.hydrophila which showed the several cellular proteins that interacted during gene expression have been reported earlier (Singh et al. 2012a, b). The initial step is required to define protein–protein interactions in cellular systems for better understanding of biological complexity (De Las and Montanillo 2010). The dynamics of large scale genetic regulatory networks of cells are an important goal in systems and synthetic biology. The system level dynamical properties of the genetic network of Escherichiacoli that regulates its metabolism and show how its design leads to biologically useful cellular properties (Samal and Jain 2008). The protein–protein interactions occur when the two or several proteins/enzymes bind to each other frequently to carry out their biological function. There are 2,709 interactions between proteins of Saccharomycescerevisiae has been reported which facilitate the establishment of a single large network of 2,358 interactions among 1,548 proteins (Schwikowski et al. 2000). It is a new approach for targeting of any gene for fine tuning of cellular mechanism by knowing the predicted proteins/enzymes involves in gene regulatory network.
Conclusions
The present study was undertaken to generate the 3-D model of α-subunit (catalytic subunit) of H+, K+-ATPase from H. sapiens and virtual screening for finding of potent drugs. Omeprazole and Rabeprazole showed better binding affinity with other drugs. The study though provides the possible way of interactions between the drug and the protein and results obtain thus could be used for further studies to get better insight.
Construction of novel ATP4A gene regulatory network in H. sapiens plays a key role for better understanding of the gene regulation for fine tuning of proton pump and ion channels. In near future, it is an urgent need to evaluate ATP4A gene network through in vitro and in vivo experiments for better control gastric ulcers.
Electronic supplementary material
Acknowledgments
Authors are grateful to A. K. Singh, Satya Prakash and Pritee Singh for providing the suggestions, encouragement and fruitful discussion during preparation of manuscript. Authors appreciate the anonymous reviewers for their valuable comments and suggestions to improve the quality of the paper.
Conflict of interest
There is no competing interest.
References
- Altschul SF, Thomas LM, Alejandro AS, Jinghui Z, Zheng Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acid Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano S, Yoshida A, Yashiro H, Kobayashi Y, Morisato A, Ogawa H, Takeguchi N, Morii M. The cavity structure for docking the K(+)-competitive inhibitors in the gastric proton pump. J Biol Chem. 2004;279(14):13968–13975. doi: 10.1074/jbc.M308934200. [DOI] [PubMed] [Google Scholar]
- Besancon M, Simon A, Sachs G, Shin JM. Sites of reaction of the gastric H, K-ATPase with extracytoplasmic thiol reagents. J Biol Chem. 1997;272(36):22438–22446. doi: 10.1074/jbc.272.36.22438. [DOI] [PubMed] [Google Scholar]
- Carafoli E. The Ca2+ pump of the plasma membrane. J Biol Chem. 1992;267:2115–2118. [PubMed] [Google Scholar]
- Chang GK, Watts JA, Watts A. Ligand docking in the gastric H+/K+-ATPase: homology modeling of reversible inhibitor binding sites. J Med Chem. 2005;48(23):7145–7152. doi: 10.1021/jm050326o. [DOI] [PubMed] [Google Scholar]
- De Las Rivas J, Montanillo C. Protein-protein interactions essentials: key concepts to building and analyzing interactome networks. PLoS Comput Biol. 2010;6:e1000807. doi: 10.1371/journal.pcbi.1000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL (2002) The PyMOL molecular graphics system. DeLano Scientific, Palo Alto, CA
- Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. A qualitative and quantitative characterization of known drug databases. J Comp Chem. 1999;1:55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- Glynn IM, Karlish SJ. Occluded cations in active transport. Annu Rev Biochem. 1990;59:171–205. doi: 10.1146/annurev.bi.59.070190.001131. [DOI] [PubMed] [Google Scholar]
- Guha R, Howard MT, Hutchison GR, Murray-Rust P, Rzepa H, Steinbeck C, Wegner J, Willighagen EL. The Blue Obelisk-interoperability in chemical informatics. J Chem Inf Model. 2006;46:991–998. doi: 10.1021/ci050400b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ino Y, Gotoh M, Sakamoto M, Tsukagoshi K, Hirohashi S. Dysadherin, a cancer-associated cell membrane glycoprotein, down-regulates E-cadherin and promotes metastasis. Proc Natl Acad Sci USA. 2002;99:365–370. doi: 10.1073/pnas.012425299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaunin P, Horisberger JD, Richter K, Good PJ, Rossier BC, Geering K. Processing, intracellular transport, and functional expression of endogenous and exogenous alpha-beta 3 Na, K-ATPase complexes in Xenopus oocytes. J Biol Chem. 1992;267:577–585. [PubMed] [Google Scholar]
- Kim CG, Watts JA, Watts A. Ligand docking in the gastric H+/K+-ATPase: homology modeling of reversible inhibitor binding sites. J Med Chem. 2005;48(23):7145–7152. doi: 10.1021/jm050326o. [DOI] [PubMed] [Google Scholar]
- Kroemer RT, Doughty SW, Robinson AJ, Richards WG. Prediction of the three-dimensional structure of human interleukin-7 by homology modeling. Protein Eng. 1996;9(6):493–498. doi: 10.1093/protein/9.6.493. [DOI] [PubMed] [Google Scholar]
- Lambrecht N, Corbett Z, Bayle D, Karlish SJ, Sachs G. Identification of the site of inhibition by omeprazole of a alpha-beta fusion protein of the H, K-ATPase using site-directed mutagenesis. J Biol Chem. 1998;273(22):13719–13728. doi: 10.1074/jbc.273.22.13719. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. a program to check the stereochemical quality of protein structure. J Appl Crystallogr. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- Law RJ, Munson K, Sachs G, Lightstone FC. An ion gating mechanism of gastric H, K-ATPase based on molecular dynamics simulations. Biophys J. 2008;95(6):2739–2749. doi: 10.1529/biophysj.107.128025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubarski I, Pihakaski-Maunsbach K, Karlish SJ, Maunsbach AB, Garty H. Interaction with the Na, K-ATPase and tissue distribution of FXYD5 (related to ion channel) J Biol Chem. 2005;280(45):37717–37724. doi: 10.1074/jbc.M506397200. [DOI] [PubMed] [Google Scholar]
- Ma J, Liu W, Yan X, Wang Q, Zhao Q, Xue Y, Ren H, Wu L, Cheng Y, Li S, Miao L, Yao L, Zhang J. Inhibition of endothelial cell profilation and tumor angiogenesis by up-regulating NDRG2 expression in breast cancer cells. PLoS ONE. 2012;7(2):e32368. doi: 10.1371/journal.pone.0032368. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Melnichouk S, Friendship RM, Dewey CE, Bildfell R. Evaluation of lansoprazole (an H+/K+-ATPase inhibitor) and azithromycin (an antibiotic) for control of gastric ulceration in swine during periods of feed deprivation. Can J Vet Res. 1999;63:248–252. [PMC free article] [PubMed] [Google Scholar]
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comp Chem. 1998;19:16–39. doi: 10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B. [DOI] [Google Scholar]
- Morth JP, Pedersen BP, Toustrup-Jensen MS, Sorensen TLM, Petersen J, Andersen JP, Vilsen B, Nissen P. Crystal structure of the sodium–potassium pump. Nature. 2007;450:1043–1049. doi: 10.1038/nature06419. [DOI] [PubMed] [Google Scholar]
- Munson K, Garcia R, Sachs G. Inhibitor and ion binding sites on the gastric H, K-ATPase. Biochemistry. 2005;44(14):5267–5284. doi: 10.1021/bi047761p. [DOI] [PubMed] [Google Scholar]
- Munson K, Law RJ, Sachs G. Analysis of the gastric H, K ATPase for ion pathways and inhibitor binding sites. Biochemistry. 2007;46(18):5398–5417. doi: 10.1021/bi062305h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi S, editor. Australian medicines handbook 2006. Adelaide: Australian Medicines Handbook; 2006. [Google Scholar]
- Sachs G, Munson K, Balaji VN, Aures-Fischer D, Hersey SJ, Hall K. Functional domains of the gastric HK ATPase. J Bioenerg Biomembr. 1989;21(5):573–588. doi: 10.1007/BF00808114. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modeling by satisfaction of spatial restrains. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Samal A, Jain S. The regulatory network of E. coli metabolism as a Boolean dynamical system exhibits both homeostasis and flexibility of response. BMC Syst Biol. 2008;2:21. doi: 10.1186/1752-0509-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwikowski B, Uetz P, Fields S. A network of protein–protein interactions in yeast. Nat Biotechnol. 2000;18(12):1257–1261. doi: 10.1038/82360. [DOI] [PubMed] [Google Scholar]
- Sekimata M, Pérez-Melgosa M, Miller SA, Weinmann AS, Sabo PJ, Sandstrom R, Dorschner MO, Stamatoyannopoulos JA, Wilson CB. CCCTC-binding factor and the transcription factor T-bet orchestrate T helper 1 cell-specific structure and function at the interferon-gamma locus. Immunity. 2009;31(4):551–564. doi: 10.1016/j.immuni.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JM, Munson K, Vagin O, Sachs G. The gastric HK-ATPase: structure, function, and inhibition. Pflugers Arch. 2009;457(3):609–622. doi: 10.1007/s00424-008-0495-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V, Somvanshi P, Rathore G, Kapoor D, Mishra BN. Gene cloning, expression and homology modeling of hemolysin gene from Aeromonas hydrophila. Protein Expr Purif. 2009;65(1):1–7. doi: 10.1016/j.pep.2008.11.015. [DOI] [PubMed] [Google Scholar]
- Singh V, Somvanshi P, Rathore G, Kapoor D, Mishra BN. Gene Cloning, expression and characterization of recombinant aerolysin from Aeromonashydrophila. Appl Biochem Biotechnol. 2010;160(7):1985–1991. doi: 10.1007/s12010-009-8752-3. [DOI] [PubMed] [Google Scholar]
- Singh V, Mani I, Chaudhary DK. Analysis of the multicopper oxidase gene regulatory network of Aeromonas hydrophila. Syst Synth Biol. 2012;6:51–59. doi: 10.1007/s11693-012-9096-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V, Chaudhary DK, Mani I. Gene network analysis of Aeromonas hydrophila for novel drug target discovery. Syst Synth Biol. 2012;6:23–30. doi: 10.1007/s11693-012-9093-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slayman CL, Zuckier GR. Differential function properties of a P-type ATPase/proton pump. Ann NY Acad Sci. 1989;574:233–245. doi: 10.1111/j.1749-6632.1989.tb25162.x. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTALX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima C, Sasabe H, Stokes DL. Three-dimensional cryo-electron microscopy of the calcium ion pump in the sarcoplasmic reticulum membrane. Nature. 1993;362:467–471. doi: 10.1038/362469a0. [DOI] [PubMed] [Google Scholar]
- Tuukkanen J, Väänänen HK. Omeprazole, a specific inhibitor of H+–K+-ATPase, inhibits bone resorption in vitro. Calcif Tissue Int. 1986;38(2):123–125. doi: 10.1007/BF02556841. [DOI] [PubMed] [Google Scholar]
- Vagin O, Munson K, Lambrecht N, Karlish SJ, Sachs G. Mutational analysis of the K+-competitive inhibitor site of gastric H, K-ATPase. Biochemistry. 2001;40(25):7480–7490. doi: 10.1021/bi0105328. [DOI] [PubMed] [Google Scholar]
- Vagin O, Turdikulova S, Tokhtaeva E. Polarized membrane distribution of potassium-dependent ion pumps in epithelial cells: different roles of the N-glycans of their beta subunits. Cell Biochem Biophys. 2007;47(3):376–391. doi: 10.1007/s12013-007-0033-6. [DOI] [PubMed] [Google Scholar]
- Wallmark B, Jaresten BM, Larsson H, Ryberg B, Brandstrom A, Fellenius E. Differentiation among inhibitory actions of omeprazole, cimetidine, and SCN- on gastric acid secretion. Am J Physiol. 1983;245:G64–G71. doi: 10.1152/ajpgi.1983.245.1.G64. [DOI] [PubMed] [Google Scholar]
- Yan D, Hu YD, Li S, Chang MS. A model of 3D structure of H+/K+-ATPase catalytic subunit derived by homology modeling. Acta Pharmacol Sin. 2004;25(4):474–479. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.