

Summary
The bed nucleus of the stria terminalis (BST) plays a prominent role in brain integration of acute responses to stressful stimuli. This study tests the hypothesis that the BST plays a complementary role in regulation of physiological changes associated with chronic stress exposure. Male Sprague-Dawley rats received bilateral ibotenate lesions or sham lesions of the posterior medial region of the BST (BSTpm), an area known to be involved in inhibition of HPA axis responses to acute stress. Chronic stress was induced by 14-day exposure to twice daily stressors in an unpredictable sequence (chronic variable stress (CVS)). On the morning after the end of CVS, stressed and non-stressed controls were exposed to a novel restraint stress challenge. As previously documented, CVS caused adrenal hypertrophy, thymic involution, and attenuated body weight gain. None of these endpoints were affected by BSTpm lesions. Chronic stress exposure facilitated plasma corticosterone responses to the novel restraint stress and elevated CRH mRNA. Lesions of the BSTpm increased novel stressor-induced plasma ACTH and corticosterone secretion and enhanced c-fos mRNA induction in the paraventricular nucleus of the hypothalamus (PVN). In addition, lesion of the BSTpm resulted in an additive increase in CVS-induced facilitation of corticosterone responses and PVN CRH expression. Collectively these data confirm that the BSTpm inhibits HPA responses to acute stress, but do not strongly support an additional role for this region in limiting HPA axis responses to chronic drive. The data further suggest that acute vs. chronic stress integration are subserved by different brain circuitry.
Keywords: BST, corticotropin releasing hormone, vasopressin, paraventricular nucleus of the hypothalamus, corticosterone, ACTH
Introduction
Activation of the hypothalamic-pituitary-adrenal (HPA) axis is a critical component of the body’s stress response. The main output of this axis is the release of glucocorticoids from the adrenal cortex into the circulation, primarily to redistribute energy resources following a real or perceived threat to homeostasis (McEwen and Stellar, 1993). The release of glucocorticoids from the adrenal glands is stimulated by circulating adrenocorticotropic hormone (ACTH) released from the anterior pituitary. Upstream, ACTH release is driven by hypophysiotrophic secretagogues, including corticotropin releasing hormone (CRH) and arginine vasopressin (AVP), which are synthesized and released by neurons in the paraventricular nucleus of the hypothalamus (PVN).
During chronic stress, prolonged or repeated activation of the HPA axis can lead to long-term changes in HPA tone and responsiveness. In particular, chronic stress can lead to potentiated basal ACTH and/or corticosterone secretion, adrenal hypertrophy and elevated PVN CRH and AVP mRNA and protein expression (Hauger et al., 1988; Herman et al., 1995; Kiss and Aguilera, 1993; Ulrich-Lai et al., 2006b). Chronic exposure to homotypic stressors typically engender some degree of HPA axis habituation (Dhabhar et al., 1997; Odio and Brodish, 1989; Pitman et al., 1988; Viau and Sawchenko, 2002). However, despite the presence of an enhanced glucocorticoid feedback signals, HPA responses to new stressors are either maintained or augmented following chronic stress a process known as stress facilitation (Armario et al., 1985; Bhatnagar and Dallman, 1998; Bhatnagar and Vining, 2003; Dallman et al., 1992; Hauger et al., 1990; Kiss and Aguilera, 1993; Ostrander et al., 2006). Additionally, chronic stress is implicated in the dysregulation of glucocorticoid secretion that is associated with many disease states such as depression, post-traumatic stress disorder (PTSD) and other anxiety disorders (see (Gold and Chrousos, 2002)).
There is considerable evidence indicating that HPA axis reactivity to stress is influenced by a number of limbic forebrain regions, including the amygdala, hippocampus and prefrontal cortex (see (Herman et al., 2005)). These limbic regions have little direct input to the medial parvocellular PVN and most likely modulate the release of CRH and AVP through synaptic relay circuits. Anatomical studies indicate that the hippocampus and amygdala mainly relay signals by heavily innervating PVN-projecting basal forebrain and hypothalamic structures. Notably, the bed nucleus of the stria terminalis (BST) is among the main extrahypothalamic regions that receives abundant input from all of the noted limbic regions (Cullinan et al., 1993; Dong et al., 2001b; Dong and Swanson, 2004; Gu et al., 2003; Sawchenko and Swanson, 1983).
Several lines of evidence mark the BST as a major mediator of the HPA axis responses to stress (Casada and Dafny, 1991; Crane et al., 2003; Dunn, 1987; Feldman et al., 1990; Forray and Gysling, 2004; Gray et al., 1993; Zhu et al., 2001) raising the possibility that the BST may play a role in HPA axis disorders mediated by upstream limbic structures. The BST can be subdivided into a number of cytoarchitecturally distinct subnuclei with distinct afferent input and efferent targets (Swanson, 1998). In particular, the posterior medial area of the BST (BSTpm) which includes the principal nucleus, is known to be a major inhibitor of neuroendocrine responses to acute stress (Choi et al., 2007; Dunn, 1987; Herman et al., 1994). However, the role of the BSTpm in chronic stress regulation remains to be determined and may provide further understanding of how adaptations in limbic BST circuitry may play a role in stress-related disease states. Since our previous work had demonstrated strong inhibition of acute HPA responses by the BSTpm (Choi et al., 2007), the current studies test the hypothesis that the BSTpm is necessary for regulating neuroendocrine and physiological responses to chronic stress. To test this hypothesis, this study uses bilateral ibotenate lesions targeting the PVN-projecting BSTpm to assess the necessity of this region for mediating central and peripheral HPA axis responses to 14 days of chronic variable stress (CVS).
Materials and Methods
Animals
Forty-eight adult male Sprague Dawley rats (275–300g Harlan, Indianapolis, IN) were used for this study. All rats were housed three per cage in conventional shoebox rat cages. Food and water were available ad libitum in a temperature and humidity controlled vivarium, maintained on a 12/12-hr light/dark cycle (lights on at 0600 hr). Upon arrival, all rats acclimated to the animal facility for at least 7 days prior to surgery. Animals were maintained in accordance with the Guide for the Care and Use of Laboratory Animals (NIH, 1996). All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Cincinnati.
Ibotenate lesions
Rats were anesthetized by intraperitoneal injections of 87 mg/kg ketamine/13 mg/kg xylazine mixture. Preemptive analgesia was administered by subcutaneous injections of 260 μg/kg butorphanol (Torbugesic). Each rat was placed in a Kopf stereotaxic apparatus upon which skulls were exposed and burr holes were drilled at the calculated coordinates. Each rat received bilateral microinjections of ibotenate (0.5 μl per side, 4.0 μg / μl) in sterile phosphate-buffered saline (pH 7.4) or 0.9% sterile saline into the posterior BST (AP −0.8 mm, ML +/−1.3 mm, DV (dura) −6.5 mm), with coordinates calculated from bregma (Paxinos and Watson, 1998). Each microinjection used a 26 gauge 1 μl Hamilton injection syringe, mounted on the stereotaxic apparatus. Injectors were lowered to the DV coordinate over a 1 min period and left in place for 1 min prior to injection. The ibotenate or saline was manually infused over a total of 5 min at a rate of 0.05 μl / 30 s, followed by another 5 min waiting period to allow diffusion and minimize dorsal spread of injection up the needle track. The syringes were raised over a 1 min period. Skull burr holes were sealed with sterile bone wax and the skin closed with wound clips. Animals recovered for at least 7 days post-surgery prior to CVS exposure. Body weights were measured on the day of surgery, on Day 1 of CVS exposure, on Day 8 of CVS exposure, and on Day 15 following restraint stress challenge and just prior to sacrifice.
Chronic variable stress (CVS) protocol
Rats were randomly assigned to ‘sham no CVS’ (n=12), ‘sham CVS’ (n=12), ‘lesion no CVS’ (n = 12) and ‘lesion CVS’ (n = 12) groups, where ‘CVS’ rats were exposed to the CVS paradigm while ‘no CVS’ rats remained in their home cages as non-handled controls. The CVS paradigm consisted of twice daily exposure to alternating stressors along with occasional overnight stressors for 14 consecutive days (Days 1–14). Morning stressors were administered between 0930 hr and 1030 hr while afternoon stressors were conducted between 1430 hr and 1530 hr. Overnight stressors began immediately after cessation of afternoon stressors and ended with initiation of the next day’s morning stressor. CVS stressors consisted of hypoxia (30 min in 8% oxygen), cold stress (1 h at 4°C, 2 rats/cage without bedding), rotation stress (1 h at 100 rpm on a platform orbital shaker), warm swim (20 min at 31–33°C); cold swim (10 min at 16–18°C), overnight social isolation (1 rat/cage) and overnight social crowding (6 rats/cage). Stressors were presented in a randomized order with each stressor (except the overnight stressors) represented an equal number of times.
Acute restraint stress protocol
All rats received restraint stress between 0830–1030 hr on Day 15, the day after the cessation of CVS exposure. Animals were placed in well-ventilated Plexiglas restraint tubes and a tail clip blood sample (250–300 μl) was immediately collected to determine basal AM plasma ACTH and corticosterone titers. All rats remained in the restrainers for 20 min at which point another blood sample was collected. Animals were then released into their home cages to recover. An additional blood sample was taken at 40 min from the onset of the restraint stress. At 60 min from the onset of restraint, the rats were immediately sacrificed by decapitation and trunk blood was collected. Brains were removed, flash frozen in isopentane on dry ice (−45°C), and stored at −80°C. Adrenal and thymus glands were also collected and weighed.
Radioimmunoassay (RIA)
Plasma corticosterone levels were measured by RIA using 125I RIA kits from MP Biomedicals (Orangeburg, NY); plasma ACTH levels were measured by RIA using an antiserum donated by Dr. William Engeland (University of Minnesota) using 125I-labeled ACTH (Amersham Biosciences, Piscataway, NJ) as tracer (Jasper and Engeland, 1991). For each hormone, all plasma samples were analyzed within the same RIA.
Lesion verification by Nissl stain and NeuN immunohistochemistry
Brains were serially sectioned at 14 μm using a Microm cryostat, mounted onto charged glass slides and stored at −20°C. Lesion sites were verified by both Nissl staining of cells and neuronal nuclei (NeuN) immunolabeling as a neuronal marker. Sections were fixed in 4% paraformaldehyde and Nissl stained with cresyl violet, dehydrated through an ascending ethanol series and xylene, and coverslipped using DPX mountant. For NeuN immunohistochemistry, circumferences of tissue sections were surrounded by a hydrophobic slide marker using Super HT PAP pen (Research Products International). The tissue sections were fixed in 4% paraformaldehyde and extensively washed with 50mM potassium phosphate buffered-saline (KPBS) between each step. Subsequently, sections were incubated in 1% H2O2 for 10 min and blocked in incubation solution (4% normal goat serum and 0.3% Triton X-100 in KPBS) for one hour. Sections were incubated overnight with a monoclonal antibody against NeuN (1:1000; Chemicon International, Temecula, CA) with Parafilm coverslips, followed by one hour incubation in biotinylated donkey anti-goat IgG (1:500; Vector Laboratories, Burlingame, CA), and one hour incubation in avidin-horseradish peroxidase complex (1:500; ABC Elite Kit, Vector Laboratories), where each step was in incubation solution. Finally, sections were incubated for 5 min in 0.02% diaminobenzidine (DAB, Sigma-Aldrich, St. Louis, MO) resulting in a brown reaction product. Slides were dehydrated through an ascending ethanol series and xylene, and coverslipped using DPX mountant.
Lesions were identified by the location of the needle track, loss of neurons, and gliosis. Lesions targeting the BSTpm were confirmed as ‘hits’ when the primary damage included the principal nucleus of the BST, with moderate damage extending into the adjacent interfascicular and transverse nuclei of the BST. BSTpm lesions that were centered outside of the primary target (principal nucleus) were considered missed lesions. Rats with bilateral damage were included in the study, while rats with only partial unilateral lesions and/or missed lesions with damage outside of the BSTpm were removed from the analysis. The final ‘n’ for experimental groups following lesion verification were: ‘Sham No CVS’ (n=12), ‘Sham CVS’ (n=12), ‘Lesion No CVS’ (n = 8) and ‘Lesion CVS’ (n = 9).
In situ hybridization
Antisense AVP, CRH, and c-fos riboprobes were generated from linearized clones by in vitro transcription using 35S labeled UTP and the appropriate RNA polymerase as previously described in detail (Choi et al., 2007). Brain tissue slides were pretreated with 4% paraformaldehyde, acetylated, delipidized in chloroform, and dehydrated through a graded ethanol series. Each riboprobe was diluted (1.0 × 106 cpm / 50 μl buffer) in hybridization buffer (50% formamide, 1X Denhardt’s Solution, 10% dextran sulfate, 200 μg/ml fish sperm ssDNA, 100 μg/ml yeast tRNA, and 20 mM DTT), applied to slides of brain sections, coverslipped, and placed in hybridization chambers over blotting paper soaked in 50% formamide and incubated overnight at 55°C. The next day coverslips were removed and slides washed in 2X standard saline citrate (SSC). Slides were subsequently incubated in 100 μg/ml RNase A for 30 min at 37°C, washed numerous times in 0.2 × SSC, once in 0.2 × SSC for 1 hr at 65 °C, and finally dehydrated through a graded ethanol series.
Image Analysis
Hybridized slides were exposed on Kodak Biomax MR film (5 h for AVP, 7 days for CRH, and 14 days for c-fos). Film images of brain sections were captured by a digital video camera. Anatomical brain regions were identified using Swanson (Swanson, 1998), and Paxinos and Watson (Paxinos and Watson, 1998) rat brain atlases. Brain sections were matched for rostrocaudal level between rats for analyses. All brain regions and nuclei were clearly distinguishable by the specificity and intensity of AVP, CRH, and c-fos expression patterns. The parvocellular and magnocellular aspects of the PVN were identified using Nissl stained sections from a parallel series of tissue. Semi-quantitative analyses of autoradiograph images were performed using Scion Image (Scion, Frederick, MD) software and hybridization signal was expressed as gray level units. The gray level signal of a hybridized tissue region of interest was corrected by subtracting the gray level signal over a non-hybridized area of tissue (white matter, corpus callosum), and expressed as corrected gray level. 14C standards were also measured using Scion Image, and transferred to Assay Zap (Biosoft and P.L. Taylor) to generate a standard curve to verify that all measured gray levels were on the linear range of the film.
Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM). To take into account the differences in body weight, adjusted thymus and adrenal gland weights were calculated as organ weight (mg) per 100 g final body weight. AVP, CRH, and c-fos mRNA expression were expressed as corrected gray level. Integrated plasma ACTH and corticosterone were calculated as the total area under the curve (AUC) for the hormone time course of the restraint challenge. Organ weights, basal AM plasma hormones, integrated plasma hormones, and in situ hybridization data were analyzed by two-way factorial ANOVA (lesion, CVS) and significant main effects were further analyzed by Fisher’s Least Significant Difference (LSD) post hoc test. Body weight and time courses of plasma ACTH and corticosterone responses to restraint were analyzed by three-way ANOVA (lesion, CVS, time) with repeated measures (time) and significant main effects were further analyzed by Fisher’s Least Significant Difference (LSD) post hoc tests. Hormone and body weight data were analyzed using GBStat (Dynamic Microsystems Inc. Silver Spring, MD), while organ weight and mRNA data were analyzed using Statview (SAS Institute Inc., Cary, NC). Statistical significance was set at p < 0.05. Tests for homogeneity of variance were performed, and when necessary log transformations were used. Detection of outliers was performed using the Dixon-Massey method, and when necessary data were reanalyzed following outlier removal and/or transformations.
Results
Verification of lesions of the posterior medial BST
Bilateral ibotenate lesions targeted the principal nucleus of the BST. Lesion location and extent was verified by NeuN immunolabeling and Nissl stain (Figure 1). Confirmed BSTpm lesions were centered approximately 0.8 mm posterior to bregma according to the Paxinos and Watson atlas. The overall size of the lesions was relatively consistent but was not large enough to destroy the entire principal nucleus, usually sparing the ventral tip, while moderate damage was observed laterally in the interfascicular/transverse nuclei. All confirmed BSTpm lesions had minimal damage laterally into the striatum and spared any rostral damage into the anterior division of the BST. Our previous study (Choi et al., 2007) using similarly-placed lesions indicated extensive loss of GABAergic neurons in the region of the posterior medial BST, further corroborating the overall efficacy of this lesion approach.
Figure 1.
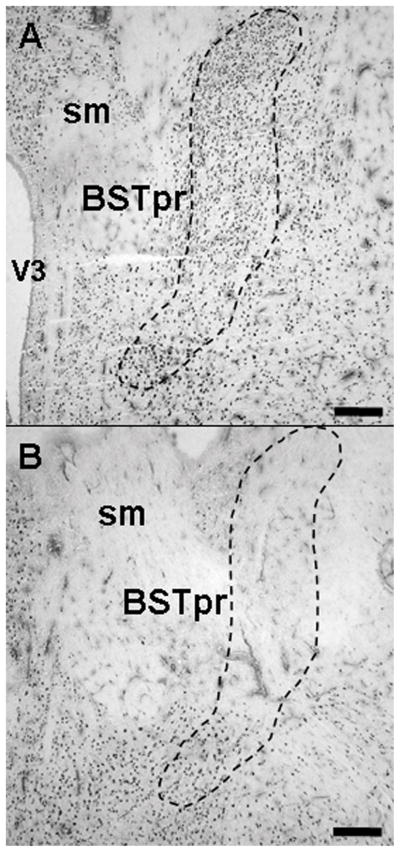
Brain tissue sections were immunolabeled for NeuN for analysis of the extent of lesions. Sham injection sites (A) targeting the posterior medial bed nucleus of the stria terminalis (BSTpm) did not cause significant neuronal cell loss, as noted by abundant NeuN staining in the region. In contrast, BSTpm lesions (B) typically damaged the vast majority of the BSTpm. Note that some staining was preserved in the ventral region of the BSTpm. Abbreviations: sm, stria medularis; V3, third ventricle. Scale bars = 300 μm. Schematic diagram (C) illustrates the extent of the spread of damage of the smallest (black shaded area) to the largest lesions (cross-hatched shaded area) confirmed as hits with primary damage of the BSTpm. Image adapted from Swanson’s Brain Maps: Structure of the Rat Brain, 2nd Edition.
Body and Organ Weights
Lesions of the BSTpm did not affect CVS-induced decreases in body weight gain. During the course of the study, analyses of body weight gain (Figure 2) indicate main effects of CVS (F1,167 = 26.28; p < 0.05) and day (F3,167 = 314.61; p < 0.05), and a CVS × day interaction (F3,167 = 63.41; p < 0.05). However, there was no effect of lesion on body weight gain and there were no other interactions. Post-hoc analyses revealed that all experimental groups gained body weight through the first day of CVS (Day 1) (p < 0.05). Upon exposure to chronic stress, ‘CVS’ groups gained less weight than their respective ‘No CVS’ groups on CVS Day 8 (p < 0.05) and CVS Day 15 (p < 0.05).
Figure 2.

Exposure to chronic variable stress (CVS) started on Day 1, and significantly decreased percent body weight gain on Days 8 and 15 regardless of lesions of the BSTpm. Data are presented as mean ± SEM, n=8–12 per group. * = p < 0.05 versus respective ‘No CVS’.
Similarly, BSTpm lesions did not affect stress induced adrenal hypertrophy or thymic atrophy. Whereas CVS caused increases in adrenal weights (Table 1) (F1,38 = 17.60; p < 0.05) and on adjusted adrenal weight/100 g body weight (Table 1) (F1,38 = 59.64; p < 0.05); there was no effect of lesion on adrenal weights and no lesion × CVS interaction. Chronic stress exposure also decreased thymus weight regardless of lesion (main effect of CVS on raw thymus weights (Table 1) (F1,38 = 13.83; p < 0.05), however, there was no effect on adjusted thymus weight/100 g body weight (Table 1)).
Table 1.
Adrenal and Thymus Weights Following BSTpm Lesions and Chronic Variable Stress
| Sham No CVS | Lesion No CVS | Sham CVS | Lesion CVS | |
|---|---|---|---|---|
| Raw Adrenal (mg) | 44.4 ± 1.4 | 45.2 ± 1.2 | 51.3 ± 1.9* | 51.9 ± 1.9* |
| Adjusted Adrenal (mg / 100g BW) | 12.5 ± 0.3 | 12.7 ± 0.4 | 15.7 ± 0.5* | 16.6 ± 0.6* |
| Raw Thymus (mg) | 371 ± 20 | 397 ± 25 | 317 ± 11* | 312 ± 14* |
| Adjusted Thymus (mg / 100g BW) | 104 ± 5.7 | 112 ± 6.7 | 97 ± 2.9 | 100 ± 4.8 |
Data are presented as mean ± SEM, n= 8–12 per group.
p < 0.05 versus respective No CVS.
Plasma ACTH and corticosterone responses to novel restraint challenge
Chronic stress caused slight but significant increases in resting AM plasma ACTH levels (significant main effect of CVS (F1,37 = 13.65; p < 0.05)) that was independent of lesion (Figure 3A). Exposure to a 20-min restraint stress increased plasma ACTH in all experimental groups (Figure 3B) (significant main effects of time (F3,167 = 95.96; p < 0.05) and lesion (F1,167 = 16.45; p < 0.05) as well as a lesion × time interaction (F3,167 = 16.51; p < 0.05) and CVS × time interaction (F3,167 = 3.92; p < 0.05)). Post-hoc analyses indicated that the posterior BST lesions potentiated plasma ACTH levels compared to respective sham control levels at the 20-min time point regardless of CVS exposure (p < 0.05). Exposure to CVS elevated ACTH levels at the 20-min time point of ‘CVS’ shams compared to ‘No CVS’ shams (p < 0.05), consistent with HPA axis facilitation (Armario et al., 1985; Bhatnagar and Dallman, 1998; Bhatnagar and Vining, 2003; Dallman et al., 1992; Hauger et al., 1990; Kiss and Aguilera, 1993a; Ostrander et al., 2006). Lesions enhanced the integrated plasma ACTH response to restraint (Figure 3C) (F1,33 = 13.55; p < 0.05) regardless of CVS exposure (p < 0.05).
Figure 3.
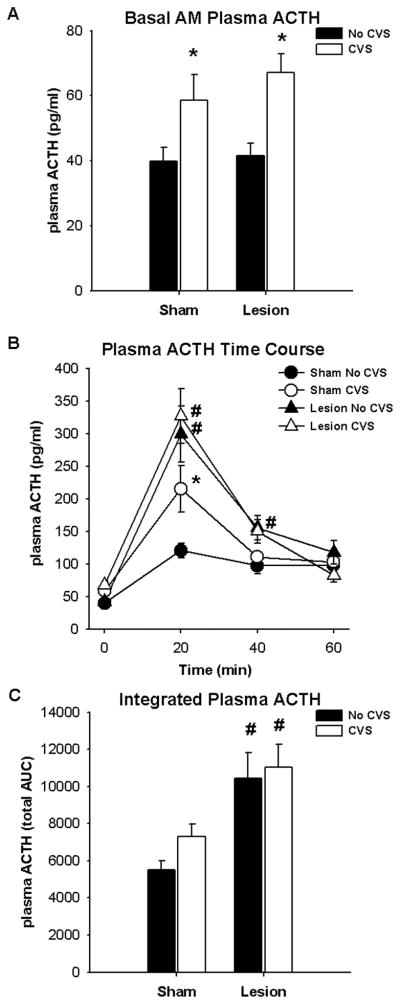
Exposure to CVS elevated basal AM plasma ACTH levels (A). Exposure to CVS also elevated the plasma ACTH response to acute restraint stress at 20 min following onset of restraint only in sham rats (B). Lesions of the BSTpm potentiated the plasma ACTH response to acute restraint stress at 20 min following onset of restraint regardless of CVS exposure, and at 40 min following onset of restraint only in rats with no exposure to CVS (B). Lesions of the BSTpm elevated the integrated plasma ACTH response regardless of CVS exposure (C). Data are presented as mean ± SEM, n = 8–12. * p < 0.05 versus respective ‘No CVS’, # p < 0.05 versus respective ‘Sham’. Abbreviations: AUC, area under the curve.
As was the case for ACTH, CVS resulted in elevated basal AM plasma corticosterone levels (Figure 4A) (F1,37 = 26.02; p < 0.05). Elevated basal plasma corticosterone was not affected by lesion. In contrast, plasma corticosterone responses to the 20-min novel restraint (Figure 4B) were affected by lesion (F1,167 = 25.88; p < 0.05) and CVS (F1,167 = 13.66; p < 0.05) and time (F3,167 = 302.19; p < 0.05), with a significant lesion × time interaction (F3,167 = 9.74; p < 0.05). Post-hoc analyses revealed that posterior BST lesions potentiated plasma corticosterone levels at the 20- and 40-min time points compared to respective sham control levels (p <0.05). Chronic stress also caused elevations in plasma corticosterone at the 40 min time point of lesion rats compared to lesion ‘No CVS’ rats (p <0.05), suggesting that CVS potentiated corticosterone secretion within the lesion group. For integrated plasma corticosterone levels (Figure 4C), there were significant main effects of lesion (F1,37 = 33.91; p < 0.05) and CVS (F1,37 = 14.90; p < 0.05), but there was no CVS × lesion interaction. Lesions elevated the integrated plasma corticosterone levels (p < 0.05), while CVS further increased integrated corticosterone levels within the lesion group (p < 0.05). Together with the time-course data, these data are consistent with an additive effect of lesions on chronic stress facilitation of corticosterone responses.
Figure 4.
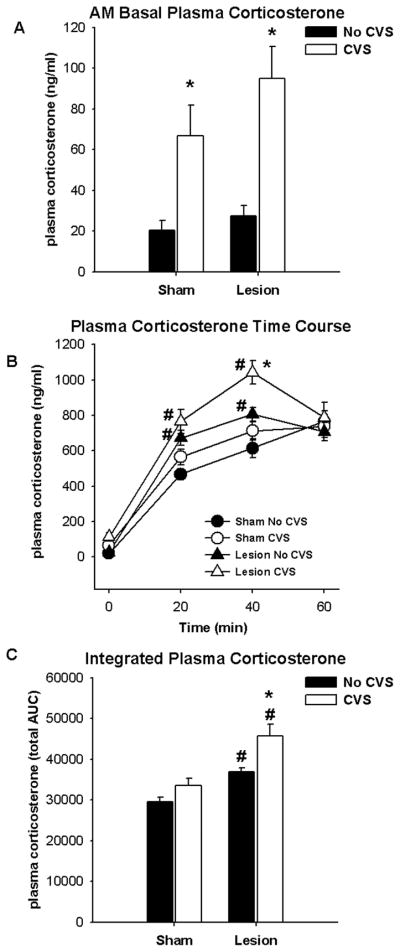
Exposure to CVS elevated basal AM plasma corticosterone levels (A). Lesions of the BSTpm potentiated plasma corticosterone response to restraint at 20 min and 40 min following onset of stress, regardless of CVS exposure (B). Exposure to CVS elevated the plasma corticosterone response at 40 min following onset of restraint only in rats with lesions. Lesions of the BSTpm enhanced the integrated plasma corticosterone response regardless of CVS exposure (C). Additionally, exposure to CVS elevated the integrated plasma corticosterone response only in rats with lesions (C). Data are presented as mean ± SEM, n = 8–12. * p < 0.05 versus respective ‘No CVS’, # p < 0.05 versus respective ‘Sham’. Abbreviations: AUC, area under the curve.
c-fos, CRH, and AVP mRNA expression in the PVN
Induction of c-fos mRNA was assessed as a measure of PVN neuronal activation by the novel restraint stress (Figure 5). Lesion significantly augmented c-fos mRNA induction in the PVN (F1,38 = 5.98; p < 0.05). However, there was no effect of CVS and no lesion × CVS interaction, indicating that c-fos mRNA induction was not sensitized by chronic stress in BST lesion rats. In fact, post-hoc analyses revealed that BST lesions potentiated PVN c-fos mRNA induction primarily in the absence of CVS exposure (p < 0.05).
Figure 5.

Semi-quantitative analysis revealed that BSTpm lesions elevated c-fos mRNA expression in the parventricular nucleus of the hypothalamus (PVN) only in rats with no exposure to CVS. Data are shown as mean ± SEM, n = 8–12. # p < 0.05 versus respective ‘Sham’.
In contrast, both lesion (F1,38 = 7.76; p < 0.05) and CVS (F1,38 = 9.65; p < 0.05) augmented CRH mRNA expression in the PVN (Figure 6). Post-hoc analyses revealed that CVS significantly elevated CRH mRNA expression in the PVN (p < 0.05). In addition, lesions of BSTpm caused a differential enhancement of PVN CRH mRNA expression in rats with prior chronic stress exposure (p < 0.05).
Figure 6.

Semi-quantitative analysis revealed that exposure to CVS elevated corticotropin releasing hormone (CRH) mRNA expression in the PVN regardless of lesions of the BSTpm. Lesions of the BSTpm elevated CRH mRNA expression in the PVN only in rats exposed to CVS. Data are shown as mean ± SEM, n = 8–12. * p < 0.05 versus respective ‘No CVS’, # p < 0.05 versus respective ‘Sham’. Abbreviations: PVNmp, medial parvocellular PVN.
There were no main effects of lesion or CVS on AVP mRNA expression in the parvocellular PVN (Figure 7), and there was no lesion × CVS interaction. Similarly, there were no differences among experimental groups in AVP mRNA levels in the magnocellular PVN or in the supraoptic nucleus (data not shown).
Figure 7.
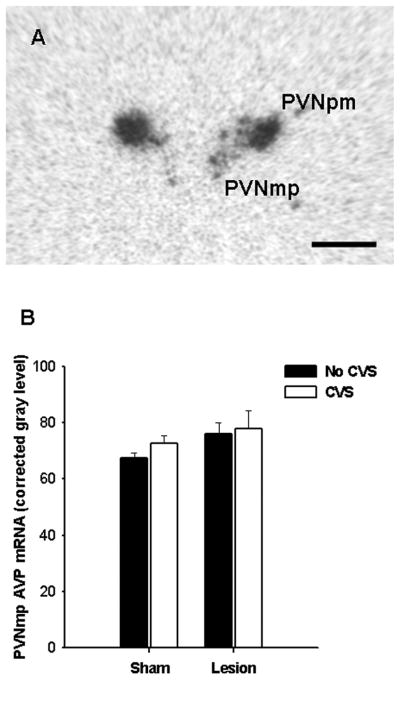
Semi-quantitative analyses revealed that neither exposure to CVS or lesions affected arginine vasopressin (AVP) mRNA expression in the PVN. Data are shown as mean ± SEM, n = 8–12. Abbreviations: PVNmp, medial parvocellular PVN.
Discussion
Consistent with our previous work, this study indicates that the BSTpm, which includes the principal nucleus of the BST, plays a major role in the inhibition of HPA axis reactivity to an acute stress challenge. The data replicate our previous findings that lesions of the posterior medial BST region enhance stress-induced PVN activation of c-fos mRNA and increase plasma ACTH and corticosterone responses to restraint stress (Choi et al., 2007). In addition, lesions of the posterior medial regions of the BST increase peak and integrated plasma corticosterone responses to novel restraint and enhance PVN CRH mRNA expression in chronically-stressed rats, lending further support to the conclusion that the BSTpm is necessary for normal inhibition of acute stress responses (Choi et al., 2007).
Exposure to CVS elevated peak and total plasma corticosterone response to novel restraint only in lesion rats. Whereas a trend was observed in the sham-lesion CVS group, post-hoc analysis did not reveal significant increases in corticosterone relative to respective non-CVS controls at any time point. Nonetheless, it should be noted that the results remain consistent with stress facilitation in this group, as previous studies note that maintained responsiveness reflects the ability to overcome the cumulative impact of chronic stress-induced increases in glucocorticoid exposure (Dallman et al., 1992). The magnitude of the facilitation effect in BSTpm lesion groups appears to be a summation of the effects of lesion and CVS, being additive rather than synergistic in nature. Thus, enhanced facilitation may be due to a lesion-induced increase in acute stress responding superimposed atop increases consequent to CVS, rather than being a separate product of chronic drive on central stress circuits.
Chronic stress exposure also elevated plasma ACTH levels in the sham rats, consistent with previous studies of chronic stress facilitation of responses to novel stressors (Dallman et al., 1992; Hauger et al., 1990). However, in contrast to the corticosterone data, CVS exposure did not further elevate the ACTH response in rats with lesions. The failure to observe facilitation of the ACTH response in lesion CVS rats is somewhat at odds with the corticosterone data. The disagreement between the plasma ACTH and corticosterone data raises the possibility that CVS is able to modulate pathways regulating the sensitivity of the adrenal to stress-induced pulses of ACTH. This possibility is supported by documented dissociations between the plasma ACTH and corticosterone responses in other chronic stress paradigms (Armario et al., 1985; Ostrander et al., 2006). There is evidence that the circadian corticosterone peak is mediated by increased sympathetic drive at the level of the adrenal (Ulrich-Lai et al., 2006a). Thus, it is possible that interruption of a general stress inhibitory circuit may enhance adrenal responsiveness via descending neural pathways modulating adrenocortical activation (Ulrich-Lai et al., 2006a). Alternatively, since the kinetics of the ACTH response are much more rapid than that for corticosterone, it is possible that sampling time points for ACTH were not optimal for detection of response sensitization. Overall, these data suggest that CVS may differentially affect stress responses at the level of the anterior pituitary vs. adrenal cortex.
Central HPA axis responses to novel restraint were also assessed using c-fos mRNA expression as an indirect marker of neuronal activation in the PVN (Cullinan et al., 1995; Imaki et al., 1992). c-fos mRNA is robustly induced in the PVN in response to restraint, with peak induction occurring near 30–60 min following stress onset (Cullinan et al., 1995; Figueiredo et al., 2003; Imaki et al., 1992). Importantly, c-fos mRNA is absent in the PVN in unstressed animals (Cullinan et al., 1995), reflecting the linkage between c-fos gene induction and neural stimulation. Consistent with our previous data (Choi et al., 2007), acute stress-induced c-fos mRNA in the PVN was significantly increased by lesions of the BSTpm only in rats with no prior exposure to CVS. However, the lesion-induced increase in PVN activation was absent in the CVS-exposed rats, suggesting an attenuation of c-fos mRNA induction by CVS. Regardless, there was no significant main effect of CVS exposure on c-fos induction in the PVN, which is in contrast with previous findings where habituation of PVN c-fos activation to restraint is observed in models of repeated restraint stress (Melia et al., 1994; Stamp and Herbert, 1999; Umemoto et al., 1994), and on occasion following CVS (Ulrich-lai et al 2007). This discrepancy may be associated with variations in time point sampling of c-fos mRNA or Fos protein, and may be related to response duration rather than peak level of Fos induction.
Based on our previous work investigating the effects of posterior medial BST lesions on acute stress, we predicted that a loss of HPA axis inhibition would generate greater HPA activity following each stress exposure during the two weeks of CVS, thereby resulting in exacerbated pathologies of chronic stress. In this study CVS exposure did evoke reduced body weight gain, adrenal enlargement, and thymic involution, as previously observed with this paradigm (Herman et al., 1995; Prewitt and Herman, 1997; Ulrich-Lai et al., 2006b). In addition, CVS exposure enhanced resting AM ACTH and corticosterone levels. However, none of these indices of chronic stress were affected by lesions of the BSTpm, suggesting that lesion rats were able to successfully adapt to the chronic stress regimen. Thus, while the BSTpm may regulate the magnitude of HPA responses to an acute stress challenge, they do not appear to be responsible for long-term adaptations to chronic stress.
It remains to be determined whether adaptations to chronic stress are regulated by individual executive regions or by multiple inputs from many areas of the brain. Possible regulators of chronic stress adaptation include regions such as the locus coeruleus, which exhibits enhanced norepinephrine sympathetic capacity following chronic stress (Pardon et al., 2003; Ziegler et al., 1999), and the posterior paraventricular thalamus, which appears to be responsible for chronic stress habituation of the HPA axis (Bhatnagar et al., 2000). In addition, there are data to support the existence of a general, CRH-driven network that can modulate responsiveness to repeated stimuli (Dallman et al., 2003).
Previous studies in our laboratory have also demonstrated that large lesions targeting the posterior region of the BST increase CRH mRNA expression in the PVN (Choi et al., 2007; Herman et al., 1994). The present study replicated the observed increases in PVN CRH mRNA expression following BSTpm lesion. In addition, CVS exposure augmented CRH mRNA only in lesion rats. These results are consistent with an additive effect of lesion and stress on production of CRH which may be relevant to the observed enhancement of corticosterone responses to chronic stress. The overall increase in expression of CRH mRNA is consistent with the enhanced response capacity seen in the posterior medial BST lesion group, and suggests that this enhancement in CRH expression is related to dynamic (i.e., stress-related HPA activation) rather than static (i.e., increases in steady-state corticosterone) HPA axis activity.
Previous work from our group demonstrated that BSTpm lesions also enhance AVP mRNA in the parvocellular PVN (Choi et al., 2007). However, changes in parvocellular AVP mRNA did not reach statistical significance in the current study. This discrepancy may be related to the difference in post-lesion survival time in the two studies (due to the additional 2 week stress period), or possibly to the increased variance associated with the inclusion of additional experimental groups in the design. In combination, the data suggest that changes in parvocellular AVP mRNA may not be a robust or enduring effect of BSTpm ablation.
Determinations of CRH and AVP mRNA were made in animals 60 min after initiation of restraint. Previous work from our group and others suggests that stress-induced elevations in CRH and AVP mRNA occur at relatively long post-stimulus latencies (> 60 minutes) following onset of restraint, for AVP, 90 min (Herman, 1995), and for CRH, 4 hours (Ma et al., 1997). Thus, our measures are highly likely to reflect baseline CRH and AVP mRNA expression.
The posterior medial BST receives input from numerous HPA axis-regulating limbic structures including the ventral subiculum, medial prefrontal cortex, and medial nucleus of the amygdala (Cullinan et al., 1993; Dong et al., 2001a; Dong and Swanson, 2004; Gu et al., 2003). The BSTpm is positioned to integrate emotional information from the basolateral amygdala and/or central nucleus of the amygdala into the stress response via the anterior lateral BST nuclei, which is known to regulate fear and anxiety-like behavior (see (Walker et al., 2003)). However it is unclear whether the posterior medial BST specifically regulates fear and anxiety responses. The excitatory effects of posterior medial BST lesions on HPA axis responses to acute stress are similar to those seen following lesions of the ventral subiculum (Herman et al., 1998) and medial prefrontal cortex (Figueiredo et al., 2003), raising the possibility that the BST may relay information from these structures to the PVN. In the case of the ventral subiculum, anatomical interconnections with the PVN via this portion of the BST have been previously established (Cullinan et al., 1993). However, it is important to note that the role of cortical and hippocampal regions in chronic stress regulation have yet to be established, thus it is unclear whether a partial role of the BST on HPA axis facilitation is mediated by these structures.
In conclusion, the BSTpm is an important inhibitor of stress responsivity to novel stimuli. However, the BSTpm does not appear to be responsible for changes in steady-state HPA activity or HPA axis facilitation that occur following chronic stress. The data suggest that acute vs. chronic response pathways are likely subserved by distinct brain circuits. The position of the BST at the interface of behavioral and neuroendocrine integration, suggests that this region may be critical for interpretation of stimuli relevant to stress-related disease states such as depression and PTSD. Therefore, the posterior medial BST is likely to be a major component of the brain circuitry involved in both normal and pathological stress adaptation.
Acknowledgments
This study was supported by MH49698 (JPH), DA16466 (MMO), DK67820 (YMU), NS07453 (NKE) and DK59803 (ARF).
Special thanks to Kenny Jones, Dr. Nancy Mueller, Ben Packard, Dr. Miyuki Tauchi, and Ingrid Thomas, for their technical assistance in this study. We would also like to thank Dr. Matthias Tschoep for the use of his microscopic and surgical equipment.
References
- Armario A, Restrepo C, Castellanos JM, Balasch J. Dissociation between adrenocorticotropin and corticosterone responses to restraint after previous chronic exposure to stress. Life Sci. 1985;36:2085–2092. doi: 10.1016/0024-3205(85)90304-2. [DOI] [PubMed] [Google Scholar]
- Bhatnagar S, Dallman M. Neuroanatomical basis for facilitation of hypothalamic-pituitary-adrenal responses to a novel stressor after chronic stress. Neuroscience. 1998;84:1025–1039. doi: 10.1016/s0306-4522(97)00577-0. [DOI] [PubMed] [Google Scholar]
- Bhatnagar S, Viau V, Chu A, Soriano L, Meijer OC, Dallman MF. A cholecystokinin-mediated pathway to the paraventricular thalamus is recruited in chronically stressed rats and regulates hypothalamic-pituitary-adrenal function. J Neurosci. 2000;20:5564–5573. doi: 10.1523/JNEUROSCI.20-14-05564.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar S, Vining C. Facilitation of hypothalamic-pituitary-adrenal responses to novel stress following repeated social stress using the resident/intruder paradigm. Horm Behav. 2003;43:158–165. doi: 10.1016/s0018-506x(02)00011-9. [DOI] [PubMed] [Google Scholar]
- Casada JH, Dafny N. Restraint and stimulation of the bed nucleus of the stria terminalis produce similar stress-like behaviors. Brain Res Bull. 1991;27:207–212. doi: 10.1016/0361-9230(91)90069-v. [DOI] [PubMed] [Google Scholar]
- Choi DC, Furay AR, Evanson NK, Ostrander MM, Ulrich-Lai YM, Herman JP. Bed nucleus of the stria terminalis subregions differentially regulate hypothalamic-pituitary-adrenal axis activity: implications for the integration of limbic inputs. J Neurosci. 2007;27:2025–2034. doi: 10.1523/JNEUROSCI.4301-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane JW, Buller KM, Day TA. Evidence that the bed nucleus of the stria terminalis contributes to the modulation of hypophysiotropic corticotropin-releasing factor cell responses to systemic interleukin-1beta. J Comp Neurol. 2003;467:232–242. doi: 10.1002/cne.10918. [DOI] [PubMed] [Google Scholar]
- Cullinan WE, Herman JP, Battaglia DF, Akil H, Watson SJ. Pattern and time course of immediate early gene expression in rat brain following acute stress. Neuroscience. 1995;64:477–505. doi: 10.1016/0306-4522(94)00355-9. [DOI] [PubMed] [Google Scholar]
- Cullinan WE, Herman JP, Watson SJ. Ventral subicular interaction with the hypothalamic paraventricular nucleus: evidence for a relay in the bed nucleus of the stria terminalis. J Comp Neurol. 1993;332:1–20. doi: 10.1002/cne.903320102. [DOI] [PubMed] [Google Scholar]
- Dallman MF, Akana SF, Scribner KA, Bradbury MJ, Walker CD, Strack AM, Cascio CS. Stress, feedback and facilitation in the hypothalamo-pituitary-adrenal axis. Journal of Neuroendocrinology. 1992;4:517–526. doi: 10.1111/j.1365-2826.1992.tb00200.x. [DOI] [PubMed] [Google Scholar]
- Dallman MF, Pecoraro N, Akana SF, La Fleur SE, Gomez F, Houshyar H, Bell ME, Bhatnagar S, Laugero KD, Manalo S. Chronic stress and obesity: a new view of “comfort food”. Proc Natl Acad Sci U S A. 2003;100:11696–11701. doi: 10.1073/pnas.1934666100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhabhar FS, McEwen BS, Spencer RL. Adaptation to prolonged or repeated stress--comparison between rat strains showing intrinsic differences in reactivity to acute stress. Neuroendocrinology. 1997;65:360–368. doi: 10.1159/000127196. [DOI] [PubMed] [Google Scholar]
- Dong HW, Petrovich GD, Swanson LW. Topography of projections from amygdala to bed nuclei of the stria terminalis. Brain Res Brain Res Rev. 2001a;38:192–246. doi: 10.1016/s0165-0173(01)00079-0. [DOI] [PubMed] [Google Scholar]
- Dong HW, Petrovich GD, Watts AG, Swanson LW. Basic organization of projections from the oval and fusiform nuclei of the bed nuclei of the stria terminalis in adult rat brain. J Comp Neurol. 2001b;436:430–455. doi: 10.1002/cne.1079. [DOI] [PubMed] [Google Scholar]
- Dong HW, Swanson LW. Projections from bed nuclei of the stria terminalis, posterior division: implications for cerebral hemisphere regulation of defensive and reproductive behaviors. J Comp Neurol. 2004;471:396–433. doi: 10.1002/cne.20002. [DOI] [PubMed] [Google Scholar]
- Dunn JD. Plasma corticosterone responses to electrical stimulation of the bed nucleus of the stria terminalis. Brain Res. 1987;407:327–331. doi: 10.1016/0006-8993(87)91111-5. [DOI] [PubMed] [Google Scholar]
- Feldman S, Conforti N, Saphier D. The preoptic area and bed nucleus of the stria terminalis are involved in the effects of the amygdala on adrenocortical secretion. Neuroscience. 1990;37:775–779. doi: 10.1016/0306-4522(90)90107-f. [DOI] [PubMed] [Google Scholar]
- Figueiredo HF, Bruestle A, Bodie B, Dolgas CM, Herman JP. The medial prefrontal cortex differentially regulates stress-induced c-fos expression in the forebrain depending on type of stressor. Eur J Neurosci. 2003;18:2357–2364. doi: 10.1046/j.1460-9568.2003.02932.x. [DOI] [PubMed] [Google Scholar]
- Forray MI, Gysling K. Role of noradrenergic projections to the bed nucleus of the stria terminalis in the regulation of the hypothalamic-pituitary-adrenal axis. Brain Res Brain Res Rev. 2004;47:145–160. doi: 10.1016/j.brainresrev.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Gold PW, Chrousos GP. Organization of the stress system and its dysregulation in melancholic and atypical depression: high vs low CRH/NE states. Mol Psychiatry. 2002;7:254–275. doi: 10.1038/sj.mp.4001032. [DOI] [PubMed] [Google Scholar]
- Gray TS, Piechowski RA, Yracheta JM, Rittenhouse PA, Bethea CL, Van de Kar LD. Ibotenic acid lesions in the bed nucleus of the stria terminalis attenuate conditioned stress-induced increases in prolactin, ACTH and corticosterone. Neuroendocrinology. 1993;57:517–524. doi: 10.1159/000126400. [DOI] [PubMed] [Google Scholar]
- Gu G, Cornea A, Simerly RB. Sexual differentiation of projections from the principal nucleus of the bed nuclei of the stria terminalis. J Comp Neurol. 2003;460:542–562. doi: 10.1002/cne.10677. [DOI] [PubMed] [Google Scholar]
- Hauger RL, Lorang M, Irwin M, Aguilera G. CRF receptor regulation and sensitization of ACTH responses to acute ether stress during chronic intermittent immobilization stress. Brain Res. 1990;532:34–40. doi: 10.1016/0006-8993(90)91738-3. [DOI] [PubMed] [Google Scholar]
- Hauger RL, Millan MA, Lorang M, Harwood JP, Aguilera G. Corticotropin-releasing factor receptors and pituitary adrenal responses during immobilization stress. Endocrinology. 1988;123:396–405. doi: 10.1210/endo-123-1-396. [DOI] [PubMed] [Google Scholar]
- Herman JP. In situ hybridization analysis of vasopressin gene transcription in the paraventricular and supraoptic nuclei of the rat: regulation by stress and glucocorticoids. J Comp Neurol. 1995;363(1):15–27. doi: 10.1002/cne.903630103. [DOI] [PubMed] [Google Scholar]
- Herman JP, Adams D, Prewitt C. Regulatory changes in neuroendocrine stress-integrative circuitry produced by a variable stress paradigm. Neuroendocrinology. 1995;61:180–190. doi: 10.1159/000126839. [DOI] [PubMed] [Google Scholar]
- Herman JP, Cullinan WE, Watson SJ. Involvement of the bed nucleus of the stria terminalis in tonic regulation of paraventricular hypothalamic CRH and AVP mRNA expression. J Neuroendocrinol. 1994;6:433–442. doi: 10.1111/j.1365-2826.1994.tb00604.x. [DOI] [PubMed] [Google Scholar]
- Herman JP, Dolgas CM, Carlson SL. Ventral subiculum regulates hypothalamo-pituitary-adrenocortical and behavioural responses to cognitive stressors. Neuroscience. 1998;86:449–459. doi: 10.1016/s0306-4522(98)00055-4. [DOI] [PubMed] [Google Scholar]
- Herman JP, Ostrander MM, Mueller NK, Figueiredo H. Limbic system mechanisms of stress regulation: hypothalamo-pituitary-adrenocortical axis. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:1201–1213. doi: 10.1016/j.pnpbp.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Imaki T, Shibasaki T, Hotta M, Demura H. Early induction of c-fos precedes increased expression of corticotropin-releasing factor messenger ribonucleic acid in the paraventricular nucleus after immobilization stress. Endocrinology. 1992;131:240–246. doi: 10.1210/endo.131.1.1612001. [DOI] [PubMed] [Google Scholar]
- Jasper MS, Engeland WC. Synchronous ultradian rhythms in adrenocortical secretion detected by microdialysis in awake rats. Am J Physiol. 1991;261:R1257–1268. doi: 10.1152/ajpregu.1991.261.5.R1257. [DOI] [PubMed] [Google Scholar]
- Kiss A, Aguilera G. Regulation of the hypothalamic pituitary adrenal axis during chronic stress: Responses to repeated intraperitoneal hypertonic saline injection. Brain Res. 1993;630:262–270. doi: 10.1016/0006-8993(93)90665-a. [DOI] [PubMed] [Google Scholar]
- Ma XM, Levy A, Lightman SL. Rapid changes in heteronuclear RNA for corticotrophin-releasing hormone and arginine vasopressin in response to acute stress. J Endocrinol. 1997;152(1):81–89. doi: 10.1677/joe.0.1520081. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Stellar E. Stress and the individual. Mechanisms leading to disease. Arch Intern Med. 1993;153:2093–2101. [PubMed] [Google Scholar]
- Melia KR, Ryabinin AE, Schroeder R, Bloom FE, Wilson MC. Induction and habituation of immediate early gene expression in rat brain by acute and repeated restraint stress. J Neurosci. 1994;14:5929–5938. doi: 10.1523/JNEUROSCI.14-10-05929.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odio M, Brodish A. Age-related adaptation of pituitary-adrenocortical responses to stress. Neuroendocrinology. 1989;49:382–388. doi: 10.1159/000125142. [DOI] [PubMed] [Google Scholar]
- Ostrander MM, Ulrich-Lai YM, Choi DC, Richtand NM, Herman JP. Hypoactivity of the hypothalamo-pituitary-adrenocortical axis during recovery from chronic variable stress. Endocrinology. 2006;147:2008–2017. doi: 10.1210/en.2005-1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardon MC, Ma S, Morilak DA. Chronic cold stress sensitizes brain noradrenergic reactivity and noradrenergic facilitation of the HPA stress response in Wistar Kyoto rats. Brain Res. 2003;971:55–65. doi: 10.1016/s0006-8993(03)02355-2. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 4. San Diego: Academic Press; 1998. [DOI] [PubMed] [Google Scholar]
- Pitman DL, Ottenweller JE, Natelson BH. Plasma corticosterone levels during repeated presentation of two intensities of restraint stress: chronic stress and habituation. Physiol Behav. 1988;43:47–55. doi: 10.1016/0031-9384(88)90097-2. [DOI] [PubMed] [Google Scholar]
- Prewitt CM, Herman JP. Hypothalamo-Pituitary-Adrenocortical Regulation Following Lesions of the Central Nucleus of the Amygdala. Stress. 1997;1:263–280. doi: 10.3109/10253899709013746. [DOI] [PubMed] [Google Scholar]
- Sawchenko PE, Swanson LW. The organization of forebrain afferents to the paraventricular and supraoptic nuclei of the rat. J Comp Neurol. 1983;218:121–144. doi: 10.1002/cne.902180202. [DOI] [PubMed] [Google Scholar]
- Stamp JA, Herbert J. Multiple immediate-early gene expression during physiological and endocrine adaptation to repeated stress. Neuroscience. 1999;94:1313–1322. doi: 10.1016/s0306-4522(99)00368-1. [DOI] [PubMed] [Google Scholar]
- Swanson LW. Brain maps: structure of the rat brain. 2. Amsterdam: Elsevier; 1998. [Google Scholar]
- Ulrich-Lai YM, Arnhold MM, Engeland WC. Adrenal splanchnic innervation contributes to the diurnal rhythm of plasma corticosterone in rats by modulating adrenal sensitivity to ACTH. Am J Physiol Regul Integr Comp Physiol. 2006a;290(4):R1128–35. doi: 10.1152/ajpregu.00042.2003. [DOI] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Figueiredo HF, Ostrander MM, Choi DC, Engeland WC, Herman J. Chronic stress induces adrenal hyperplasia and hypertrophy in a subregion-specific manner. Am J Physiol Endocrinol Metab. 2006b doi: 10.1152/ajpendo.00070.2006. [DOI] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Ostrander MM, Thomas IM, Packard BA, Furay AR, Dolgas CM, Van Hooren DC, Figueiredo HF, Mueller NK, Choi DC, Herman JP. Daily limited access to sweetened drink attenuates hypothalamic-pituitary-adrenocortical axis stress responses. Endocrinology. 2007 doi: 10.1210/en.2006-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemoto S, Noguchi K, Kawai Y, Senba E. Repeated stress reduces the subsequent stress-induced expression of Fos in rat brain. Neurosci Lett. 1994;167:101–104. doi: 10.1016/0304-3940(94)91037-5. [DOI] [PubMed] [Google Scholar]
- Viau V, Sawchenko PE. Hypophysiotropic neurons of the paraventricular nucleus respond in spatially, temporally, and phenotypically differentiated manners to acute vs. repeated restraint stress: rapid publication. J Comp Neurol. 2002;445:293–307. doi: 10.1002/cne.10178. [DOI] [PubMed] [Google Scholar]
- Walker DL, Toufexis DJ, Davis M. Role of the bed nucleus of the stria terminalis versus the amygdala in fear, stress, and anxiety. Eur J Pharmacol. 2003;463(1–3):199–216. doi: 10.1016/s0014-2999(03)01282-2. [DOI] [PubMed] [Google Scholar]
- Zhu W, Umegaki H, Suzuki Y, Miura H, Iguchi A. Involvement of the bed nucleus of the stria terminalis in hippocampal cholinergic system-mediated activation of the hypothalamo--pituitary--adrenocortical axis in rats. Brain Res. 2001;916:101–106. doi: 10.1016/s0006-8993(01)02871-2. [DOI] [PubMed] [Google Scholar]
- Ziegler DR, Cass WA, Herman JP. Excitatory influence of the locus coeruleus in hypothalamic-pituitary-adrenocortical axis responses to stress. J Neuroendocrinol. 1999;11:361–369. doi: 10.1046/j.1365-2826.1999.00337.x. [DOI] [PubMed] [Google Scholar]