

Summary
DNA mismatch repair (MMR) ensures replication fidelity by correcting mismatches generated during DNA replication. Although human MMR has been reconstituted in vitro, how MMR occurs in vivo is unknown. Here, we show that an epigenetic histone mark, H3K36me3, is required in vivo to recruit the mismatch recognition protein hMutSα (hMSH2-hMSH6) onto chromatin through direct interactions with the hMSH6 PWWP domain. The abundance of H3K36me3 in G1 and early S phases ensures that hMutSα is enriched on chromatin before mispairs are introduced during DNA replication. Cells lacking the H3K36 tri-methyltransferase SETD2 display microsatellite instability (MSI) and an elevated spontaneous mutation frequency, characteristic of MMR-deficient cells. This work reveals that a histone mark regulates MMR in human cells and explains the long-standing puzzle of MSI-positive cancer cells that lack detectable mutations in known MMR genes.
Introduction
DNA mismatch repair (MMR) maintains genome stability primarily by correcting base- base and small insertion/deletion (ID) mispairs generated during DNA replication (Kolodner, 1996; Kunkel and Erie, 2005; Li, 2008; Modrich and Lahue, 1996). In human cells, these mispairs are recognized by hMSH2-hMSH6 (hMutSα) and hMSH2-hMSH3 (hMutSβ). Normally, cells express more hMSH6 than hMSH3, leading to a hMutSα:hMutSβ ratio of ~10:1 (Drummond et al., 1997; Marra et al., 1998). Despite their redundant activities in mismatch recognition, both complexes are required for MMR, and defective or abnormal expression of hMSH6 or hMSH3 leads to a mutator phenotype (Drummond et al., 1997; Drummond et al., 1995; Harrington and Kolodner, 2007; Marsischky et al., 1996). Previous studies have shown that genetic and epigenetic modifications that impair the expression of these and other MMR genes, especially hMSH2, hMSH6 and hMLH1, cause susceptibility to certain types of cancer, including hereditary non-polyposis colorectal cancer (HNPCC) (Fishel and Kolodner, 1995; Kane et al., 1997; Modrich and Lahue, 1996).
At the cellular level, defects in MMR cause a mutator phenotype, which can be readily detected in eukaryotic cells as instability in simple repetitive DNA sequences called microsatellites. Thus, microsatellite instability (MSI) is regarded as a hallmark of MMR deficiency (Kolodner, 1996; Kunkel and Erie, 2005; Li, 2008; Modrich and Lahue, 1996). However, a significant fraction of MSI-positive colorectal cancers express MMR genes at normal levels and do not carry a detectable mutation in or hypermethylation of known MMR genes (Peltomaki, 2003). Similarly, certain non-colorectal cancer cells with MSI also appear to be proficient in MMR (Gu et al., 2002; Wang et al., 2011). The molecular mechanism underlying MSI in these cases is obscure.
The MMR capacity of mammalian cells has typically been evaluated using a functional assay that measures in vitro repair of a naked model DNA heteroduplex (Holmes et al., 1990; Thomas et al., 1991; Zhang et al., 2005). This assay has helped identify MMR defects in HNPCC and other MSI-positive cancers (Parsons et al., 1993; Umar et al., 1994) and has been invaluable in characterizing the MMR pathway in human cells in great molecular detail (reviewed by (Li, 2008)). However, increasing evidence suggests that a mismatch assembled into nucleosomes is a poor substrate for the in vitro MMR system. Li et al. (Li et al., 2009) showed that nucleosomes derived from recombinant histones and a mismatch-containing DNA diminished the mismatch binding, ATPase and DNA sliding activities of hMutSα, which are required for MMR. Schopf et al. (Schopf et al., 2012) demonstrated that hMutSα failed to restore MMR to an hMSH6-deficient nuclear extract when DNA heteroduplexes were assembled into nucleosomes by preincubating with the extract. These observations suggest that additional factors and/or mechanisms are needed for MMR in vivo, possibly by disrupting nucleosomes or timely recruiting MMR proteins, or both. Consistent with this hypothesis, histone modifications and chromatin remodeling factors have been implicated in MMR (Javaid et al., 2009; Kadyrova et al., 2011) and MMR has been show to couple with DNA replication (Hombauer et al., 2011a; Simmons et al., 2008), during which nucleosomes are disrupted. More strikingly, the hMSH6 subunit of hMutSα contains a Pro-Trp-Trp-Pro (PWWP) domain (Laguri et al., 2008) and this domain, which is present in many chromatin-associated proteins, was recently identified as a ‘reader’ of trimethylated Lys36 in histone 3 (H3K36me3) (Dhayalan et al., 2010; Vermeulen et al., 2010; Vezzoli et al., 2010). However, it is not yet known whether the H3K36me3 mark plays a role in MMR.
Here, we demonstrate that H3K36me3 interacts specifically with the hMSH6 PWWP domain of hMutSα in vitro and in vivo, and that the histone methyltransferase SETD2, which is responsible for trimethylation of H3K36 (Edmunds et al., 2008), is required for human MR in vivo. Consistent with this, cells depleted of SETD2 and H3K36me3 display a mutator phenotype characterized by MSI and an elevated mutation frequency at the HPRT locus. The data presented here strongly suggest that the H3K36me3 histone mark regulates human MMR in vivo by recruiting hMutSα onto chromatin to be replicated. We therefore propose that the status of H3K36me3 in a specific gene or intergenic region could potentially influence the local mutation rate in that region of the chromosome.
Results
The hMSH6 PWWP domain interacts with H3K36me3 and is essential for hMutSα binding to chromatin
The hMSH6 subunit of hMutSα contains a PWWP domain (Laguri et al., 2008) and this conserved domain has recently been proposed to interact specifically with H3K36me3 (Dhayalan et al., 2010; Vermeulen et al., 2010; Vezzoli et al., 2010). Figure 1A shows an alignment of the hMSH6 PWWP domain with 5 other PWWP domains, including that of BRPF1, the only PWWP domain for which an atomic resolution structure of the complex with H3K36me3 is available (Vezzoli et al., 2010; Wu et al., 2011). Interestingly, the co-crystal structures of the BRPF1 PWWP and H3K36me3 peptide (Vezzoli et al., 2010; Wu et al., 2011) indicate that three residues in the PWWP domain form an aromatic cage surrounding the H3K36me3 (Figure 1B, left). Consistent with this, our alignment of PWWP domains shows that the proposed ‘cage’ residues are highly conserved (Figure 1A, blue dots). Based on these data, we generated a model of hMSH6 bound to the H3K36me3 peptide (Figure 1B, right) by superimposing the PWWP domains of hMSH6 and BRPF1 (Laguri et al., 2008).
Figure 1. The PWWP domain and structural models for interaction with H3K36me3.

(A) Domain structure of hMSH6 and sequence alignment of PWWP modules. Upper panel shows the domain structure of hMSH6. PIP, PCNA-interacting protein motif; NLS, nuclear localization signal. Lower panel shows alignment of representative human PWWP domains. The blue dots indicate residues that form an aromatic cage and bind to H3K36me3. Aligned proteins are hMSH6, BRPF1, WHSC1_N, WHSC1_C, ZDWPW1, and HDGF.
(B) Interactions between PWWP domains and the H3K36me3-containing H3 peptide. The crystal structure of BRPF1 PWWP domain bound to the H3K36me3 peptide (PDB: 2X4Y) is shown in the left panel. BRPF1 does not have the Trp residues and instead contains a PSYP sequence (shown in yellow). The structure of the hMSH6 PWWP domain, which has the authentic PWWP motif, was determined by NMR (Laguri et al., 2008) in the absence of an H3 peptide (PDB: 2GFU). The H3 peptide in the BRPF1 complex is shown with human hMSH6 (right panel) after superimposing the conserved PWWP domains of the two proteins. The aromatic cage (colored blue) encloses the trimethylated Lys. The rotamer conformations of W106 and F133 in hMSH6 likely have to adjust upon binding of the H3K36me3.
The above data prompted us to ask whether the H3K36me3 mark modulates the interaction between hMutSα and chromatin and whether such an interaction involves the hMSH6 PWWP domain. To answer these questions, a glutathione-S-transferase (GST) fusion protein including hMSH6 residues 89 to 194 was used to pull down histone octamers isolated from HeLa cells (carrying ‘native’ histone modifications) or assembled using recombinant histones. The results show that the hMSH6 PWWP domain efficiently pulls down histone octamers from HeLa cells, but it pulls down recombinant histone octamers with very low efficiency (Figure 2A). This suggests a specific interaction between the hMSH6 PWWP domain and an epigenetic histone signature.
Figure 2. The MSH6 PWWP domain interacts with histone octamers containing H3K36me3.
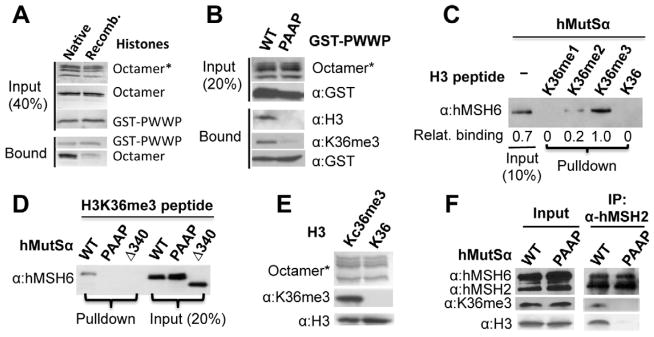
(A) GST pull-down assay for interaction of wild type GST-PWWP peptide with native histone octamer (purified from HeLa cells) or recombinant histone octamer. Unless otherwise mentioned, octamer marked with asterisk in this figure was detected by staining with Coomassie blue, and proteins not marked were detected by silver staining or Western blot using antibodies against the indicated components.
(B) GST pull-down assay for interaction between the native histone octamer and recombinant wild type (WT) or mutant (PAAP) PWWP peptide.
(C) hMutSα pull-down assay by biotin-labeled H3 peptides containing various forms of methylation in K36, as indicated. Bound hMutSα was eluted and detected by an hMSH6 antibody. Relative hMutSα binding was determined using the level of hMutSα in the K36me3 peptide-containing reaction as a reference.
(D) Interaction of H3 peptide containing K36me3 with hMutSα with wild type or a mutant PWWP domain, as indicated. The experiment was performed as in (C).
(E) Recombinant histone H3 with a chemically installed analog of tri-methyllysine at position 36 (H3Kc36me3/Kc36me3) forms histone octamers with other histone proteins (i.e., H2A, H2B, and H4) as efficient as recombinant wild type histone H3 (K36).
(F) Co-immunoprecipitation of histone octamer containing H3Kc36me3 and hMutSα with wild type (WT) or mutant (PAAP) PWWP domain. hMSH2 antibody was used for pulldown. The presence of H3Kc36me3 in the pulldown was detected using an H3K36me3-specific antibody.
The specificity of the interaction between the hMSH6 PWWP domain and natively modified octamers was examined by the following experiments. First, the same pull-down assay was performed using native histone octamers purified from HeLa cells (Rodriguez-Collazo et al., 2009) and wild type or a mutant hMSH6 PWWP fusion protein in which W105 and W106 are replaced by two alanines (PAAP), and the histone octamers bound to the GST-fusion proteins were detected by an H3K36me3-specific antibody. As shown in Figure 2B, wild type (WT) but not the mutant GST-hMSH6 PWWP selectively binds native histone octamers containing H3K36me3. Similar results were also obtained with a PWWP mutant containing an Y103A mutation (data not shown). Second, when H3 peptides containing no, mono-, di-, or tri-methylated K36 were incubated with hMutSα in the pull-down assay (Figure 2C), little interaction was detected between hMutSα and the peptide containing no (K36) or mono- (K36me1) methylations, but hMutSα was pulled down by di- (H3K36me2) and tri- (K36me3) methylated peptides, with approximately 5-fold more hMutSα co-precipitating with trimethylated peptide than with dimethylated peptide. These results suggest that hMutSα preferentially binds to H3K36me3.
In the following experiments, the interaction between the trimethylated peptide from H3 and several PWWP-mutated hMutSα variants was examined. One mutant hMutSα contained a PAAP motif (hMutSα[PAAP]) instead of the PWWP motif and another had a deletion of the first 340 amino acid residues (Δ340), including the PWWP domain, in hMSH6 (hMutSα[Δ340]). As shown in Figure 2D, the H3K36me3 peptide pulled down wild type hMutSα but did not pull down PWWP-deficient hMutSα proteins. Finally, co-immunoprecipitation was performed to confirm the interaction between the hMutSα PWWP domain and the H3K36me3-containing histone octamer. Here, recombinant histone H3 with an analog of trimethylated K36 (Kc36me3) (Simon et al., 2007) was used to assemble histone octamers; the octamers were recognized by antibody specific to H3K36me3 (Figure 2E), indicating that H3Kc36me3 is structural mimic of H3K36me3. The resulting H3Kc36me3 octamers were then incubated with hMutSα or hMutSα[PAAP]. As shown in Figure 2F, hMSH2 antibody only co-immunoprecipitated wild type hMutSα and H3Kc36me3-containing octamers. Thus, these data support the idea that the hMSH6 PWWP domain interacts specifically with trimethylated H3K36 in vitro and suggest that a similar specific interaction between hMutSα and H3K36me3 might occur on chromatin in vivo.
PWWP domain is dispensable for MMR in vitro, but essential for hMSH6 interaction with chromatin
The role of the hMSH6 PWWP domain in MMR was tested by analyzing the ability of wild type hMutSα or PWWP-deficient hMutSα to restore MMR to a nuclear extract from a MSH2-deficient leukemia cell line, NALM6 (Gu et al., 2002), using a functional in vitro assay (Zhang et al., 2005). Surprisingly, both PWWP-deficient proteins, hMutSα[PAAP] and hMutSα[Δ3 40], could efficiently restore MMR to NALM6 extracts (Figure 3A), suggesting that the hMSH6 PWWP domain is not essential for MMR in vitro.
Figure 3. The role of hMSH6 PWWP domain and its interaction with H3K36me3 in MMR.

(A) In vitro MMR assay to determine hMutSα with wild type (WT) or a mutant (PAAP or Δ340) PWWP domain for its ability to restore MMR to hMSH2-deficient NALM6 nuclear extracts as described (Zhang et al., 2005). HeLa nuclear extracts (HL) were used as a positive control. Arrows indicate repair products.
(B) Fluorescence imaging of cells expressing EGFP-hMSH6 containing WT or a mutant PWWP domain in hMSH6-deficient DLD-1 cells in S phase.
(C) Number of hMSH6 foci per nucleus in DLD-1 cells transfected with EGFP-hMSH6 containing WT or a mutant PWWP domain. The “n” value indicates the total number of nuclei analyzed in a given case. The error bars represent SD.
(D) Western blot analysis showing the expression of the indicated proteins in DLD-1 cells stably transfected with a scrambled shRNA (control) or a SETD2-specific shRNA.
(E) Fluorescence imaging of SETD2-knockdown or control DLD-1cells expressing wild type EGFP-hMSH6 in S phase.
The role of the hMSH6 PWWP domain in localizing hMSH6 to chromatin was examined in hMSH6-deficient DLD-1 cells expressing enhanced green fluorescent protein (EGFP)-tagged wild type or mutant hMSH6. Three hMSH6 mutants were generated, which contained alanine substitutions at the aromatic cage residues W105 and W106 (PAAP), Y103 (Y103A) or F133 (F133A). DLD-1 cells expressing EGFP-tagged wild type hMSH6, hMSH6(Y103A), hMSH6(PAAP) or hMSH6(F133A) were arrested in S phase and then analyzed by fluorescent confocal microscopy. The results show that hMSH6 clearly formed foci in cells expressing wild type hMSH6, but significantly (p<0.001) fewer hMSH6 foci were found in cells expressing the mutant hMSH6 proteins (Figures 3B and 3C); instead, fluorescence remained evenly distributed in the nucleus in the latter cells (Figure 3B). Together with the data shown in Figures 1 and 2, these observations imply that the hMSH6 PWWP domain, although not required for MMR in vitro, is required to recruit hMSH6 to chromatin. This supports the idea that the hMSH6 PWWP domain ‘reads’ the H3K36me3 mark.
H3K36me3 facilitates localization of hMutSα to chromatin in vivo
SETD2 is a methyltransferase responsible for H3K36 trimethylation (Duns et al., 2010; Edmunds et al., 2008; Yoh et al., 2008). To determine if hMSH6 chromatin localization relies on H3K36me3, we performed SETD2 knockdown by shRNA in DLD-1 cells, which apparently led to a targeted depletion of SETD2, as the same vector containing a scrambled shRNA (control) did not reduce expression of SETD2 (Figure 3D). As expected, the SETD2-knockdown cells expressed a low level of H3K36me3 (Figure 3D). Interestingly, hMSH6 formed distinct foci in control DLD-1 cells, but it formed fewer foci and was distributed more evenly within the nucleus in shSETD2-DLD-1 cells (Figure 3E). These results suggest that H3K36me3 influences the distribution of hMSH6 in chromatin.
This idea was further tested with the endogenous hMSH6 in MMR-proficient HeLa cells with or without a targeted SETD2 knockdown by shRNA (Figure 4A). After synchronization of control HeLa (transfected with a scrambled shRNA) and SETD2-depleted HeLa in S or G2/M phase, chromatin localization of endogenously expressed hMSH6 was monitored by immunofluorescence using an antibody to hMSH6. The results show that during S phase, significantly fewer hMSH6 foci were observed in the SETD2/H3K36me3-depleted HeLa cells than in control HeLa cells (Figures 4B and 4C), further supporting the idea that localization of hMSH6 to chromatin is facilitated by H3K36me3. In addition, ~70% of hMSH6 foci appeared to colocalize with H3K36me3 in control HeLa cells but not in SETD2/H3K36me3-depleted cells (Figure 4B). Similar results were also obtained using an antibody to hMSH2 (Supplemental Figure S1A), indicating that the effect is for the whole hMutSα complex. These results were confirmed in another HeLa clone whose SEDT2 was stably knocked down by a second SETD2 shRNA (Figure S1B). Interestingly, the number of hMSH6 foci (Figures 4B and 4C) was very low in control G2/M HeLa cells. However, this has nothing to do with the amount of hMSH6 in these cells because hMSH6 in G2/M is as abundant as in other cell cycle phases (Figures 4D and 4E).
Figure 4. SETD2/H3K36me3 depletion alters hMSH6 nuclear localization.

(A) Western blot analysis showing the expression of the indicated proteins in HeLa cells stably transfected with a scrambled shRNA (control) and two different SETD2-specific shRNAs (SETD2).
(B) hMSH6 and H3K36me3 foci formation and colocalization in control- and shSETD2-HeLa cells in S or G2/M phase.
(C) The number of hMSH6 foci per nucleus in control- and shSETD2-HeLa cells. The “n” value indicates the total number of nuclei analyzed in a given case. The error bars represent SD.
(D) Western blot analysis showing the abundance of hMSH6 and H3K36me3 in G1, S, or G2/M phase in control HeLa cells. S phase was divided into early (E), middle (M), and late (L) sub-phases.
(E) Quantification of hMSH6 or H3K36me3 expression in different cell cycle phases from three independent Western blots. The intensity of each protein in G1 was used as a reference after correction for loading (i.e., the intensity of tubulin). The error bars represent SD.
Also see Figs. S1 and S2.
To determine if this phenomenon is directly correlated with the abundance of H3K36me3, H3K36me3 and H3 were quantified in HeLa cells arrested at different stages of the cell cycle. The results show that H3K36me3 reaches a maximum abundance in early S phase, declines to a very low level by the end of S phase and G2/M, and begins to increase in abundance in G1, even though the amount of histone H3 is relatively constant throughout the cell cycle (Figures 4D and 4E). We also observed abundant hMSH6 foci and their partial (~80%) colocalization with H3K36me3 in G1 phase (Figure S1C). Together, these results suggest that H3K36me3 recruits hMutSα to chromatin in vivo before and during early S phase, which could increase the efficiency of MMR in actively replicating chromatin.
Although the interaction between hMSH6 and chromatin appears to be facilitated by H3K36me3, not all hMSH6 foci colocalize with H3K36me3 foci in S-phase HeLa cells (Figures 4B). These results may suggest one or both of the following possibilities: 1) not all H3K36me3 marks recruit hMutSα; and 2) after binding, hMutSα disassociates from H3K36me3 during DNA replication, possibly due to specific interactions with the replication machinery, as previously reported (Hombauer et al., 2011a; Kleczkowska et al., 2001) (see Discussion for more details).
To further determine that the H3K36me3-PWWP interaction is specific for recruiting hMSH6, we measured nuclear distribution of the hMSH3 subunit of hMutSβ and its colocalization with H3K36me3. We show that hMSH3, which lacks a PWWP domain, does not interact with H3K36me3 and its nuclear localization is independent of H3K36me3 (Figure S2), consistent with the notion that human cells utilize different mechanisms for hMutSα and hMutSβ recruitments (Hong et al., 2008).
HeLa cells with SETD2 knockdown display a mutator phenotype
As suggested above, if the hMSH6-H3K36me3 interaction recruits hMutSα to chromatin in vivo, then it could be essential for MMR in vivo. If this prediction is correct, cells lacking or depleted for H3K36me3 will be MMR-deficient and have an increased mutation frequency. To explore this prediction, control and SETD2-depleted HeLa cells were tested for MSI at four microsatellite loci as described (Parsons et al., 1993). The results (Figure 5A) show no MSI in control HeLa cells, while 4 out of 14 (28.6%) subclones from SETD2/H3K36me3-depleted HeLa cells showed either new microsatellite species (asterisk) or deletion of a microsatellite mark (Δ). As a positive control, MSI was also analyzed in hMSH6-deficient DLD-1 cells, and the results show new repeat species in 6 out of 15 (40%) subclones (Figure S3). Despite a difference in the percentage of subclones showing new repeat species, the data clearly demonstrate that like hMSH6-deficient DLD-1 cells, SETD2/H3K36me3-depleted HeLa cells display MSI.
Figure 5. SETD2-deficient HeLa cells display a mutator phenotype.

(A) Analysis of PCR product patterns of four microsatellite markers in subclones derived from control- and shSETD2-HeLa cells. The black Δ or asterisk shows the clone exhibiting deletion of a repeat mark or new repeat species, respectively.
(B) HPRT mutability in control- and shSETD2- HeLa and DLD-1 cells. Data are presented as mean +/− SD. Fold-increase in mutation frequency was calculated using the mutation frequency of control HeLa as a reference, and t-test was used to determine the p value.
(C) In vitro MMR assay using nuclear extracts isolated from control- and shSETD2-HeLa cells.
Also see Fig. S3.
To further confirm the mutator phenotype in SETD2/H3K36me3-depleted cells, we measured the spontaneous forward mutation frequency in the hypoxanthine-guanine phosphoribosyltransferase (HPRT) gene (Kat et al., 1993) in SETD2/H3K36me3-depleted and control HeLa cells. As shown in Figure 5B, the spontaneous mutation frequency in SETD2/H3K36me3-depleted HeLa cells had an 18-fold increase (1.2 × 10−5, p<0.05) compared to that in control HeLa cells (6.9 × 10−7), indicating that SETD2/H3K36me3 depletion causes a mutator phenotype. When the same analysis was performed in hMSH6-deficient shSETD2-DLD-1 and control DLDL-1 cells, the mutation frequency essentially remained unchanged (Figure 5B). This result suggests that SETD2 depletion only alters the mutation frequency in hMSH6-competent cells, which is consistent with the idea that H3K36me3 recruits hMutSα. Interestingly, the mutation frequency in SETD2/H3K36me3-depleted HeLa cells is ~10-fold lower than that in hMSH6-deficient DLD-1 cells, which is likely due to both the efficiency of SETD2 depletion and the huge difference in the total passage number between these two cells. In addition, H3K36me3 may not be the only mechanism for hMSH6 recruitment.
To determine if SETD2/H3K36me3 modulates the enzymatic function(s) of MMR proteins, the in vitro MMR activity of SETD2/H3K36me3-depleted HeLa cells was examined. Consistent with our model (see below), these cells are not defective in MMR in vitro (Figure 5C). This observation indicates that SETD2 and H3K36me3 are not physically involved in MMR, and that depletion of SETD2/H3K36me3 does not alter the expression or function of MMR genes.
Cancer cells deficient in SETD2 display MSI and fail to recruit hMutSα to chromatin
Recent studies identified SETD2 as a tumor suppressor for clear cell renal cell carcinoma (ccRCC) (Dalgliesh et al., 2010; Duns et al., 2010; Gerlinger et al., 2012; Varela et al., 2011), but the mechanism linking SETD2 deficiency to ccRCC tumorigenesis remains unknown. We hypothesize that defective MMR may contribute to tumorigenesis in SETD2-deficient ccRCC patients. To test this hypothesis, we screened several ccRCC cell lines for SETD2 mutations and identified a SETD2-deficient ccRCC cell line, UOK143, which carries A5197 →G and T5306 →C mutations, leading to N1734D and S1769P amino acid substitutions in SETD2, respectively (Figure S4A). Western blot analysis shows that UOK143 cells express undetectable amounts of SETD2, which is readily detected in the SETD2-proficient ccRCC cell line, UOK121 (Figure 6A). As expected, the amount of H3K36me3 is much lower in UOK143 cells than in UOK121 cells (Figure 6A). We then analyzed the distribution of hMSH6 and H3K36me3 in these ccRCC cells. H3K36me3 was barely detectable in UOK143 cells by immunofluorescence, but was relatively more abundant in UOK121 cells (Figure 6B). Correspondingly, in S-phase UOK121 cells, hMSH6 foci were abundant and partially (~70%) colocalized with H3K36me3, while significantly fewer and smaller hMSH6 foci were observed in S-phase UOK143 cells (Figures 6B and 6C), which is similar to what was observed in SETD2/H3K36me3-depleted HeLa (Figure 4B) and DLD-1 (Figure 3E) cells. As noted previously, these results suggest that H3K36me3 facilitates localization of hMSH6 (hMutSα) to chromatin.
Figure 6. Cancer cells defective in SETD2 exhibit classical MMR-deficient phenotype.

(A) Western blot analysis of SETD2 and H3K36me3 in ccRCC cell lines UOK121 and UOK143.
(B) Nuclear localization of H3K36me3 and hMSH6 in ccRCC cell lines UOK121 and UOK143 in S phase.
(C) The number of hMSH6 foci per nucleus in UOK121 and UOK143 cells. The “n” value indicates the total number of nuclei analyzed in each case. The error bars represent SD.
(D) PCR analysis of microsatellites BAT25, BAT26, and D5S346 in subclones of UOK121 and UOK143 cells. The red Δ or asterisk shows the clone exhibiting deletion of a repeat mark or new repeat species, respectively.
(E) Nuclear localization of H3K36me3 and hMSH6 in UOK143 cells transfected with or without yeast Set2 in S phase.
Also see Fig. S4.
To determine if the failure to recruit hMutSα confers an MMR-deficient phenotype to UOK143 cells, we examined MSI in UOK143 and UOK121 cells. The results revealed no MSI in UOK121 cells and that UOK143 cells exhibited mono- and di-nucleotide repeat instability, as all subclones, except clone 1, exhibit either new repeat species or deletion of a microsatellite marker (Figure 6D). To rule out the possibility that UOK143 cells carry a defective MMR component, we measured in vitro MMR activity of UOK143 and UOK121, observing the same normal levels of MMR activity and proteins in extracts from both cell lines (data not shown). These results suggest that MSI in UOK143 cells is caused by loss of SETD2 activity, not loss of MMR capacity.
We previously characterized a Burkitt’s lymphoma cell line, NAMALWA, that displays MSI but is proficient in MMR in vitro (Gu et al., 2002). We therefore analyzed if this cell line is defective in SETD2. The result shows that NAMALWA cells carry a heterozygous deletion of exon 8 in SETD2 (Figure S4B), which also alters the reading frame of the gene. Consistent with this result, NAMALWA cells express a much lower level of SETD2 and have a lower level of H3K36me3 than SETD2-proficient cells such as NALM6 cells (Figure S4C). These observations suggest that depletion of H3K36me3 promotes ccRCC and could play a role in promoting other cancers (see Discussion).
Restoration of H3K36me3 in SETD2-deficient cells restores hMutSα chromatin localization
To further confirm that H3K36me3 facilitates recruitment of hMutSα onto chromatin, we transiently transfected UOK143 cells with the yeast Set2 gene, which is known to efficiently promote H3K36 trimethylation in human cells (Luco et al., 2010). Compared with control cells transfected with an empty vector, Set2-expressing UOK143 cells exhibited higher levels of H3K36me3, larger and more hMSH6 foci, and partial colocalization of H3K36me3 and hMSH6. These observations are consistent with the hypothesis that H3K36me3 recruits hMutSα onto chromatin in vivo.
Discussion
In this study, we demonstrate that H3K36me3 recruits hMutSα to chromatin in a cell cycle-dependent manner, such that H3K36me3 and the histone methyltransferase SETD2 are required for MMR in vivo. The finding provides a molecular explanation for the lack of concurrence between the MSI phenotype and the MMR genotype among human cancer cells.
Although human MMR has been reconstituted in vitro (Constantin et al., 2005; Zhang et al., 2005), little is known how the MMR machinery operates on chromatin in vivo. The identification of the hMSH6 PWWP domain as a reader of H3K36me3 prompted us to hypothesize that H3K36me3 recruits hMutSα via this domain onto chromatin. We show here that 1) wild type but not mutant hMSH6 PWWP domain interacts with H3K36me3-containing histone octamers (Figure 2); 2) the hMSH6 PWWP domain is essential for hMutSα chromatin localization (Figure 3B); 3) cells depleted with H3K36me3 fail to recruit hMutSα (Figures 3D, 4B, and 6B); 4) restoration of the H3K36me3 signal also restores hMutSα chromatin localization in SETD2-deficient cells (Figure 6E); and 5) SETD2/H3K36me3-depleted cells display an MMR-deficient mutator phenotype (Figures 5 and 6D). Therefore, H3K36me3 and the PWWP domain regulate human MMR in vivo.
In support of this conclusion, the abundance of hMSH6 (and hMutSα) foci in the nucleus of HeLa cells correlates with the abundance of H3K36me3 during the cell cycle. The level of H3K36me3 is highest in the early S phase, significantly declines in the middle S phase, and little remains in late S phase and G2/M (Figure 4D), consistent with published results (Bonenfant et al., 2007; Ryba et al., 2010). Correspondingly, hMSH6 foci are readily seen in S phase, but are rarely seen in G2/M (Figure 4C). These results suggest that MMR is mainly active in S phase, when it corrects DNA replication-associated nucleotide misincorporations (Hombauer et al., 2011a; Hombauer et al., 2011b; Simmons et al., 2008), but that hMutSα is likely recruited to chromatin before DNA replication initiates. This appears to be consistent with a recent yeast study. Despite that yeast MSH6 does not possess a PWWP domain, and is probably not recruited to chromatin by H3K36me3, Hombauer et al. (Hombauer et al., 2011a) showed that yeast MutSα is present at the replication fork, independent of the presence of mispaired bases. However, we provide evidence that localizing hMutSα to chromatin, although essential for MMR in vivo, is not sufficient to trigger or facilitate the biochemical reaction of MMR in the context of chromatin, as judged by the fact that a mismatch located between two histone octamers bearing the H3K36me3 signature could not be corrected by MMR-competent nuclear extracts (Figure S5), which also contain all chromatin remodeling/modifying factors. This observation suggests that the hMutSα recruitment to chromatin by H3K36me3 only sets up an on-call system for MMR, which is ready whenever it is needed, but triggering the MMR reaction needs both specific mismatch signal and an environment of DNA replication, which in part includes disassembly of nucleosome structure.
Based on previously published data and the results presented here, we propose a model for the initiation of MMR in human cells in vivo (Figure 7). First, the SETD2 methyltransferase converts H3K36me2 to H3K36me3 either before or in early S phase. Then, H3K36me3 helps recruit hMutSα onto chromatin through its interaction with the hMSH6 PWWP domain. During DNA replication, nucleosomes are dynamically assembled and disassembled, such that nucleosomes ahead of the replication fork are disrupted and those behind the replication fork are rapidly re-assembled (Ransom et al., 2010). Nucleosome disassembly provides the replication machinery access to DNA, and at the same time, disrupts the H3K36me3-PWWP interaction, thereby releasing hMutSα from histone octamers. The released hMutSα can then readily attach to temporarily histone-free nascent DNA through its strong DNA binding activity and/or by interacting with PCNA via the hMSH6 PIP (PCNA interacting protein) box (Clark et al., 2000; Flores-Rozas et al., 2000). hMutSα then slides along the DNA helix (Gorman et al., 2007; Gradia et al., 1997; Mendillo et al., 2005) to locate mispairs, which triggers downstream events in the MMR pathway. However, mismatches assembled in the nascent nucleosomes behind the replication fork will not be repaired (Figure S5).
Figure 7. Model for hMutSα recruitment to chromatin and mismatched DNA.

In the late G1 and/or early S phase, SETD2 converts H3K36me2 to H3K36me3. H3K36me3 recruits hMutSα onto chromatin via its interaction with the hMSH6 PWWP domain before DNA replication initiates. During DNA replication, Nucleosome disassembly exposes naked DNA to the replication machinery and also disrupts the H3K36me3-PWWP interaction, which releases hMutSα from histone octamers. hMutSα, which has a strong DNA binding activity, can readily bind to nascent DNA with or without an interaction with PCNA. Once a mismatch is introduced, hMutSα can quickly identify the mismatch to initiate the MMR reaction. Also see Fig. S5.
It is worth mentioning that both the human and yeast MSH6 PIP boxes have been shown to be required for MutSα colocalization with replication factories (Hombauer et al., 2011a; Kleczkowska et al., 2001). Interestingly, depletion of the PIP box only moderately (~10–15%) reduces MMR activity in yeast (Hombauer et al., 2011a; Shell et al., 2007) and does not abolish hMSH6 foci formation in human cells (Kleczkowska et al., 2001). These observations indicate that PIP-defective MutSα can still be efficiently recruited to chromatin. We therefore propose that in human cells, the hMSH6 PIP box and PWWP domain are likely to play different but complementary roles in MMR. One possibility is that the PWWP domain localizes hMutSα to H3K36me3-containing chromatin before replication initiates and then the PIP box helps localize hMutSα to newly-formed mispairs through its interaction with PCNA during DNA replication. This could also explain the presence of a fraction of hMSH6 foci that do not colocalize with H3K36me3 foci in S phase (Figures 4B, 6B, 6E and S1). Further investigations are needed to explore this and other possibilities.
The data presented here suggest that tumors defective in SETD2 fail to recruit hMutSα to chromatin and therefore are MMR-deficient in vivo. Recently, a number of exome sequencing studies have identified SETD2 mutations in ccRCC (Dalgliesh et al., 2010; Duns et al., 2010; Gerlinger et al., 2012; Varela et al., 2011), lung cancer (Govindan et al., 2012; Imielinski et al., 2012), and hematological maliganencies (Zhang et al., 2012). These SETD2-deficient cancers have no detectable mutations in MMR genes; however, results presented here are consistent with the idea that SETD2 mutations are responsible for an MMR-defect in these cells. Notably, all SETD2-deficient ccRCC tumors have a high frequency of small insertion/deletion mutations and dominant single-nucleotide substitutions (Dalgliesh et al., 2010; Gerlinger et al., 2012), which are hallmarks of MMR deficiency. In addition, a subset of gastric cancers that display MSI have no mutations in known MMR genes, but are defective in SETD2 (Wang et al., 2011). A similar situation may exist in some MSI-positive colorectal cancer cells (Peltomaki, 2003).
H3K36me3 interacts with multiple PWWP-containing proteins in vivo, and the importance of H3K36me3 in transcriptional regulation is well documented (Musselman et al., 2012). How is the dual impact of H3K36me3 on MMR and transcription regulated or partitioned? Because MMR is not active in G1, it remains to be investigated if the observed hMSH6-H3K36me3 interaction in G1 (Figure S1B) facilitates transcription or occurs just before cells enter S phase (note that double-thymidine treatment arrests the cell cycle at the G1-S boundary). Since MMR is coupled to DNA replication, MMR and transcription could only compete with one another for H3K36me3 during S phase. Although an as-yet-unknown mechanism may avoid such competition, Wansink et al. (Wansink et al., 1994) have already shown that transcription and replication rarely occur at the same time in the same place in S-phase nuclei. If universally true, then there is little risk of conflict between roles for H3K36me3 in MMR and transcription.
In summary, we demonstrate here that H3K36me3 regulates MMR in vivo. This striking finding underscores the importance of the histone code in maintaining genome stability. Recent studies reveal that mutation rates in cancer genomes are closely related to histone modification-directed chromatin organization (Schuster-Bockler and Lehner, 2012). In that regard, future studies may reveal that the abundance of H3K36me3 in a gene or gene regulatory region plays a role in determining the mutability of that segment of the human genome.
Experimental procedures
Cell lines
HeLa, NALM6 and NAMALWA cells were grown in RPMI1640 medium supplemented with 10% fetal bovine serum (FBS). UOK121, UOK143 and DLD-1 cells were maintained in DMEM with 10% FBS. The stable SETD2-knockdown HeLa (shSETD2-HeLa) and SETD2 knockdown DLD-1 lines were created by lentivirus (Sigma) transfection under puromycin selection, according to the manufacturer’s instructions.
Protein and peptide preparations
Human wild type and mutant MutSα proteins were expressed and purified as described (Zhang et al., 2005). The hMSH6 gene used for mutagenesis was a gift of Dr. Richard Kolodner. For expression of GST-PWWP fusion peptides, wild type and mutant PWWP domain fragments of hMSH6 were PCR-amplified and cloned into pGEX4T2 (GE Healthcare Life Sciences). After verification by sequencing, the resulting plasmids were used for protein expression and purification from an E. coli Rosetta (DE3) strain (Novagen). Recombinant H2A, H2B, H3 and H4 were obtained and histone octamers were assembled as described (Li et al., 2009). Native histone octamers were isolated from HeLa cells as described (Rodriguez-Collazo et al., 2009). EGFP-hMSH6 expression vector (pEGFP-C1-hMSH6, a gift of Dr. Akira Yasui) was used to generate the expression vectors of EGFP-hMSH6-PAAP, EGFP-hMSH6-Y103A, and EGFP-hMSH6-F133A. Histone H3 peptides (ARKSAPATGGVK36KPHRYRP) containing various forms of K36 methylation were commercially synthesized (GenScript, Piscataway, New Jersey).
MSI and HPRT mutability analyses
For each cell line tested for MSI, independent single cell colonies were isolated in 96-well microtiter plates and genomic DNA was isolated. Four microsatellite markers (BAT25, BAT26, D2S123, D5S346) were used to for MSI analysis (Parsons et al., 1993).
The HPRT mutation assay was conducted as described (Kat et al., 1993). Cells (5×105) were seeded in triplicate 100-mm Petri dishes for 12 h and fed with complete medium containing 5 μM freshly prepared 6-thioguanine (6-TG). The plating efficiency was determined by culturing 5×102 cells similarly in the absence of 6-TG. After culturing a 10 day-culturing, cell colonies were visualized by staining with 0.05% crystal violet. The mutation frequency was determined by dividing the number of 6-TG resistant colonies by the total number of cells plated after being corrected for the colony-forming ability.
Cell synchronization and cell cycle analysis
Cell synchronization was performed as described (Stojic et al., 2004). Cells were arrested at G1/S by culturing for 18 h in complete medium containing 2 mM thymidine, 10 h in thymidine-free medium, and then thymidine-containing medium for an additional 15 h before release into complete medium. Cells were harvested at 0 h (G1 phase), 1 h (early S), 2.5 h (middle S), 4h (late S), and 8h (G2/M). Cell cycle status was confirmed by flow cytometry.
Microscopy and immunofluorescence analysis
Immunofluorescence analysis was performed essentially as described (Kleczkowska et al., 2001). Fluorescence images were obtained and analyzed using an FV-1000 Olympus confocal scanning laser microscopy system. The percentage of colocalized H3K36me3 and hMSH6 foci was quantified using the Olympus FV10-ASW2.1 software, based on analyzing the Pearson correlation coefficient as described (Adler and Parmryd, 2010).
Mismatch repair assay
In vitro MMR assays were performed as described (Holmes et al., 1990; Zhang et al., 2005). Unless otherwise specified, MMR activity was determined in a 20-μL reaction containing 50 μg of nuclear extracts, 100 ng of a circular DNA substrate containing a G-T mismatch in the presence or absence of hMutSα. The reaction was incubated at 37 °C for 15 min, and repair was scored and analyzed by restriction enzyme digestions and agarose gel electrophoresis.
Western blot, co-immunoprecipitation and pull-down assays
Antibodies used in this study were from Santa Cruz Biotechnology (MSH2, MSH3, histone H3, GST and tubulin), BD Biosciences (MSH6), Sigma (SETD2), and Cell Signaling Technology (SETD2 and H3K36me3).
Statistical analysis
All statistical assays, Student’s t-test and One-way analysis of variance (ANOV) with Tukey’s multiple comparison, were performed using GraphPad Prism 5.0 (GraphPad Software, Inc.). Data were considered statistically significant if p-values were less than 0.05 or 0.001, as indicated.
Supplementary Material
Highlights.
Mismatch repair protein MutSα (MSH2-MSH6) binds H3K36me3 via the MSH6 PWWP domain.
MutSα localizes to chromatin in G1 to early S phase before DNA replication.
Depletion of H3K36 methyltransferase SETD2 leads to a mutator phenotype.
SETD2 deficiency explains microsatellite instability in MMR-positive cancer cells.
Acknowledgments
We thank Richard Kolodner, Tom Misteli, Jeffrey Parvin, Youfeng Yang, and Akira Yasui for reagents, and Yang Shi, Dangsheng Li, Charles Ensor and Nathan Vanderford for stimulating discussions and helpful comments. The work was supported in part by NIH grants CA167181, CA115942 and GM089684 (to G.-M.L.), CA104333 (to L.G.), and the NIDDK intramural research fund (to W.Y.), and a grant from the Kentucky Lung Cancer Research Program. G.-M.L. holds the James-Gardner Chair in Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adler J, Parmryd I. Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander’s overlap coefficient. Cytometry A. 2010;77:733–742. doi: 10.1002/cyto.a.20896. [DOI] [PubMed] [Google Scholar]
- Bonenfant D, Towbin H, Coulot M, Schindler P, Mueller DR, van Oostrum J. Analysis of dynamic changes in post-translational modifications of human histones during cell cycle by mass spectrometry. Mol Cell Proteomics. 2007;6:1917–1932. doi: 10.1074/mcp.M700070-MCP200. [DOI] [PubMed] [Google Scholar]
- Clark AB, Valle F, Drotschmann K, Gary RK, Kunkel TA. Functional interaction of proliferating cell nuclear antigen with MSH2-MSH6 and MSH2-MSH3 complexes. J Biol Chem. 2000;275:36498–36501. doi: 10.1074/jbc.C000513200. [DOI] [PubMed] [Google Scholar]
- Constantin N, Dzantiev L, Kadyrov FA, Modrich P. Human mismatch repair: Reconstitution of a nick-directed bidirectional reaction. J Biol Chem. 2005;280:39752–39761. doi: 10.1074/jbc.M509701200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, Jeltsch A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. The Journal of biological chemistry. 2010;285:26114–26120. doi: 10.1074/jbc.M109.089433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond JT, Genschel J, Wolf E, Modrich P. DHFR/MSH3 amplification in methotrexate-resistant cells alters the hMutSalpha/hMutSbeta ratio and reduces the efficiency of base-base mismatch repair. Proc Natl Acad Sci U S A. 1997;94:10144–10149. doi: 10.1073/pnas.94.19.10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond JT, Li GM, Longley MJ, Modrich P. Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science. 1995;268:1909–1912. doi: 10.1126/science.7604264. [DOI] [PubMed] [Google Scholar]
- Duns G, van den Berg E, van Duivenbode I, Osinga J, Hollema H, Hofstra RM, Kok K. Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer research. 2010;70:4287–4291. doi: 10.1158/0008-5472.CAN-10-0120. [DOI] [PubMed] [Google Scholar]
- Edmunds JW, Mahadevan LC, Clayton AL. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. The EMBO journal. 2008;27:406–420. doi: 10.1038/sj.emboj.7601967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishel R, Kolodner RD. Identification of mismatch repair genes and their role in the development of cancer. Curr Opin Genet Dev. 1995;5:382–395. doi: 10.1016/0959-437x(95)80055-7. [DOI] [PubMed] [Google Scholar]
- Flores-Rozas H, Clark D, Kolodner RD. Proliferating cell nuclear antigen and Msh2p-Msh6p interact to form an active mispair recognition complex. Nat Genet. 2000;26:375–378. doi: 10.1038/81708. [DOI] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England journal of medicine. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman J, Chowdhury A, Surtees JA, Shimada J, Reichman DR, Alani E, Greene EC. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Mol Cell. 2007;28:359–370. doi: 10.1016/j.molcel.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindan R, Ding L, Griffith M, Subramanian J, Dees N, Walker KL, Maher CA, Fulton R, Fulton L, Wallis J, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–1134. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradia S, Acharya S, Fishel R. The human mismatch recognition complex hMSH2-hMSH6 functions as a novel molecular switch. Cell. 1997;91:995–1005. doi: 10.1016/s0092-8674(00)80490-0. [DOI] [PubMed] [Google Scholar]
- Gu L, Cline-Brown B, Zhang F, Qiu L, Li GM. Mismatch repair deficiency in hematological malignancies with microsatellite instability. Oncogene. 2002;21:5758–5764. doi: 10.1038/sj.onc.1205695. [DOI] [PubMed] [Google Scholar]
- Harrington JM, Kolodner RD. Saccharomyces cerevisiae Msh2-Msh3 acts in repair of base-base mispairs. Mol Cell Biol. 2007;27:6546–6554. doi: 10.1128/MCB.00855-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes J, Jr, Clark S, Modrich P. Strand-specific mismatch correction in nuclear extracts of human and Drosophila melanogaster cell lines. Proc Natl Acad Sci U S A. 1990;87:5837–5841. doi: 10.1073/pnas.87.15.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombauer H, Campbell CS, Smith CE, Desai A, Kolodner RD. Visualization of eukaryotic DNA mismatch repair reveals distinct recognition and repair intermediates. Cell. 2011a;147:1040–1053. doi: 10.1016/j.cell.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombauer H, Srivatsan A, Putnam CD, Kolodner RD. Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science. 2011b;334:1713–1716. doi: 10.1126/science.1210770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Z, Jiang J, Hashiguchi K, Hoshi M, Lan L, Yasui A. Recruitment of mismatch repair proteins to the site of DNA damage in human cells. J Cell Sci. 2008;121:3146–3154. doi: 10.1242/jcs.026393. [DOI] [PubMed] [Google Scholar]
- Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javaid S, Manohar M, Punja N, Mooney A, Ottesen JJ, Poirier MG, Fishel R. Nucleosome remodeling by hMSH2-hMSH6. Molecular cell. 2009;36:1086–1094. doi: 10.1016/j.molcel.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadyrova LY, Blanko ER, Kadyrov FA. CAF-I-dependent control of degradation of the discontinuous strands during mismatch repair. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:2753–2758. doi: 10.1073/pnas.1015914108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–811. [PubMed] [Google Scholar]
- Kat A, Thilly WG, Fang WH, Longley MJ, Li GM, Modrich P. An alkylation-tolerant, mutator human cell line is deficient in strand-specific mismatch repair. Proc Natl Acad Sci U S A. 1993;90:6424–6428. doi: 10.1073/pnas.90.14.6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleczkowska HE, Marra G, Lettieri T, Jiricny J. hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes Dev. 2001;15:724–736. doi: 10.1101/gad.191201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodner R. Biochemistry and genetics of eukaryotic mismatch repair. Genes Dev. 1996;10:1433–1442. doi: 10.1101/gad.10.12.1433. [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- Laguri C, Duband-Goulet I, Friedrich N, Axt M, Belin P, Callebaut I, Gilquin B, Zinn-Justin S, Couprie J. Human mismatch repair protein MSH6 contains a PWWP domain that targets double stranded DNA. Biochemistry. 2008;47:6199–6207. doi: 10.1021/bi7024639. [DOI] [PubMed] [Google Scholar]
- Li F, Tian L, Gu L, Li GM. Evidence that nucleosomes inhibit mismatch repair in eukaryotic cells. J Biol Chem. 2009;284:33056–33061. doi: 10.1074/jbc.M109.049874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra G, Iaccarino I, Lettieri T, Roscilli G, Delmastro P, Jiricny J. Mismatch repair deficiency associated with overexpression of the MSH3 gene. Proc Natl Acad Sci U S A. 1998;95:8568–8573. doi: 10.1073/pnas.95.15.8568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsischky GT, Filosi N, Kane MF, Kolodner R. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 1996;10:407–420. doi: 10.1101/gad.10.4.407. [DOI] [PubMed] [Google Scholar]
- Mendillo ML, Mazur DJ, Kolodner RD. Analysis of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 and MLH1-PMS1 complexes with DNA using a reversible DNA end-blocking system. J Biol Chem. 2005;280:22245–22257. doi: 10.1074/jbc.M407545200. [DOI] [PubMed] [Google Scholar]
- Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- Musselman CA, Lalonde ME, Cote J, Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nature structural & molecular biology. 2012;19:1218–1227. doi: 10.1038/nsmb.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons R, Li GM, Longley MJ, Fang WH, Papadopoulos N, Jen J, de la Chapelle A, Kinzler KW, Vogelstein B, Modrich P. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell. 1993;75:1227–1236. doi: 10.1016/0092-8674(93)90331-j. [DOI] [PubMed] [Google Scholar]
- Peltomaki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. Journal of clinical oncology. 2003;21:1174–1179. doi: 10.1200/JCO.2003.04.060. [DOI] [PubMed] [Google Scholar]
- Ransom M, Dennehey BK, Tyler JK. Chaperoning histones during DNA replication and repair. Cell. 2010;140:183–195. doi: 10.1016/j.cell.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Collazo P, Leuba SH, Zlatanova J. Robust methods for purification of histones from cultured mammalian cells with the preservation of their native modifications. Nucleic acids research. 2009;37:e81. doi: 10.1093/nar/gkp273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, Schulz TC, Robins AJ, Dalton S, Gilbert DM. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010;20:761–770. doi: 10.1101/gr.099655.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopf B, Bregenhorn S, Quivy JP, Kadyrov FA, Almouzni G, Jiricny J. Interplay between mismatch repair and chromatin assembly. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:1895–1900. doi: 10.1073/pnas.1106696109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster-Bockler B, Lehner B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012 doi: 10.1038/nature11273. [DOI] [PubMed] [Google Scholar]
- Shell SS, Putnam CD, Kolodner RD. The N terminus of Saccharomyces cerevisiae Msh6 is an unstructured tether to PCNA. Mol Cell. 2007;26:565–578. doi: 10.1016/j.molcel.2007.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons LA, Davies BW, Grossman AD, Walker GC. Beta clamp directs localization of mismatch repair in Bacillus subtilis. Mol Cell. 2008;29:291–301. doi: 10.1016/j.molcel.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MD, Chu F, Racki LR, de la Cruz CC, Burlingame AL, Panning B, Narlikar GJ, Shokat KM. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell. 2007;128:1003–1012. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojic L, Mojas N, Cejka P, Di Pietro M, Ferrari S, Marra G, Jiricny J. Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev. 2004;18:1331–1344. doi: 10.1101/gad.294404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DC, Roberts JD, Kunkel TA. Heteroduplex repair in extracts of human HeLa cells. J Biol Chem. 1991;266:3744–3751. [PubMed] [Google Scholar]
- Umar A, Boyer JC, Thomas DC, Nguyen DC, Risinger JI, Boyd J, Ionov Y, Perucho M, Kunkel TA. Defective mismatch repair in extracts of colorectal and endometrial cancer cell lines exhibiting microsatellite instability. J Biol Chem. 1994;269:14367–14370. [PubMed] [Google Scholar]
- Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, Davies H, Jones D, Lin ML, Teague J, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–542. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142:967–980. doi: 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- Vezzoli A, Bonadies N, Allen MD, Freund SM, Santiveri CM, Kvinlaug BT, Huntly BJ, Gottgens B, Bycroft M. Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat Struct Mol Biol. 2010;17:617–619. doi: 10.1038/nsmb.1797. [DOI] [PubMed] [Google Scholar]
- Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, Chan TL, Kan Z, Chan AS, Tsui WY, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43:1219–1223. doi: 10.1038/ng.982. [DOI] [PubMed] [Google Scholar]
- Wansink DG, Manders EE, van der Kraan I, Aten JA, van Driel R, de Jong L. RNA polymerase II transcription is concentrated outside replication domains throughout S-phase. J Cell Sci. 1994;107(Pt 6):1449–1456. doi: 10.1242/jcs.107.6.1449. [DOI] [PubMed] [Google Scholar]
- Wu H, Zeng H, Lam R, Tempel W, Amaya MF, Xu C, Dombrovski L, Qiu W, Wang Y, Min J. Structural and histone binding ability characterizations of human PWWP domains. PloS one. 2011;6:e18919. doi: 10.1371/journal.pone.0018919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoh SM, Lucas JS, Jones KA. The Iws1:Spt6:CTD complex controls cotranscriptional mRNA biosynthesis and HYPB/Setd2-mediated histone H3K36 methylation. Genes & development. 2008;22:3422–3434. doi: 10.1101/gad.1720008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yuan F, Presnell SR, Tian K, Gao Y, Tomkinson AE, Gu L, Li GM. Reconstitution of 5′-directed human mismatch repair in a purified system. Cell. 2005;122:693–705. doi: 10.1016/j.cell.2005.06.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.