

Abstract
Understanding the molecular basis of within and between species phenotypic variation is one of the main goals of Biology. In Drosophila, most of the work regarding this issue has been performed in D. melanogaster, but other distantly related species must also be studied to verify the generality of the findings obtained for this species. Here, we make the case for D. americana, a species of the virilis group of Drosophila that has been diverging from the model species, D. melanogaster, for approximately 40 Myr. To determine the suitability of this species for such studies, polymorphism and recombination estimates are presented for D. americana based on the largest nucleotide sequence polymorphism data set so far analyzed (more than 100 data sets) for this species. The polymorphism estimates are also compared with those obtained from the comparison of the genome assembly of two D. americana strains (H5 and W11) here reported. As an example of the general utility of these resources, we perform a preliminary study on the molecular basis of lifespan differences in D. americana. First, we show that there are lifespan differences between D. americana populations from different regions of the distribution range. Then, we perform five F2 association experiments using markers for 21 candidate genes previously identified in D. melanogaster. Significant associations are found between polymorphism at two genes (hep and Lim3) and lifespan. For the F2 association study involving the two sequenced strains (H5 and W11), we identify amino acid differences at Lim3 and Hep that could be responsible for the observed changes in lifespan. For both genes, no large gene expression differences were observed between the two strains.
Keywords: Drosophila americana genome, polymorphism, recombination, lifespan evolution
Introduction
Understanding the molecular basis of within- and between-species phenotypic variation is one of the main goals of Biology, but in Drosophila, it has been determined in a few cases only (Catania et al. 2004; Schlenke and Begun 2004; Aminetzach 2005; Pool and Aquadro 2007; Bono et al. 2008; Fry et al. 2008; Matzkin 2008). Moreover, most of the work regarding this issue has been done in model species, such as D. melanogaster and closely related species. Therefore, it is unclear to what extent what is known for model species can be generalized to other nonmodel species. It should be noted that even within a single genus, such as Drosophila, cases of lineage-specific adaptive evolution have been found (Barbash et al. 2004; Presgraves and Stephan 2007; Bachtrog 2008; Llopart and Comeron 2008; Morales-Hojas et al. 2009), and this could be an indication that the genetic basis of phenotypic variation might be different in distantly related Drosophila groups. Moreover, genes that have been reported as harboring variability that explains within species phenotypic variation in D. melanogaster, have been found to be missing in distantly related Drosophila species (Reis et al. 2011).
Ideally, the Drosophila species to be chosen for the needed comparative studies should have the following features: be distantly related to D. melanogaster, be easy to collect across the entire range of the distribution and easy to maintain in the laboratory, present a large amount of genotypic and phenotypic variation, show evidence for a large amount of historical recombination, little evidence of population subdivision or chromosomal polymorphisms, and have at least one genome available.
Several Drosophila species are likely suitable for the needed comparative work but here, we make the case for D. americana that belongs to the virilis group of species (Drosophila subgenus). Although different age estimates have been obtained for the divergence of the Drosophila and Sophophora (to which D. melanogaster belongs) subgenera, recent work using a relaxed molecular clock, multiple calibration points, multiple genes, and many species suggests a divergence time of approximately 40 Myr (see Morales-Hojas and Vieira [2012] for a detailed discussion on the age of the two subgenera). Drosophila virilis, a species that has been diverging from D. americana for approximately 4.1 Myr (Morales-Hojas et al. 2011) has its genome already sequenced (Drosophila 12 Genomes et al. 2007). This species is native to the eastern Palearctic and Oriental realms (Alexander 1976), and thus, this is where most phenotypic and genotypic variation should be found (Vieira and Charlesworth 1999). Only a few wild-caught D. virilis individuals from these realms are, however, available.
Drosophila americana is native to the United States where it has been independently evolving for approximately 1 Myr (Caletka and McAllister 2004; Morales-Hojas et al. 2008). This species is widely distributed, across the Central and Eastern regions of the United States from the South (Texas to the states around the Gulf of Mexico) to the North of the country (from Montana to Maine) (Patterson and Stone 1952). This species can be easily collected along the margins of marshes, lakes, and rivers, especially those where there is a high density of Salix species (Throckmorton 1982), and in recent years, several articles were published using hundreds of wild-caught D. americana individuals from different populations (Vieira et al. 2001; McAllister 2002; McAllister et al. 2008; Reis et al. 2008).
Drosophila americana is thought to present a large amount of genotypic variation, low levels of population structure and a stable historical population size (Schäfer et al. 2006; Morales-Hojas et al. 2008). The phenotypic variation of this species regarding ecologically relevant traits is already being explored (Wittkopp et al. 2011; Reis et al. 2011).
Polymorphic chromosomal rearrangements can be problematic when performing association studies (one of the main tools when addressing the molecular basis of phenotypic variation), because they may create linkage disequilibrium over large physical distances, and thus, in principle it is best to choose a species without chromosomal polymorphism. In D. americana, however, there is one chromosomal fusion and six inversions with estimated frequency higher than 5% (X/4 fusion, Xc, 2b, 4a, 4b, 5a, and 5b, on Muller’s elements A/B, A, E, B, B, C, and C, respectively [Hsu 1952]).
Extensive sampling across the D. americana distribution range has shown that the X/4 fusion is present as a shallow cline being frequent in the north of the geographic distribution and almost absent in the south of the distribution (Vieira et al. 2001; McAllister 2002; McAllister et al. 2008). The X/4 fusion is no more than 29,000 years old (Vieira et al. 2006) and arose on a Xc-inverted chromosome (Vieira et al. 2001, 2006; McAllister 2002). The Xc inversion is in between 0.27 and 1.6 Myr old (Hsu 1952; Spicer and Bell 2002; Caletka and McAllister 2004; Vieira et al. 2006; Morales-Hojas et al. 2008, 2011). It should be noted that, according to Hsu (1952), 97.5% of the X/4 fusion chromosomes harbor the Xc inversion, whereas only 7.5% of the nonfusion chromosomes show the Xc inversion. To have a stable X/4 fusion–Xc gradient, at the molecular level, the selection target(s) must be completely associated with the X/4–Xc chromosomal arrangement.
The frequency of the polymorphic 2b, 4a, 4b, 5a, and 5b inversions is different in the north and south of the distribution. It should be noted that 84.5% of 4a inversion chromosomes show the 4b inversion, whereas only 3.2% of 4b-inverted chromosomes do not show the 4a inversion (Hsu 1952). There is no physical overlap between inversions 5a and 5b but the two inversions are never found on the same chromosome, although a large number of individuals from the center of the D. americana distribution show both inversions (Hsu 1952). There are no chromosomes without both 5a and 5b (Hsu 1952).
Molecular markers are available for the X/4 fusion, and 4ab and 5b inversions (Vieira et al. 2001; Evans et al. 2007; Reis et al. 2008, 2011). Perfect markers could be developed for 4a and 5a inversions because the breakpoints of these inversions have been determined at the molecular level (Evans et al. 2007; Fonseca et al. 2012). Therefore, if needed, individuals (or strains) can be easily surveyed for their karyotype before using them in association studies.
To show that D. americana is suitable for comparative studies on the molecular basis of phenotypic variation, in the first sections of this article, we provide the most detailed estimates for this species, regarding polymorphism levels and patterns, based on 110 nucleotide gene sequence data sets (including 34 new data sets), as well as, on genome data for two D. americana strains genomes. The D. americana genomes here reported are the first ones for this species. Moreover, we provide the most detailed estimate for the historical recombination rate for this species, based on the 110 nucleotide gene sequence data sets here analyzed. Finally, to show that D. americana presents a large amount of phenotypic variation, and that the genome data here made available can greatly speed up the research on the molecular basis of phenotypic variation, we perform a preliminary study on the molecular basis of lifespan differences in D. americana. We show significant differences in lifespan between different D. americana populations and perform five F2 association studies for lifespan, using 21 candidate genes. For the two genes showing an association with lifespan (hep and Lim3), we address whether large differences in expression levels and/or amino acid differences could explain the observed lifespan differences.
Materials and Methods
Polymorphism and Recombination Analyses Based on Multiple Individuals
For the 34 new gene sequence data sets, genomic DNA of wild-caught D. americana males, previously characterized for the presence of the Xc inversion and X/4 fusion using molecular markers (see Reis et al. [2008] for details) was used for polymerase chain reaction (PCR) amplification. The amplification products were isolated from a 1.2–1.5% agarose gel using the QIAEXII Gel Extraction Kit (QIAGEN, CA, USA). Then, they were cloned using the TOPO-TA Cloning Kit for Sequencing from Invitrogen (Invitrogen, Spain). DNA sequencing was performed with the ABI PRISM BigDye cycle-sequencing kit version 1.1 (Perkin Elmer, CA, USA), using the universal primers for the priming sites present in the vector arms. Sequencing runs were performed by STAB VIDA (Lisbon, Portugal). For each individual, at least three colonies were analyzed to correct for possible PCR errors. For each gene, the primers, PCR amplification conditions used, as well as, accession numbers for the new DNA sequences, can be found in supplementary table S1, Supplementary Material online. The remaining 76 data sets are from previous studies on D. americana (Hilton and Hey 1996; McAllister and Charlesworth 1999; McAllister and McVean 2000; Vieira et al. 2001, 2003, 2006; Begun and Whitley 2002; McAllister 2003; Maside et al. 2004; McAllister and Evans 2006; Evans et al. 2007; Morales-Hojas et al. 2008, 2009; Betancourt et al. 2009; Wittkopp et al. 2009, 2011; Reis et al. 2011). Polymorphism (π and θ values) estimates were obtained using the DnaSP software (Librado and Rozas 2009). The average polymorphism data set is 967-bp long (the median is 823 bp).
Maximum likelihood estimates of polymorphism and recombination levels were obtained using LAMARCK version 2.1.6 (http://evolution.gs.washington.edu/lamarc/index.html, last accessed March 26, 2013; Kuhner 2009) under models assuming constant population size and models allowing for population size changes. Gene conversion tracts were identified using the method of Betran et al. (1997), as implemented in DnaSP (Librado and Rozas 2009). The number of fixed, shared, and polymorphic mutations in one chromosomal background that are monomorphic in the other chromosomal background were also obtained using DnaSP.
Genome Sequencing
Genomic DNA was isolated using the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Illumina paired-end libraries were generated following the instructions of the Illumina Paired-End Sample Preparation protocol. For the isofemale strain H5 established using a single inseminated female collected in 2004 at Lake Hurricane, Mississippi, two libraries with different fragment sizes (220 and 480 bp) were generated for paired-end sequencing, while for the isofemale strain W11 established using a single inseminated female collected in 2004 at Lake Wappapelo, Missouri, only a single library (fragment size 280 bp) was used for paired-end sequencing. Each of the libraries was run on a single GAIIx lane using the paired-end read (2 × 101 bp) protocol. Cluster generation and sequencing were performed using the Illumina Paired-End Cluster Generation Kit version 4 and the Sequencing Kit version 4 according to the manufacturer’s instructions.
Using D. virilis as a reference, the inferred (using chromosomal inversion molecular markers) basic chromosomal formula for the two sequenced D. americana strains is Xabc, 2a, 3, 5a, and 6 (Muller’s elements A, E, D, C and F, respectively) (Fonseca et al. 2012). Inversions Xa, Xb, and 2a are fixed between D. virilis and D. americana (Hsu 1952). Inversion Xa occurred in the virilis lineage (Fonseca et al. 2012) and not in the americana lineage as originally reported (Hsu 1952).
Assembly of the D. americana Genomes
We started with approximately 180 million reads produced from 3 paired-end sequencing libraries with insert sizes of approximately 220, 280, and 480 bp, encompassing a total of approximately 18 GB of sequence with an average read length of 100 bp. Reads with an average read quality below 10 were discarded (∼4% of the total). There is no evidence for contamination with bacterial DNA in the assembled genomes.
A de novo assembly of each strain was performed independently using Abyss version 1.2.6 (Simpson et al. 2009). The resulting contigs were scaffolded using SSPACE (Boetzer et al. 2011) and Minimus2 (Sommer et al. 2007). Small scaffolds with less than 200 bp were discarded. Finally, using the D. virilis genome (release 1.2, February 2010) as a reference, the scaffolds were clustered into the six Muller's elements and then ordered and oriented using Mauve (Rissman et al. 2009; Darling et al. 2010). When the contigs/scaffolds of known position are used, the mean coverage for strains H5 and W11 is 75× and 60×, respectively. These numbers were estimated by mapping back the reads to the assemblies using the Bowtie aligner (Langmead et al. 2009). The SAM file produced by Bowtie was processed using SAM tools (Li et al. 2009) to obtain the mean coverage of the positions covered by reads. The alignments generated by Mauve were edited and used to estimate polymorphism and divergence values between D. americana and D. virilis along chromosomes using Variscan (Vilella et al. 2005).
Lifespan
To determine the average lifespan of D. americana flies from different regions of the species range, the lifespan of a single male and a single virgin female from the following 65 isofemale strains was used: (from the very north of the distribution: O27, O28, O29, O30, O31, O32, O33, O34, O35, O36, O37, O38, O39, O40, O42, O43, O45, O47, O50, O53, O57, O61, O62, O64, O66, O67, and O69; from the center of the distribution: HI1, HI13, HI14, HI15, HI18, HI23, HI25, HI27, HI29, W4, W10, W11, W18, W23, W25, W26, W27, W28, W29, W33, W36, W37, W42, and W46 [see Reis et al. {2008} for details]; from the very south of the distribution: CB05.08, CB05.10, CB05.20, CB05.22, CI05.02, CI05.28, CI05.30, CI05.36, RB10.10, RB10.12, RB10.14, RB10.16, RB10.20, and RB10.22; the strains from the south of the distribution have been donated by Bryant McAllister from Iowa University; details can be found at http://www.biology.uiowa.edu/mcallister/bfm_flies.html, last accessed March 26, 2013). The O strains have been collected in 2008, the HI and W strains in 2004, the CB and CI in 2005 and the RB strains in 2010 (see Reis et al. [2008] and http://www.biology.uiowa.edu/mcallister/bfm_flies.html, last accessed March 26, 2013 for details). Single individuals were kept in a vial containing standard food (10% [mass/volume] yeast, 4% [mass/volume] wheat flour, 8% [mass/volume] sugar, 0.4% [mass/volume] salt diet, 1% agar [mass/volume], and 0.5% propionic acid [volume/volume]), at 25 °C constant temperature.
To gain further insight into the differences found between D. americana populations, we first crossed a male and a virgin female from the same strain (for both O57 and W29) to ensure that, by typing these individuals with molecular markers, we knew the karyotype of all the progeny that is used in the crosses we set up. According to molecular markers, the O57 individuals used show the D. americana polymorphic chromosome rearrangements X/4 fusion, Xc, 4ab, and 5b inversions (Muller’s elements A/B, A, B and C, respectively), while the W29 individuals used only show the 5a inversion (Muller’s element C). To synchronize the whole experiment, at the F1 generation, brother–sister mating, as well as, crosses between a single O57 male and a single W29 female, and a single O57 female and a single W29 male were performed. Approximately 50 males and 50 females were collected from each cross, in a total of approximately 400 individuals. Single flies were kept in individual flasks containing standard medium. All flies were checked every 2 days and the vials were changed every week (25 °C) and every 2 weeks (18 °C) until they were dead. These experiments were performed in climate chambers both at 18 and 25 °C (400 individuals for each temperature tested) with cycles of 12 h of light and 12 h of dark to address how temperature influences lifespan. Summary statistics and nonparametric association tests were performed using the software SPSS Statistics version 17.0 (SPSS Inc., Chicago, IL, USA).
Likelihood Tests of Selection
The random-sites models implemented in the PAML software (Yang 2007) have been used. The likelihoods estimated using neutral (M7) and positive selection (M8) models were compared using a Likelihood Ratio Test. For 46 lifespan candidate genes (Paaby and Schmidt 2009), we attempted to retrieve sequences from the 12 publicly available annotated Drosophila species (D. melanogaster, D. simulans, D. sechellia, D. yakuba, D. erecta, D. ananassae, D. persimilis, D. pseudoobscura, D. willistoni, D. grimshawi, D. mojavensis, and D. virilis) from Flybase (http://flybase.org/, last accessed March 26, 2013), although this was not always possible due to nonannotation, missannotation and unclear orthology. The phylogenetic trees used as input in the codeml analysis were estimated using MrBayes (Ronquist and Huelsenbeck 2003). The GTR model of sequence evolution was used, allowing for among-site rate variation and a proportion of invariable sites. Third codon positions are allowed to have a gamma distribution shape parameter that is different from that of first and second codon positions. Two simultaneous and completely independent analyzes, starting from random trees, were run for 500,000 generations (each with one cold and three heated chains). Samples were taken every 100th generation. The first 1,250 samples were discarded (burn-in).
Genotype-Phenotype Association Studies
Five isofemale strains (H5 from Lake Hurricane, Mississippi, originally collected in 2004; W11, W29, and W46 from Lake Wappapelo, Missouri, originally collected in 2004; and O57 from Fremont, Nebraska, originally collected in 2008) were used to establish five crosses (F0) between a single male and a single female (H5♂ × W11♀; W11♂ × W46♀; W29♂ × O57♀; O57♂ × H5♀; and W46♂ × W29♀). These strains were selected, because they were established with flies originated from distinct regions of the distribution, and present different chromosome arrangements. After mass breeding between F1 males and females, for each cross, approximately 100 F2 males were individually collected in single vials after eclosion and maintained at 25 °C under 12 h of light and 12 h of dark cycles, until they were dead. Vials were checked every 2 days and changed every week. Based on the genome information on D. americana strains H5 and W11, markers were developed for a set of 21 lifespan candidate genes. For each gene, information on the primers, restriction enzymes, PCR amplification conditions used, as well as, the single nucleotide polymorphism (SNP) that was typed can be found in supplementary table S2, Supplementary Material online. Genotype–phenotype associations were tested using nonparametric tests as implemented in SPSS Statistics version 17.0 (SPSS Inc.,Chicago, IL, USA). The sequential Bonferroni correction for multiple testing has been used. Using the same software, linear regression analyses (including a constant) were performed, to estimate the amount of phenotypic variation explained by variation in candidate genes.
Gene Expression Analyses
Expression levels for genes showing a statistically significant association after Bonferroni correction in the H5♂ × W11♀ cross were determined in sets of three male individuals 0, 10, 30, and 60 days old from strains H5 and W11, to account for the possibility that gene expression changes are observed in some adult stages only. These sets of three individuals were maintained in single vials at 25 °C, under 12 h of light and 12 h of dark cycles and food vials were changed every week until they have reached the age required to perform the experiments. For both hep and Lim3, differences in expression levels are apparently highest at day 0 (see Results). To verify whether the differences are statistically significant, the expression levels of both genes were determined separately in three male individuals 0 day old from strains H5 and W11.
Living individuals were frozen in liquid nitrogen and total RNA was extracted from each set of individuals using TRIzol Reagent (Invitrogen, Spain) according to the manufacturer’s instructions and treated with DNase I (RNase-Free) (Ambion, Portugal). cDNA was synthesized by reverse transcription with SuperScript III First-Strand Synthesis SuperMix for quantitative reverse transcriptase-PCR (qRT-PCR) (Invitrogen, Spain) using random primers. Reactions where template was not added, and reactions with RNA that was not reverse transcribed were performed to confirm the absence of genomic DNA contamination.
Highly efficient specific primers (supplementary table S3, Supplementary Material online) were used to perform qRT-PCR experiments using the synthesized cDNA and every experiment was performed in duplicate. Amplification efficiency of each primer pair was checked with serial dilutions of cDNA (data not shown). qRT-PCR was performed with the iQ SYBR Green Supermix (Bio-Rad, Portugal) according to the manufacturer’s instructions in a Bio-Rad iCycler with the following program: 3 min at 95 °C; 40 cycles of 30 s at 94 °C, 30 s at 56 °C and 30 s at 72 °C followed by a standard melting curve. The endogenous Ribosomal protein L32 (RpL32) was used as the reference gene. Fold change in expression was calculated using the 2−ΔΔCT method (Livak and Schmittgen 2001).
Results
Characterization of Levels and Patterns of Nucleotide Variation Using Gene Sequences
Species with high polymorphism levels are best suited for comparative studies on the molecular basis of phenotypic variation. Therefore, to address the suitability of D. americana as a species for comparative studies on the molecular basis of phenotypic variation, we first characterize levels and patterns of nucleotide variation across its genome, using both nucleotide sequence data from many unrelated individuals (fig. 1 and supplementary table S4, Supplementary Material online), as well as, the genome sequence data of two unrelated strains (discussed later).
Fig. 1.—

Per site polymorphism (θ) estimates along the Drosophila americana genome using multiple individuals and all (black bar), silent (dark blue bar), synonymous (cyan bar), intron (green bar), and nonsynonymous (red bar), sites. The average per site polymorphism value estimated from the gene sequence data sets for that region is represented by black (all sites), dark blue (silent sites), cyan (synonymous sites), green (intron sites), and red (nonsynonymous) lines. The dotted black line represents the average per site polymorphism value estimated from the genome data for the chromosome (all sites).
When using nucleotide sequence data from many unrelated individuals, regions that were a priori predicted to have different polymorphism values than the average, such as, the base of the X chromosome (Muller’s element A) (Vieira et al. 2006) or regions influenced by chromosomal inversions (Evans et al. 2007) were treated separately. Levels of synonymous and intron variability are similar (nonparametric sign test; P > 0.05), whereas silent and nonsynonymous variability levels are statistically different (nonparametric sign test; P < 0.001). On average silent site variability levels are 13.1 times higher than nonsynonymous site variability levels.
For chromosome 3 (Muller’s element D), 20 sequence sets from genes equally spaced along the chromosome are now available. It should be noted that this chromosome shows almost no inversion polymorphism (only one polymorphic inversion with an estimated frequency lower than 1% [Hsu 1952]) that could affect polymorphism levels. For this chromosome, the average level of silent site diversity is 1.876% (supplementary table S4, Supplementary Material online). The effective population size (Ne) of a species is proportional to the level of intraspecific neutral or nearly neutral polymorphism, and thus Ne can be estimated if the mutation rate is known (Kimura 1985; Ohta 1992). On the other hand, the mutation rate can be estimated from per site silent divergence values between species, if the divergence time and the number of generations per year are known. For chromosome 3 genes, the average per site silent site divergence (with Jukes–Cantor correction) between D. americana and D. virilis is 0.08705 (data not shown; the two species have been diverging for 4.1 Myr [Morales-Hojas et al. 2011]). When using D. americana flies from the south of the distribution, at 25 °C, the average of the minimum time of egg to adult is 16.8 days (Vieira J., Reis M., Vieira C.P., unpublished data). Moreover, in this species, at 25 °C, it takes at least four additional days to fully develop the ovaries (Vieira J., Reis M., Vieira C.P., unpublished). Therefore, the minimum generation time for this species is 21 days. In the south of the distribution, given the monthly average temperatures, there are likely 6 months when breeding conditions are adequate. Thus, it is conceivable that in the south of the distribution there are at least eight generations every year. Therefore, the mutation rate is estimated to be 1.3 × 10−9 (0.08705/[2 × 4.1 × 10−6 × 8]) per site and per year, under the assumption of equal mutations rates in the virilis and americana lineages. This mutation rate is similar to previous estimates for D. virilis (0.9–1.4 × 10−9 per site and per year [Vieira and Charlesworth 1999]) and D. melanogaster (2 × 10−9 per site and per year [Aquadro et al. 1994]). The inferred D. americana effective population size is thus 3.6 million individuals. This is the geometric average of the D. americana historical effective population size over a time period on the order of 4Ne generations (Kimura 1985; Ohta 1992), and thus, in this species, levels of variability likely reflect its history over a period of more than 1 Myr, that is, approximately the time D. americana has been independently evolving (Caletka and McAllister 2004; Morales-Hojas et al. 2008).
Unexpectedly, given that the effective population size of the X chromosome should be three-fourth of that inferred for autosomes (Kimura 1985; Ohta 1992), levels of polymorphism are similar at the 3rd chromosome and X chromosome regions unaffected by segregating chromosomal rearrangements. Such a pattern has been also observed in D. melanogaster, where it has been argued that background selection (the hitchhiking effect of deleterious mutations) is more effective on the autosomes than on the X chromosome, because of the lack of crossing over in male Drosophila (Charlesworth 2012).
When comparing the same X chromosome region in between pros28.1 and Yp1, levels of variability are almost the double in nonfusion chromosomes than in X/4 (Muller’s elements A/B) fusion Xc-inverted chromosomes. This suggests that the two regions are evolving separately. Indeed, the survey of 14,903 bp in this region (fused1–Yp1 region), revealed 119 apparent fixed nucleotide differences (including five that encode for apparent amino acid differences) between X/4 fusion Xc-inverted and standard chromosomes (table 1). Given the estimated polymorphism levels for X standard chromosomes, approximately 20 apparent fixations are expected, immediately after the appearance of the Xc-inverted chromosomes (estimate obtained using the approach of Vieira et al. [2001]; data not shown). All other fixations are expected to have occurred after the appearance of the Xc inversion. Moreover, in this region only 5% of all polymorphisms are shared between chromosomal types. It should be noted that these estimates are obtained after removing sequences that showed evidence for rare recombination events between the two chromosomal backgrounds (gene conversion tracts). There is an excess of derived mutations in X/4 fusion Xc-inverted chromosomes because of the unique origin of these rearrangements (table 2). Nevertheless, there are also regions (graf and Trf4-1) where too many derived fixed silent mutations are detected in the standard chromosomal arrangement (table 2). This could be an indication of positive selection operating in these regions on standard chromosomes. The Tajima’s D value for graf, when using all variants and only chromosomes with the standard chromosomal arrangement, indicates, however, no departure from neutrality (D = +0.06; P > 0.05). For Trf4-1, the Tajima’s D value, when using all variants and only chromosomes with the standard chromosomal arrangement (D = −1.75; P < 0.05), indicates a departure from neutrality. It should be noted that for Trf4-1 there is a derived amino acid fixed difference in the standard chromosomal arrangement. Trf4-1 is involved in the polyadenylation-mediated degradation of snRNAs (Nakamura et al. 2008). Further studies are needed to address the possibility that positive selection is partially responsible for the observed divergence between X/4 fusion and nonfusion chromosomes in the fused1–Yp1 region.
Table 1.
Differentiation between X/4 Fusion Xc-Inverted and Standard Chromosomes
| Gene | PSa | PFa | Fa | Sa | GCT |
|---|---|---|---|---|---|
| Dpr8 | 1 | 1 (1) | 0 | 0 | |
| nej | 5 | 2 | 0 | 0 | |
| CG18543 | 17 (3) | 4 (2) | 3 (1) | 4 | 1 |
| CG18543b | 21 (3) | 2 (2) | 3 (1) | 0 | |
| para | 16 | 18 | 0 | 4 | |
| Fused1 | 43 (6) | 8 (5) | 7 (1) | 1 | |
| Rhp | 15 (1) | 15 | 0 | 2 | 1 |
| Rhpb | 16 (1) | 6 | 0 | 1 | |
| CG1657 | 27 | 8 | 6 | 1 | |
| Org1 | 54 | 25 | 1 | 9 | 1 |
| Org1b | 62 | 14 | 6 | 1 | |
| Cp36 | 2 | 1 | 0 | 0 | |
| CG32632 | 34 | 4 | 13 | 0 | |
| App1 | 32 | 24 | 0 | 16 (1) | 2 |
| App1b | 10 | 28 | 0 | 13 (1) | |
| Or1a | 74 | 10 | 20 | 2 | 2 |
| Or1ab | 67 | 12 | 28 | 2 | |
| Graf | 18 (1) | 6 (1) | 10 | 2 | 1 |
| Grafb | 20 (1) | 5 (1) | 11 | 0 | |
| Nep3 | 55 | 12 | 16 (1) | 1 | 1 |
| Nep3b | 49 | 12 | 17 (1) | 1 | |
| CG8949 | 82 | 16 | 0 | 30 | 2 |
| CG8949b | 84 | 26 | 0 | 16 | |
| Trf4-1 | 27 | 27 (4) | 13 (1) | 1 (1) | 2 |
| Trf4-1b | 27 | 12 (4) | 18 (1) | 1 (1) | |
| Sp1 | 27 | 20 | 7 | 1 | 2 |
| Sp1b | 24 | 17 | 9 | 1 | |
| Yp1 | 14 (2) | 9 | 4 | 2 (1) | |
| Totalb | 525 (13) | 185 (13) | 122 (6) | 44 (3) |
Note.—PS, total number of mutations polymorphic in standard chromosomes, but monomorphic in X/4 fusion Xc-inverted chromosomes; PF, total number of mutations polymorphic in X/4 fusion Xc-inverted chromosomes, but monomorphic in standard chromosomes; F, fixed mutations between standard and X/4 fusion Xc-inverted chromosomes; S, shared mutations between standard and X/4 fusion Xc-inverted chromosomes; GCT, number of sequences showing gene conversion tracts.
aValues in parentheses are the number of amino acid replacements.
bAfter excluding sequences showing gene conversion tracts.
Table 2.
Apparently Fixed Silent Mutations between X/4 Fusion Xc-Inverted and Standard Chromosomes
| Gene | Silent Fixations | Derived Silent Fixations in the X/4 Fusion Xc- Inverted and Standard Chromosomesa | Expected Number of Silent Apparent Fixations due to the Single Origin of X/4 Fusion Xc-Inverted Chromosomesb | Corrected Valuesc |
|---|---|---|---|---|
| Fused1 | 6 | 6 and 0 | 1.36 | 4.64 and 0* |
| Rhp | 0 | 0 and 0 | 0.52 | 0 and 0 |
| CG1657 | 6 | 4 and 1 | 0.90 | 3.1 and 1 |
| Org1 | 6 | 2 and 2 | 2.02 | 0 and 2 |
| Cp36 | 0 | 0 and 0 | 0.22 | 0 and 0 |
| CG32632 | 13 | 6 and 6 | 0.97 | 5.03 and 6 |
| App1 | 0 | 0 and 0 | 1.54 | 0 and 0 |
| Or1a | 28 | 12 and 3 | 2.44 | 9.56 and 3 |
| Graf | 11 | 1 and 6 | 0.61 | 0.39 and 6* |
| Nep3 | 16 | 9 and 4 | 1.80 | 7.2 and 4 |
| CG8949 | 0 | 0 and 0 | 3.60 | 0 and 0 |
| Trf4-1 | 17 | 3 and 12 | 0.87 | 2.13 and 12*** |
| Sp1 | 9 | 4 and 2 | 0.93 | 3.07 and 2 |
| Yp1 | 4 | 3 and 1 | 1.42 | 1.58 and 1 |
aThe ancestral state, when using D. virilis gene sequences as the outgroup, could not be determined for all apparently fixed mutations between the two chromosomal backgrounds.
bAccording to Vieira et al. (2001).
cFor the X/4 fusion Xc-inverted chromosomal background this may be an overcorrection, because it is not possible to infer the ancestral state, when using D. virilis gene sequences as an outgroup, for all variants that are apparently fixed between the two chromosomal backgrounds.
*P < 0.05.
***P < 0.01.
The fused1–Yp1 region is approximately 2.9-Mb long and thus, given the large number of fixed differences observed between the two chromosomal arrangements, it is estimated to harbor tens of thousands apparent fixed mutations (including hundreds of fixed amino acid differences and mutations in regulatory regions) between the two chromosomal backgrounds. Therefore, it can in principle harbor the causative mutations responsible for phenotypic differences between populations from the north and the south of the D. americana distribution. It is likely that the region where significant differentiation is found between the two chromosomal backgrounds is larger than the fused1–Yp1 region.
Although levels of variability are similar at chromosome 2 (Muller’s element E) and 3 (Muller’s element D), levels of variability seem to be higher for chromosome 4 (Muller’s element B). As reported previously, variability levels for chromosome 6 (Muller’s element F) are much lower than for the remaining of the genome (Betancourt et al. 2009), and are 7.6 times lower than for chromosome 3.
Characterization of Levels and Patterns of Nucleotide Variation Using Genome Data
Although the use of nucleotide sequence data from multiple unrelated individuals from different populations across the species distribution allows the generalization of the conclusions to the whole species, it is necessarily limited to a small fraction of the genome. Therefore, we also estimate levels of nucleotide variability using genome data that we acquired for two D. americana strains. These are the first genomes for this species.
Using a de novo approach (see Materials and Methods), it was possible to assemble the genome of the D. americana strains H5 and W11 into 24,251 and 34,687 scaffolds, respectively (table 3). In some cases, the same genome region may be represented by different scaffolds, as the result of the assembly process, likely due to polymorphism segregating within strains, the use of short reads or both. Even a human-curated analysis of every genome region may not be enough to fully address this issue. For strains H5 and W11, more than 50% of the scaffolds are larger than 28,025 and 19,358 bp, respectively, and more than 90% of the scaffolds are larger than 11,961 and 8,256 bp, respectively. Within scaffolds, there are very few (<0.5%) undetermined positions. In D. melanogaster, there are 22,509 annotated proteins (including multiple isoforms produced by the same gene; FlyBase version FB2010_08, released 13 October 2010). Using tblastx, after discarding the matches with an alignment smaller than 50% of the total length of the protein, and considering as a hit only those entries that produce an E value smaller than 1E−10, 70.2% of the annotated D. melanogaster proteins have a hit in both D. americana genomes (15,308 hits; fig. 2), whereas 5.8% and 6.2% show a hit only in the H5 and W11 genomes, respectively. Nevertheless, when using the same approach, 84.1% of the annotated D. melanogaster proteins have a hit in the D. virilis genome (18,934 hits; fig. 2). The D. virilis genome is a high coverage genome (Drosophila 12 Genomes et al. 2007). Therefore, it seems likely that approximately 15.9% of the orthologous D. melanogaster protein-coding genes cannot be recognized using this approach. Under the assumption that a similar fraction of genes cannot be recognized in D. americana, it is likely that approximately 83.5% (0.702/0.841) of the D. americana genes are present in the H5 and W11 genomes. The genome data here reported is thus useful for most gene-centered studies. Blast searches of the H5 and W11 genomes can be performed at http://cracs.fc.up.pt/∼nf/dame/index.html, last accessed March 26, 2013. The assembled scaffolds can be also downloaded from this site.
Table 3.
Summary Statistics for the Two D. americana Genomes
| Strain | H5 | W11 |
|---|---|---|
| Size (kb) | 163,287 | 166,080 |
| Scaffolds | 24,251 | 34,687 |
| Max. (bp) | 229,288 | 135,613 |
| Mean (bp) | 6,733 | 4,788 |
| Min. (bp) | 200 | 200 |
| N50 length (bp) | 28,025 | 19,358 |
| N50 | 1,328 | 1,939 |
| N90 length (bp) | 11,961 | 8,256 |
| N90 | 4,083 | 5,955 |
| GC% | 40.57 | 40.42 |
| Ns | 732,237 | 641,609 |
| Ns% | 0.45 | 0.39 |
Note.—Size of the genome in thousand base pairs (kb), number of scaffolds, maximum (Max), mean and minimum (Min) scaffold size in bp. N50 length is the length of the shortest scaffold in an assembly such that the sum of scaffolds of equal length or longer is at least 50% of the total length of all scaffolds. N50 is the ordinal of the shortest scaffold in an assembly such that the sum of scaffolds of equal length or longer is at least 50 of the total length of all scaffolds. The percentage of GC content (GC%) and the number of unknown nucleotides in the scaffolds (Ns) and the respective percentage in the scaffolds (Ns%).
Fig. 2.—
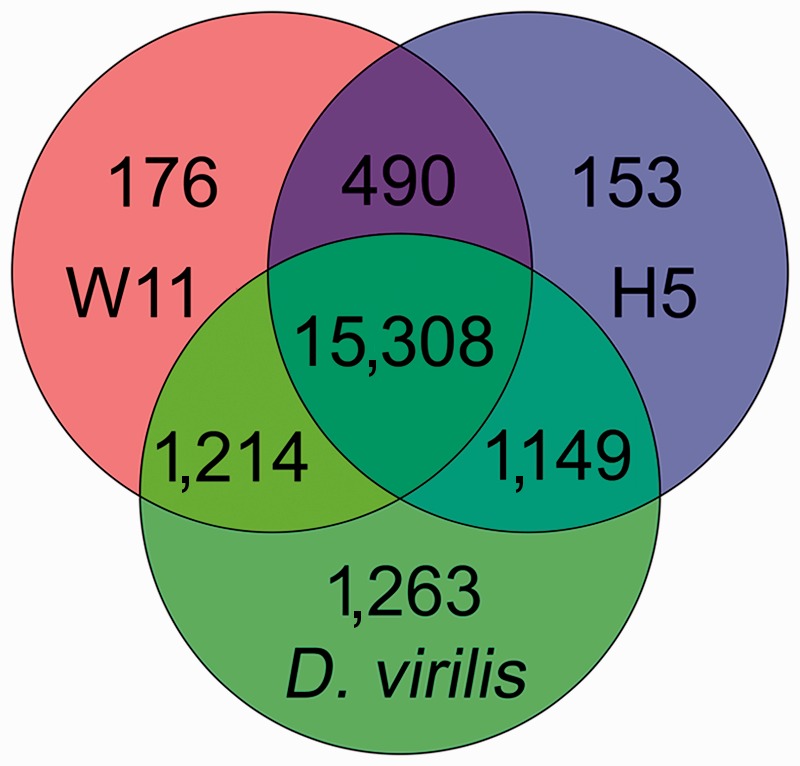
Venn diagram showing the number of Drosophila melanogaster coding sequences that produce a hit in the D. virilis scaffolds of known location, the H5 and W11 D. americana genomes. The D. melanogaster gene annotation was obtained from FlyBase (version FB2010_08, released 13 October 2010). We blasted D. melanogaster coding sequences (22,509 coding sequences, including all different isoforms) against the D. americana genomes (using tblastx with E-value lower than 1E−10 and discarding the matches with an alignment inferior to 50%).
The D. americana scaffolds were aligned with the large D. virilis scaffolds of known cytological location using Mauve (Rissman et al. 2009), and this alignment (after removing D. americana scaffolds that could not be aligned with the D. virilis scaffolds) was used to estimate D. americana polymorphism levels, as well as, divergence levels between D. americana and D. virilis, along chromosomes (fig. 3). When compared with the estimates obtained from gene sequences data sets, the average level of variability is higher for Muller’s elements B, D, E, and F (but not for Muller’s element A). Such an observation suggests that intergenic regions (poorly represented in the gene sequence data sets) are more variable than gene regions. As expected, for all chromosomes, variability levels are lower near telomeres and at the base of the chromosomes. Low variability valleys are observed in Muller’s elements A and C but these do not correspond to inversion breakpoint regions (fig. 3).
Fig. 3.—

Per site polymorphism estimates along Muller’s elements. The telomere is always on the left of the figure. It should be noted that the gene order presented in the figure is that of Drosophila virilis. The location of the breakpoints of derived inversions in the americana lineage is also shown. Each yellow dot corresponds to a polymorphism estimate using a sliding window with a width of 10,000 bp (the slide is 5,000 bp), and a minimum of 100 sites (after excluding gapped positions). Each pink dot corresponds to a D. americana–D. virilis total divergence estimate using the same criteria. Blue dots indicate the value of the polymorphism/divergence ratio. The average D. americana polymorphism estimate, the average D. americana–D. virilis divergence estimate and the total number of sites analyzed is also given.
The X chromosome (Muller’s element A) shows the second highest average divergence. In contrast to the observation made using polymorphism data sets (discussed earlier), after correcting for divergence, this chromosome shows the expected three-fourth reduction in polymorphism levels relative to autosomes. It should be noted that both strains H5 and W11 have X/4 fusion chromosomes. The observation made above that levels of variability are higher for chromosome 4 (Muller’s element B) than for the other large autosomes is supported by the genome data. For chromosomes 2, 3, and 5 (Muller’s elements E, D, and C, respectively), the average variability levels are 1.1%, 1.3%, and 1.2%, respectively, whereas for chromosome 4 it is 1.5%. Therefore, polymorphism levels for chromosome 4 seem to be higher than the genome average (1.28%), possibly due to a lower average degree of purifying selection acting on chromosome 4. Indeed, when correcting by the average divergence level between D. americana and D. virilis, the values for these chromosomes become more similar (the ratio polymorphism/divergence is 0.19, 0.23, 0.24, and 0.21 for chromosomes 2, 3, 4, and 5, respectively). Chromosome 6 (Muller’s element F) variability levels are at least 10.2 times lower than for the other autosomes. This difference becomes even more significant if polymorphism values are corrected by divergence levels (the average is 15.5), because chromosome 6 presents the highest divergence levels.
Divergence levels for Muller’s elements C, D, and E (chromosomes 5, 3, and 3, respectively) are very similar (5.8%, 5.6%, and 6.0%, respectively), as well as, their polymorphism levels (1.2%, 1.3%, and 1.1%, respectively). Knowing that D. americana and D. virilis have been diverging for 4.1 Myr (Morales-Hojas et al. 2011), that the average divergence and polymorphism for these chromosomes is 5.8% and 1.2%, respectively, and under the assumption of eight generations a year (discussed earlier), an effective population size of approximately 3.5 million individuals can be estimated. This value is similar to the 3.6 million individuals estimate obtained in the previous section using data for chromosome 3 and silent sites only.
Recombination Rates
Genotype–phenotype association experiments are one major tool to address the molecular basis of phenotypic variation. The use of unrelated individuals from species that show high historical recombination rates across its genome allows the fine mapping of the mutations responsible for the observed phenotypic variation. Therefore, species that show high historical recombination rates are well suited for comparative studies on the molecular basis of phenotypic variation.
Likelihood estimates of the per site recombination rate were obtained for different regions of the genome, using the gene sequence data sets. Regions, that a priori were postulated to have different recombination rates, such as the base of the X chromosome (Vieira et al. 2006) or regions influenced by chromosomal inversions (Evans et al. 2007) were treated separately. Results are shown in table 4. The estimates obtained under a constant size model and under a population size change model are similar. Therefore, the per site recombination rate estimates are not dependent on the assumption of a constant population size. Most estimates are within the 0.56–0.69 interval. The highest per site recombination estimate is obtained for the base of the X chromosome of X/4 fusion chromosomes. Nevertheless, the global estimate that is shown is based on four gene data sets only and the estimate for different gene regions from the same data set varies widely (from 0.19 to 27.17; data not shown). Therefore, it is likely that the global estimate does not reflect the true per site recombination rate for this region. The per site recombination estimate for the base of the X chromosome of nonfusion chromosomes is similar to that obtained for other genomic regions. Therefore, it is conceivable that recombination is not highly suppressed in this 2 Mb region. It is possible that there is a slight effect of the 4ab inversion polymorphism on the levels of recombination experienced by the base of chromosome 4. The per site recombination estimate obtained for free 4th chromosomes from the north of the D. americana distribution is high but this estimate is based on two genes only. The per site recombination estimate obtained for chromosome 6 is based on a large number of genes (14 data sets), but it is higher than anticipated, because this chromosome harbors little variation (discussed earlier), and thus it is expected to show little evidence for recombination. Nevertheless, convergence could not be achieved when a model assuming population size changes is implemented, and thus the estimate obtained for chromosome 6 should be viewed with caution. The best estimate is that obtained for chromosome 3 that shows almost no inversion polymorphism in D. americana, and that is based on 20 genes equally spaced along the chromosome (the population per site recombination estimate is in between 0.69 and 0.72 depending on the model used). Therefore, D. americana shows high recombination levels across the major chromosomes.
Table 4.
Overall Maximum Likelihood Estimate of the Per-Site Recombination Rate for Different Regions of the D. americana Genome Under a Constant Size Population Model and Under a Model Allowing for Population Size Changes
| Constant Population Size Model (99% Lower and Upper Confidence Limits) | Population Size Change Model (99% Lower and Upper Confidence Limits) | Exponential Growth Rate (g)a | |
|---|---|---|---|
| Base of the standard X chromosome | 0.66 (0.41–0.96) | 0.86 (0.55–1.16) | −55, +90 |
| Base of the X chromosome of X/4 fusion | 1.18 (0.59–2.22) | 1.76 (0.75–2.88) | −298, +143 |
| Standard X chromosome (excluding the base of the chromosome) | 0.56 (0.48–0.64) | 0.36 (0.31–0.43) | +6, +81 |
| X/4 fusion chromosome (excluding the base of the chromosome) | 0.69 (0.55–0.85) | 0.63 (0.56–0.70) | −8, −5 |
| X Chromosome | 0.28 (0.24–0.31) | 0.30 (0.27–0.34) | +13, +77 |
| 2nd Chromosome | 0.69 (0.53–0.89) | 0.82 (0.62–1.00) | −33, +30 |
| 3rd Chromosome | 0.69 (0.60–0.78) | 0.72 (0.63–0.81) | −7, −4 |
| 4th 4ab-inverted chromosomes | 0.43 (0.31–0.59) | 0.55 (0.44–0.66) | −8, −5 |
| 4th Standard chromosome | 0.59 (0.51–0.68) | 0.85 (0.75–0.95) | −3, −2 |
| Free 4th chromosome from the north of the distribution | 0.78 (0.54–1.10) | 1.40 (1.13–1.72) | +1,058, +1,417 |
| 6th Chromosome | 0.38 (0.13–0.86) | n.a. | n.a. |
Note.—n.a., not applicable.
ag is defined in the following equation, where t is time before the present: θt = θpresent time exp(−gt). Positive values of g indicate population growth while negative values indicate population decline (Kuhner 2009).
Lifespan Differences between D. americana Populations
The previously published literature shows that D. americana is distantly related to the model species D. melanogaster, that it is widely distributed, and that it can be easily collected and maintained in the laboratory (see Introduction). Moreover, in the previous sections, it is shown that D. americana presents high levels of nucleotide variation and high historical recombination rates across most of the genome, two additional useful features when choosing a species for comparative analyzes on the molecular basis of phenotypic variation. Moreover, the two D. americana genomes that are here reported, clearly speed up the analyses that are needed to address this issue. Indeed, they allow the design of better primers than when using the available D. virilis genome and give insight into the location of D. americana SNPs and indel variation that can be used as markers in association studies. Nevertheless, it remains to be shown that it shows plenty of phenotypic variation regarding multiple phenotypic traits, and that the chromosomal polymorphism that is segregating in natural populations (and that may be maintained because of linkage to locally advantageous mutations; see Introduction), is not a problem. To get insight into the two latter issues, we performed a preliminary study on the molecular basis of lifespan differences that is presented later.
The first step of this preliminary study is to characterize the average lifespan in different D. americana populations, to determine whether there are significant differences between populations. When performing the experiment at 25 °C with a 12 h of light and dark cycles, the average lifespan of D. americana individuals from the north (O population; for males 50.3 ± 19.6 days; for virgin females 57.4 ± 19.6 days; for either N = 27), center (W + HI populations; for males 67.7 ± 17.6 days; for virgin females 67.7 ± 20.9 days; for either N = 23) and south (RB + CB + CI populations; for males 64.3 ± 15.7 days; for virgin females 78.0 ± 15.9 days; for either N = 14) populations show differences as large as 20.6 days (when comparing males from northern and southern populations), and significant associations are detected between population of origin and lifespan (nonparametric Kruskal–Wallis test; for males and virgin females P < 0.005 and P < 0.01, respectively). Significant differences are detected when looking at the lifespan of males from the north and center of the distribution (nonparametric Mann–Whitney test; P < 0.005), the north and south of the distribution (nonparametric Mann–Whitney test; P < 0.05) but not between the center and south of the distribution (nonparametric Mann–Whitney test; P > 0.05). The results remain significant after Bonferroni correction for multiple testing. For virgin females, significant differences are detected when comparing the north and south of the distribution (nonparametric Mann–Whitney test; P < 0.001), and are borderline nonsignificant when comparing the north and center of the distribution (nonparametric Mann–Whitney test; P = 0.051), but are nonsignificant when comparing the center and south of the distribution (nonparametric Mann–Whitney test; P > 0.05). The significant comparison remains significant after Bonferroni correction for multiple testing. On average, at 25 °C, males and virgin females from center and southern populations live 16.1 and 14.2 days more than males and virgin females from the northern population, respectively. It should be noted that not all strains are of the same age. Therefore, it could be argued that the observed differences are due to the accumulation of slightly deleterious mutations in the oldest strains. Nevertheless, this is not the case. The short-lived northern strains are amongst the most recently collected ones (see Material and Methods).
To gain further insight into the lifespan differences observed between males and females, as well as, when comparing different populations, D. americana individuals with the typical X/4 fusion, Xc, 4ab, and 5b inversions (strain O57) and individuals without these chromosomal rearrangements, and thus with inversion 5a (see Introduction; strain W29), have been crossed in both directions. The whole experiment was carried out at 25 °C and at 18 °C, to see how temperature could affect our conclusions. The results of these experiments are summarized in figure 4. For any given cross and temperature (with the exception of the W29♂ × W29♀ cross at 18 °C), females live longer than males, and this difference is more apparent at 25 °C than at 18 °C (females live on average from 11.3 to 17.8% longer than males at 18 °C, and from 22.8% to 98.7% longer than males at 25 °C). Therefore, in natural populations from the north of the D. americana distribution, males and females may show smaller lifespan differences than in the south of the distribution. We also note that at 18 °C, the average lifespan is always increased in comparison with the average lifespan for the same cross at 25 °C (ranging from 2.1 to 4.0 times more for males and 1.6 to 2.6 times more for females, depending on the comparison being performed).
Fig. 4.—

Schematic representation of average lifespan values (in days) for flies with different genomic backgrounds (progeny of crosses O57♂ × O57♀ [NN], O57♂ × W29♀ [NT], W29♂ × O57♀ [TN], and W29♂ × W29♀ [TT]) kept at 25 °C and 18 °C. Average lifespan values inside the same box are statistically similar (Mann–Whitney test, P > 0.05) and the distances between boxes (black lines) are proportional to the differences between the averages. LS, lifespan.
The observation that heterozygous F1 individuals have a tendency to live more than the F0 individuals (although this is not always the case; fig. 4) could be easily explained if laboratory strains become homozygous for recessive slightly deleterious mutations. Therefore, in what follows we will be comparing F1 individuals only. At 18 °C, male individuals with the X/4 fusion, Xc, 4ab, and 5a/5b inversions live 30.3% longer than male individuals with the 5a/5b inversions. Nevertheless, at 25 °C, male individuals with the 5a/5b inversion live 17.6% longer than males with the X/4 fusion, Xc, 4ab, and 5a/5b inversions. This observation suggests that there is a variant(s) affecting lifespan located on chromosomes X and/or 4, a maternal (possibly mitochondrial) or a paternal (Y chromosomal) effect. A similar pattern is observed for females. At 18 °C, females that get the X/4 fusion, Xc, 4ab, and 5b inversions from the F0 female live on average 26.4% longer than females that obtained these chromosomal rearrangements from the F0 male, pointing to a possible maternal (possibly mitochondrial) effect but such an effect is not, however, observed at 25 °C (supplementary fig. S1, Supplementary Material online shows how the survival curves, rather than the averages, change with temperature). One possible explanation is that the mitochondria from northern strain O57 are adapted to low temperatures, although other explanations cannot be ruled out at this moment.
F2 Association Studies
As there is significant variation regarding lifespan when comparing different D. americana populations, it makes sense to proceed to a classical F2 association study. Five F2 association studies were performed involving strains H5, W11, W46, W29, and O57, namely H5♂ × W11♀, W11♂ × W46♀, W29♂ × O57♀, O57♂ × H5♀, and W46♂ × W29♀. Five moderate-sized association studies were performed rather than a single large-sized association study because our goal was to infer the location of common variants with a large effect on the phenotypic trait, because these are the ones that more likely explain most of the phenotypic variation found in natural populations. Indeed, it is likely that many genes underlie differences in lifespan (Paaby and Schmidt 2009). The main features of the five crosses are shown in table 5. Seven out of the 10 possible pairwise comparisons involving different crosses are statistically significant (nonparametric Mann–Whitney U test; P < 0.05; supplementary table S5, Supplementary Material online), although only four are significant after applying the sequential Bonferroni correction for multiple testing (supplementary table S5, Supplementary Material online).
Table 5.
Lifespan Summary Statistics for the F2 Association Individuals Studied
| Cross | N | Mean | Median | Range | DRTNa (%) |
|---|---|---|---|---|---|
| H5♂ × W11♀ | 89 | 54.91 | 51.00 | 22–111 | +8.3 |
| W11♂ × W46♀ | 75 | 64.81 | 62.00 | 35–124 | +27.8 |
| W29♂ × O57♀ | 87 | 54.59 | 56.00 | 12–105 | +7.7 |
| O57♂ × H5♀ | 94 | 49.67 | 47.50 | 12–111 | −2.0 |
| W46♂ × W29♀ | 89 | 46.47 | 49.00 | 13–88 | −8.3 |
aDeviation (in percentage) relative to the D. americana Nebraska male population (the population with the shortest lifespan under the conditions here used; see Results) that lived on average 50.3 days.
Associations between Lifespan and Chromosomal Rearrangements
In D. americana natural populations, there are seven polymorphic chromosomal rearrangements (one fusion and six inversions) that show very different frequencies in the north and south of the distribution (see Introduction). Therefore, it is very likely that we have chromosomal rearrangements segregating in the F2 crosses that we set up. To address the possible effect of such common chromosomal rearrangements on lifespan, using the available molecular markers for the X/4 fusion, Xc, 4ab, and 5a/5b inversions (Vieira et al. 2001; Evans et al. 2007; Reis et al. 2008, 2011), we genotyped the F0 of all five association crosses, as well as, the F2 individuals from the crosses where such chromosomal rearrangements are segregating. There are only two significant associations between chromosomal rearrangements and lifespan involving the W29♂ × O57♀ cross (male individuals that are hemizygous for the X/4 fusion–Xc inversion and male individuals that are heterozygous for the 4ab inversion live ∼20% longer; table 6). The two significant associations are not, however, independent because of the X/4 fusion. This is not unexpected, given the results obtained in the previous section, regarding these two strains, and points out to the presence of a variant(s) influencing lifespan in the X and/or 4th chromosomes. Nevertheless, it should be noted that these associations are not significant after applying the sequential Bonferroni correction for multiple testing (assuming independence between tests which is not the case).
Table 6.
Associations between Chromosomal Rearrangements and Lifespan
| Cross | Na | Average Lifespan (Days) | Association (P Value) |
|---|---|---|---|
| W11♂ × W46♀ | 43 | 66.1 (Hemizygous X/4–Xc); | >0.05 |
| 31 | 63.9 (Hemizygous standard) | ||
| W29♂ × O57♀ | 49 | 58.7 (Hemizygous X/4–Xc); | <0.05 |
| 35 | 48.7 (Hemizygous standard) | ||
| W29♂ × O57♀ | 50 | 58.2 (Heterozygous 4ab) | <0.05 |
| 35 | 48.7 (Homozygous standard) | ||
| W29♂ × O57♀ | 22 | 55.5 (Homozygous 5a) | >0.05 |
| 18 | 51.1 (Homozygous 5b) | ||
| 47 | 55.5 (Heterozygous 5a/5b) | ||
| O57♂ × H5♀ | 50 | 49.6 (Homozygous 4ab) | >0.05 |
| 40 | 49.2 (Homozygous standard) | ||
| O57♂ × H5♀ | 16 | 54.9 (Homozygous 5a) | >0.05 |
| 21 | 51.8 (Homozygous 5b) | ||
| 55 | 46.9 (Heterozygous 5a/5b) | ||
aSample size.
Candidate Gene Approach
To identify genes that may be segregating for variants that significantly affect lifespan at the population level, we took a candidate gene approach, taking advantage of the two D. americana genomes here reported, to identify SNP markers in candidate genes that could be easily followed by the use of the appropriate restriction enzymes (supplementary table S2, Supplementary Material online). Out of the 48 candidate genes reported by Paaby and Schmidt (2009), we choose 21 genes (their predicted genome location in the H5 and W11 strains is shown in fig. 5), according to the following criteria: all genes showing evidence for an association between naturally occurring variation in D. melanogaster and lifespan were included (seven genes: Catsup, Ddc, Dox-A2, Lim3, ms(2)35Ci, stc, and tup [Paaby and Schmidt 2009]). Moreover, we looked at evidence for adaptive amino acid variation at the Drosophila genus level using a phylogenetic approach and the parameter rich models M7 and M8 (supplementary table S6, Supplementary Material online). Because of difficulties in inferring orthologies in the 12 Drosophila species analyzed, the DTS-3, hsp70Bb, and hsp70Bc genes were not analyzed. Moreover, mth gene was not analyzed because it was found to be missing in species outside the melanogaster species group (Patel et al. 2012). Evidence for amino acid adaptive evolution was found for seven genes (supplementary table S6, Supplementary Material online), and these were also included in our study. The genes showing evidence for amino acid adaptive evolution are not those showing the highest silent site divergence estimates between D. melanogaster and D. virilis, and thus, in principle, saturation at silent sites is not an issue (data not shown). Finally, seven randomly chosen D. melanogaster lifespan candidate genes were selected.
Fig. 5.—

Predicted location of the 21 D. americana genes used in the F2 association studies in strain H5 and W11.
Most of the variants that we decided to follow are also segregating in crosses other than the H5♂ × W11♀ cross, thus showing that this type of information is not useful for crosses involving the two strains only (table 7). Even when a single marker per candidate gene is used, as we did here, in crosses where strains H5 and W11 are not involved, on average 36% of the markers developed are segregating. Ten (hep, dFOXO, filamin, dilp2, Cat, SOD, PTEN, Dox-A2, Ddc, and Lim3) out of the 21 (48%) genes studied did show a significant association in at least one cross (one, two, three, and four genes on chromosomes X, 2, 3, and 4, respectively; table 7). Two genes (dilp2 and filamin) showed a significant association in two independent crosses. Finding associations is equally likely in every cross (Fisher exact test; after correction for multiple testing; P > 0.05). For the genes showing associations, the difference between extreme classes ranges from 6.7 to 23.1 days, and explain in between 1.3% and 11.1% of the total variation regarding lifespan (table 8). For the H5♂ × W11♀ cross, in every case, the allele that is associated with short lifespan comes from strain H5 (data not shown). When all markers for genes showing a significant association in the H5♂ × W11♀ cross are used, 22.4% of the lifespan variation observed in this cross is explained (Pearson correlation coefficient = 0.558; R2 = 0.311; Adjusted R2 = 0.224; N = 72). It should, however, be noted that only two associations (hep and Lim3, both in the H5♂ × W11♀ cross) are significant after applying the Bonferroni correction for multiple comparisons, and thus, only these have been further studied.
Table 7.
Associations between Candidate Gene Markers and Lifespan
| Gene | Chr | F2 Association Crossa |
||||
|---|---|---|---|---|---|---|
| H5♂ × W11♀ | W11♂ × W46♀ | W29♂ × O57♀ | O57♂ × H5♀ | W46♂ × W29♀ | ||
| ovo | X | 0.116 | 0.378 | n.a. | n.a. | n.a. |
| hep | X | 0.001b | 0.796 | n.a. | 0.543 | 0.490 |
| dFOXO | 2 | 0.018 | 0.473 | 0.828 | 0.728 | n.a. |
| filamin | 2 | 0.024 | n.a. | 0.015 | 0.758 | 0.508 |
| fwd | 2 | 0.862 | 0.580 | n.a. | n.a. | n.a. |
| InR | 2 | 0.864 | 0.593 | 0.618 | 0.425 | n.a. |
| puc | 2 | 0.524 | 0.229 | n.a. | 0.209 | n.a. |
| dilp2 | 3 | 0.003 | n.a. | 0.985 | 0.011 | 0.185 |
| Cat | 3 | 0.009 | n.a. | n.a. | 0.144 | n.a. |
| SOD | 3 | 0.184 | n.a. | n.a. | 0.044 | n.a. |
| PTEN | 4 | 0.771 | 0.732 | 0.043 | n.a. | 0.413 |
| mei-41 | 4 | 0.511 | 0.435 | 0.604 | 0.061 | n.a. |
| ms(2)35Ci | 4 | 0.124 | 0.984 | 0.644 | 0.159 | n.a. |
| tup | 4 | 0.081 | n.a. | n.a. | 0.542 | n.a. |
| Dox-A2 | 4 | 0.034 | 0.745 | n.a. | n.a. | n.a. |
| Catsup | 4 | 0.067 | 0.369 | n.a. | 0.172 | 0.917 |
| stc | 4 | 0.151 | 0.829 | n.a. | n.a. | 0.405 |
| Ddc | 4 | 0.022 | 0.909 | n.a. | n.a. | n.a. |
| Lim3 | 4 | 0.002b | n.a. | n.a. | 0.431 | n.a. |
| EF-1α48D | 5 | 0.629 | 0.742 | n.a. | n.a. | n.a. |
| magu | 5 | 0.339 | n.a. | 0.325 | 0.153 | 0.926 |
Note.—Chr, Chromosome; n.a. crosses without allelic segregation for the selected polymorphism.
aCandidate genes showing a statistically significant association (P < 0.05) are in bold.
bSignificant after applying the sequential Bonferroni correction.
Table 8.
Summary of the Crosses Showing Significant Associations
| Gene | Chr | F2 Cross | 0/0 | 1/0 | 1/1 | DBEC | R2 (%) |
|---|---|---|---|---|---|---|---|
| hep | X | H5♂ × W11♀ | 49.0 | 61.8 | 12.8 (26.1%) | 1.3 | |
| dFOXO | 2 | H5♂ × W11♀ | 62.3 | 50.9 | 49.0 | 13.3 (27.1%) | 10.0a |
| filamin | 2 | H5♂ × W11♀ | 36.7 | 56.6 | 59.0 | 22.3 (60.8%) | 9.3a |
| W29♂ × O57♀ | 54.7 | 62.0 | 44.6 | 17.4 (39.0%) | 4.3b | ||
| dilp2 | 3 | H5♂ × W11♀ | 39.8 | 55.3 | 60.9 | 21.1 (53.0%) | 11.1 |
| O57♂ × H5♀ | 44.1 | 54.2 | 10.1 (22.9%) | 8.5 | |||
| Cat | 3 | H5♂ × W11♀ | 60.4 | 55.1 | 37.3 | 23.1 (61.9%) | 9.9b |
| SOD | 3 | O57♂ × H5♀ | 45.1 | 54.2 | 9.1 (20.2%) | 7.0 | |
| PTEN | 4 | W29♂ × O57♀ | 48.7 | 58.2 | 9.5 (19.5%) | 6.8 | |
| Dox-A2 | 4 | H5♂ × W11♀ | 52.0 | 58.7 | 6.7 (12.9%) | 3.4 | |
| Ddc | 4 | H5♂ × W11♀ | 51.4 | 59.6 | 8.2 (16.0%) | 4.9 | |
| Lim3 | 4 | H5♂ × W11♀ | 60.6 | 50.0 | 10.6 (21.2%) | 8.4 |
Note.—Chr, chromosome; DBEC, difference between extreme classes in days and in percentage (within brackets).
aAssuming that 0 is recessive over 1.
bAssuming that 1 is recessive over 0.
Using the D. americana H5 and W11 genomes, for genes showing significant associations in the H5♂ × W11♀ cross (hep and Lim3), it is possible to gain insight into whether the putative causative mutation of the observed differences in lifespan could be an amino acid substitution. Hep shows four amino acid polymorphisms, although many more could be present because this region of the genome is not well represented in either the H5 or W11 strains. For three amino acid polymorphisms, the derived state could be inferred. There is one derived nonconservative change in strain W11 (an Alanine by a Threonine at position 123), and in strain H5, one derived nonconservative change (an Alanine by a Valine at position 703) and one conservative derived change (an Alanine by a Glycine at position 1,028). Lim3 shows three amino acid polymorphisms: two derived nonconservative (according to Livingstone and Barton [1993]) amino acid changes in strain H5 (a Valine by an Alanine at position 102 and a Proline by an Alanine at position 540), and a derived nonconservative amino acid change in strain W11 (a Proline by a Serine at position 536).
To address possible large gene expression differences as the reason for the observed association with lifespan, a preliminary survey of the hep and Lim3 gene expression levels was performed using H5 and W11 males. It could be argued that such gene expression changes are observed in some adult stages only, and thus, we first addressed the expression of these genes in sets of three individuals per strain that are 0, 10, 30, and 60 days old, (fig. 6). As we have a single measurement for each time point, it is impossible to determine whether the observed differences in fold changes are statistically significant. Nevertheless, in our experiment, fold changes are highest at day 0, and thus we addressed the expression of these genes, using three individual males from each strain, at day 0. No significant differences were observed when the expression levels between the two strains are compared (Student’s t test P > 0.05; fig. 6). We cannot, however, exclude the possibility of small changes in expression differences between the strains, that can only be confidently addressed using a much larger number of individuals per strain, several reference genes, and multiple primers for each gene.
Fig. 6.—

Gene expression data for the two candidate genes with a significant association after Bonferroni correction in the strains H5 (dark blue) and W11 (light blue) in sets of three individuals with different ages (a) and in three 0-day old flies from both strains (b). On the y axis is represented the normalized expression levels (fold changes relative to RpL32) for each set (a) and the means, as well as, the SEM values obtained for the three 0-day old flies (b). Inside the boxes is shown the fold-change value between the two D. americana strains (H5 relatively to W11) and the t test significance value (only in [b]).
Discussion
Here, we show that D. americana is a suitable species for comparative studies on the molecular basis of phenotypic variation. Indeed, D. americana has been diverging from D. melanogaster for at least 40 Myr (see Morales-Hojas and Vieira [2012] for a detailed discussion) and can be easily collected and maintained in the laboratory (see Introduction). Moreover, the estimate here reported for the average level of silent site polymorphism (1.876%), implies an effective population size of over 3.6 million individuals. This variability level reflects the history of this species over a period of more than 1 Myr, that is, approximately the time D. americana has been independently evolving (Caletka and McAllister 2004; Morales-Hojas et al. 2008). Therefore, there is plenty of intraspecific genetic variation. The average recombination rates are, as well, high for this species (see Results), a desirable feature when performing the needed phenotype–genotype studies.
It should be noted that in D. americana, there is no evidence for marked population structure (Schäfer et al. 2006; Morales-Hojas et al. 2008) besides that created by polymorphic chromosomal rearrangements. In this species, there is one fusion and six inversions with estimated frequency higher than 5% (X/4 fusion, Xc, 2b, 4a, 4b, 5a, and 5b [Hsu 1952]) that could complicate the interpretation of phenotype–genotype association studies. Nevertheless, there are molecular markers for the X/4 fusion, Xc, 4ab, and 5b chromosomal arrangements (Vieira et al. 2001; Evans et al. 2007; Reis et al. 2008, 2011), and perfect markers could be developed for 4a and 5a because their breakpoint sequences have been already determined (Evans et al. 2007; Fonseca et al. 2012).
The D. americana X/4 fusion Xc inversion is present as a shallow cline, being frequent in the north of the geographic distribution and almost absent in the south of the distribution (Vieira et al. 2001; McAllister 2002). Here, we show that, at the base of the X chromosome, in the region in between fused1 and Yp1, there are likely tens of thousands apparent fixations, including hundreds of apparent amino acid fixations, and fixations in regulatory regions, between X/4 fusion Xc-inverted and standard X chromosomes (see Results). These mutations have accumulated over a period of approximately 0.4–1.6 Myr (Spicer and Bell 2002; Caletka and McAllister 2004; Vieira et al. 2006; Morales-Hojas et al. 2008, 2011). As this is a highly differentiated region of the genome between individuals from the north and south of the distribution, it can in principle harbor the causative mutations responsible for many phenotypic differences between these populations. McAllister et al. (2008) have already shown a correlation between local temperature and local X/4 fusion frequency.
North–south differences regarding lifespan are here reported, thus further supporting the view that there is plenty of phenotypic variation in D. americana. Clines for lifespan have been observed in D. melanogaster as well (Paaby et al. 2010). In contrast to what has been suggested by Paaby and Schmidt (2009), in D. americana, at 25 °C, on average, flies from warmer places live longer than flies from colder places. However, caution should be exercised when extrapolating results from the laboratory to the field, because, as we here show, when the same experiment is performed at different temperatures the relative outcome differs.
In D. melanogaster and other animals, mitochondria are likely major regulators of longevity, although their exact role in aging is not fully understood (Stefanatos and Sanz 2011), and in our controlled crosses, we find some evidence for mitochondrial variant(s) that seem to extend lifespan at low temperatures but have no or a slight deleterious effect at higher temperatures. Nevertheless, this does not exclude the possibility of the contribution of variants at nuclear genes as well. As expected, virgin females live longer than males (Iliadi et al. 2009).
Having two assembled reference genomes greatly facilitates the development of markers (either anonymous or for candidate genes) for the needed lifespan-genotype association studies, and eases the identification of putative causative polymorphisms. In D. americana, besides lifespan, there are also north–south differences regarding other phenotypic traits, such as, developmental time (as large as 2 days, at 25 °C), or the propensity to enter diapause (Vieira J., Reis M., Vieira C.P., unpublished data), and the tools here reported will be useful to address those traits as well.
In a preliminary study, aimed at showing the usefulness of D. americana as a species for comparative studies on the molecular basis of phenotypic differences, five F2 association crosses involving five different strains were performed. The average lifespan of the F2 individuals from the five association crosses here reported varies by as much as 39.5%. Although it is true that the genome information here reported is most useful for association studies involving the H5 and W11 strains, even when a single marker per candidate gene is used, when strains H5 and W11 are not involved, on average, 36% of the markers developed are segregating. Two (hep and Lim3) out of the 21 candidate genes for lifespan here studied show a significant association with lifespan. Hep is necessary for actin-cable assembly and actin-based cell process formation in leading edge cells during dorsal closure (Kaltschmidt et al. 2002), whereas Lim3 encodes an RNA polymerase II transcription factor with the potential to regulate gene transcription in a variety of tissues (Rybina and Pasyukova 2010).
It is conceivable that a gene in the vicinity of the candidate gene showing association with lifespan is the one that harbors variation that influences lifespan. Nevertheless, Lim3 is one out of the seven D. melanogaster genes showing associations between naturally occurring variation and lifespan (using deficiency complementation tests). Moreover, both Hep and Lim3 show nonconservative amino acid differences between strain H5 and W11 that could be the causative mutations of the observed lifespan differences. For both genes, no large changes in expression levels were detected between the two strains.
In conclusion, D. americana is an excellent species for comparative studies on the molecular basis of phenotypic variation. The availability of two genome sequences greatly facilitates such studies, as well as, other studies using this species. For instance, D. americana can be crossed with two closely related species, namely D. novamexicana and D. virilis (whose genome is also sequenced). In the D. americana/D. virilis hybrids, developmental problems have been reported that could shed light on how speciation happened (Heikkinen 1992; Heikkinen and Lumme 1998; Nickel and Civetta 2009; Sweigart 2010a, 2010b). The availability of the genome sequence for both species will certainly speed up such studies, as well.
Supplementary Material
Supplementary tables S1–S6 and figure S1 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported by FEDER Funds through the Operational Competitiveness Programme – COMPETE; National Funds through FCT – Fundação para a Ciência e a Tecnologia under the projects FCOMP-01-0124-FEDER-008916 (PTDC/BIA-BEC/099933/2008), FCOMP-01-0124-FEDER-008916 (PTDC/EIA-EIA/100897/2008), and the project FCOMP-01-0124-FEDER-022718 (PEST-C/SAU/LA0002/2011); and a PhD grant attributed by FCT with the reference SFRH/BD/61142/2009 to M.R.
Literature Cited
- Alexander ML. The genetics of Drosophila virilis. In: Ashburner M, Novitsky E, editors. The genetics and biology of Drosophila. New York: Academic Press; 1976. pp. 1365–1419. [Google Scholar]
- Aminetzach YT. Pesticide resistance via transposition-mediated adaptive gene truncation in Drosophila. Science. 2005;309:764–767. doi: 10.1126/science.1112699. [DOI] [PubMed] [Google Scholar]
- Aquadro CF, Begun DJ, Kindahl EC. Selection, recombination and DNA polymorphism in Drosophila. In: Golding B, editor. Non-neutral evolution: theories and molecular data. New York: Chapman & Hall; 1994. pp. 46–56. [Google Scholar]
- Bachtrog D. Similar rates of protein adaptation in Drosophila miranda and D. melanogaster, two species with different current effective population sizes. BMC Evol Biol. 2008;8:334. doi: 10.1186/1471-2148-8-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbash DA, Awadalla P, Tarone AM. Functional divergence caused by ancient positive selection of a Drosophila hybrid incompatibility locus. PLoS Biol. 2004;2:e142. doi: 10.1371/journal.pbio.0020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun DJ, Whitley P. Molecular population genetics of Xdh and the evolution of base composition in Drosophila. Genetics. 2002;162:1725–1735. doi: 10.1093/genetics/162.4.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancourt AJ, Welch JJ, Charlesworth B. Reduced effectiveness of selection caused by a lack of recombination. Curr Biol. 2009;19:655–660. doi: 10.1016/j.cub.2009.02.039. [DOI] [PubMed] [Google Scholar]
- Betran E, Rozas J, Navarro A, Barbadilla A. The estimation of the number and the length distribution of gene conversion tracts from population DNA sequence data. Genetics. 1997;146:89–99. doi: 10.1093/genetics/146.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27:578–579. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- Bono JM, Matzkin LM, Castrezana S, Markow TA. Molecular evolution and population genetics of two Drosophila mettleri cytochrome P450 genes involved in host plant utilization. Mol Ecol. 2008;17:3211–3221. doi: 10.1111/j.1365-294X.2008.03823.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caletka BC, McAllister BF. A genealogical view of chromosomal evolution and species delimitation in the Drosophila virilis species subgroup. Mol Phylogen Evol. 2004;33:664–670. doi: 10.1016/j.ympev.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Catania F, et al. World-wide survey of an Accord insertion and its association with DDT resistance in Drosophila melanogaster. Mol Ecol. 2004;13:2491–2504. doi: 10.1111/j.1365-294X.2004.02263.x. [DOI] [PubMed] [Google Scholar]
- Charlesworth B. The role of background selection in shaping patterns of molecular evolution and variation: evidence from variability on the Drosophila X chromosome. Genetics. 2012;191:233–246. doi: 10.1534/genetics.111.138073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosophila 12 Genomes C, et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Evans AL, Mena PA, McAllister BF. Positive selection near an inversion breakpoint on the Neo-X chromosome of Drosophila americana. Genetics. 2007;177:1303–1319. doi: 10.1534/genetics.107.073932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca NA, Vieira CP, Schlötterer C, Vieira J. The DAIBAM MITE element is involved in the origin of one fixed and two polymorphic Drosophila virilis phylad inversions. Fly. 2012;6:71–74. doi: 10.4161/fly.19423. [DOI] [PubMed] [Google Scholar]
- Fry SN, Rohrseitz N, Straw AD, Dickinson MH. TrackFly: virtual reality for a behavioral system analysis in free-flying fruit flies. J Neurosci Methods. 2008;171:110–117. doi: 10.1016/j.jneumeth.2008.02.016. [DOI] [PubMed] [Google Scholar]
- Heikkinen E. Genetic basis of reduced eyes in the hybrids of Drosophila virilis phylad species. Hereditas. 1992;117:275–285. doi: 10.1111/j.1601-5223.1992.tb00025.x. [DOI] [PubMed] [Google Scholar]
- Heikkinen E, Lumme J. The Y chromosomes of Drosophila lummei and D. novamexicana differ in fertility factors. Heredity. 1998;81:505–513. doi: 10.1046/j.1365-2540.1998.00422.x. [DOI] [PubMed] [Google Scholar]
- Hilton H, Hey J. DNA sequence variation at the Period locus reveals the history of species and speciation events in the Drosophila virilis group. Genetics. 1996;144:1015–1025. doi: 10.1093/genetics/144.3.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu TC. Chromosomal variation and evolution in the virilis group of Drosophila. Vol. 5204. Hudson (Texas): University of Texas Publications; 1952. pp. 35–72. [Google Scholar]
- Iliadi KG, Iliadi NN, Boulianne GL. Regulation of Drosophila life-span: effect of genetic background, sex, mating and social status. Exp Gerontol. 2009;44:546–553. doi: 10.1016/j.exger.2009.05.008. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt JA, et al. Planar polarity and actin dynamics in the epidermis of Drosophila. Nat Cell Biol. 2002;4:937–944. doi: 10.1038/ncb882. [DOI] [PubMed] [Google Scholar]
- Kimura M. The neutral theory of molecular evolution. Cambridge: Cambridge University Press; 1985. [Google Scholar]
- Kuhner MK. LAMARC: Estimating population genetic parameters from molecular data. In: Lemey P, Salemi M, Vandamme A-M, editors. The phylogenetic handbook: a practical approach to phylogenetic analysis and hypothesis testing. Cambridge: Cambridge University Press; 2009. p. 750. [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The Sequence Alignment/Map (SAM) format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Livingstone CD, Barton GJ. Protein sequence alignments: a strategy for the hierarchical analysis of residue conservation. Comput Appl Biosci. 1993;9:745–756. doi: 10.1093/bioinformatics/9.6.745. [DOI] [PubMed] [Google Scholar]
- Llopart A, Comeron JM. Recurrent events of positive selection in independent Drosophila lineages at the spermatogenesis gene roughex. Genetics. 2008;179:1009–1020. doi: 10.1534/genetics.107.086231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maside X, Lee AW, Charlesworth B. Selection on codon usage in Drosophila americana. Curr Biol. 2004;14:150–154. doi: 10.1016/j.cub.2003.12.055. [DOI] [PubMed] [Google Scholar]
- Matzkin LM. The molecular basis of host adaptation in cactophilic Drosophila: molecular evolution of a Glutathione S-transferase gene (GstD1) in Drosophila mojavensis. Genetics. 2008;178:1073–1083. doi: 10.1534/genetics.107.083287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister BF. Chromosomal and allelic variation in Drosophila americana: selective maintenance of a chromosomal cline. Genome. 2002;45:13–21. doi: 10.1139/g01-112. [DOI] [PubMed] [Google Scholar]
- McAllister BF. Sequence differentiation associated with an inversion on the neo-X chromosome of Drosophila americana. Genetics. 2003;165:1317–1328. doi: 10.1093/genetics/165.3.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister BF, Charlesworth B. Reduced sequence variability on the Neo-Y chromosome of Drosophila americana americana. Genetics. 1999;153:221–233. doi: 10.1093/genetics/153.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister BF, Evans AL. Increased nucleotide diversity with transient Y Linkage in Drosophila americana. PLoS One. 2006;1:e112. doi: 10.1371/journal.pone.0000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister BF, McVean GAT. Neutral evolution of the sex-determining gene transformer in Drosophila. Genetics. 2000;154:1711–1720. doi: 10.1093/genetics/154.4.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister BF, Sheeley SL, Mena PA, Evans AL, Schlötterer C. Clinal distribution of a chromosomal rearrangement: A precursor to chromosomal speciation? Evolution. 2008;62:1852–1865. doi: 10.1111/j.1558-5646.2008.00435.x. [DOI] [PubMed] [Google Scholar]
- Morales-Hojas R, Reis M, Vieira CP, Vieira J. Resolving the phylogenetic relationships and evolutionary history of the Drosophila virilis group using multilocus data. Mol Phylogen Evol. 2011;60:249–258. doi: 10.1016/j.ympev.2011.04.022. [DOI] [PubMed] [Google Scholar]
- Morales-Hojas R, Vieira CP, Reis M, Vieira J. Comparative analysis of five immunity-related genes reveals different levels of adaptive evolution in the virilis and melanogaster groups of Drosophila. Heredity. 2009;102:573–578. doi: 10.1038/hdy.2009.11. [DOI] [PubMed] [Google Scholar]
- Morales-Hojas R, Vieira CP, Vieira J. Inferring the evolutionary history of Drosophila americana and Drosophila novamexicana using a multilocus approach and the influence of chromosomal rearrangements in single gene analyses. Mol Ecol. 2008;17:2910–2926. doi: 10.1111/j.1365-294X.2008.03796.x. [DOI] [PubMed] [Google Scholar]
- Morales-Hojas R, Vieira J. Phylogenetic patterns of geographical and ecological diversification in the subgenus Drosophila. PLoS One. 2012;7:e49552. doi: 10.1371/journal.pone.0049552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura R, et al. TRF4 is involved in polyadenylation of snRNAs in Drosophila melanogaster. Mol Cell Biol. 2008;28:6620–6631. doi: 10.1128/MCB.00448-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel D, Civetta A. An X chromosome effect responsible for asymmetric reproductive isolation between male Drosophila virilis and heterospecific females. Genome. 2009;52:49–56. doi: 10.1139/G08-102. [DOI] [PubMed] [Google Scholar]
- Ohta T. The nearly neutral theory of molecular evolution. Annu Rev Ecol Syst. 1992;23:263–286. [Google Scholar]
- Paaby AB, Blacket MJ, Hoffmann AA, Schmidt PS. Identification of a candidate adaptive polymorphism for Drosophila life history by parallel independent clines on two continents. Mol Ecol. 2010;19:760–774. doi: 10.1111/j.1365-294X.2009.04508.x. [DOI] [PubMed] [Google Scholar]
- Paaby AB, Schmidt PS. Dissecting the genetics of longevity in Drosophila melanogaster. Fly. 2009;3:29–38. doi: 10.4161/fly.3.1.7771. [DOI] [PubMed] [Google Scholar]
- Patel MV, et al. Dramatic expansion and developmental expression diversification of the methuselah gene family during recent Drosophila evolution. J Exp Zool B Mol Dev Evol. 2012;318:368–387. doi: 10.1002/jez.b.22453. [DOI] [PubMed] [Google Scholar]
- Patterson JT, Stone WS. Evolution in the genus Drosophila. New York: Macmillan; 1952. [Google Scholar]
- Pool JE, Aquadro CF. The genetic basis of adaptive pigmentation variation in Drosophila melanogaster. Mol Ecol. 2007;16:2844–2851. doi: 10.1111/j.1365-294X.2007.03324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presgraves DC, Stephan W. Pervasive adaptive evolution among interactors of the Drosophila hybrid inviability gene, Nup96. Mol Biol Evol. 2007;24:306–314. doi: 10.1093/molbev/msl157. [DOI] [PubMed] [Google Scholar]
- Reis M, et al. A comparative study of the short term cold resistance response in distantly related Drosophila species: the role of regucalcin and Frost. PLoS One. 2011;6:e25520. doi: 10.1371/journal.pone.0025520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis M, Vieira CP, Morales-Hojas R, Vieira J. An old bilbo-like non-LTR retroelement insertion provides insight into the relationship of species of the virilis group. Gene. 2008;425:48–55. doi: 10.1016/j.gene.2008.08.010. [DOI] [PubMed] [Google Scholar]
- Rissman AI, et al. Reordering contigs of draft genomes using the Mauve Aligner. Bioinformatics. 2009;25:2071–2073. doi: 10.1093/bioinformatics/btp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Rybina OY, Pasyukova EG. A naturally occurring polymorphism at Drosophila melanogaster Lim3 locus, a homolog of human LHX3/4, affects Lim3 transcription and fly lifespan. PLoS One. 2010;5:e12621. doi: 10.1371/journal.pone.0012621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer MA, Orsini L, McAllister BF, Schlotterer C. Patterns of microsatellite variation through a transition zone of a chromosomal cline in Drosophila americana. Heredity. 2006;97:291–295. doi: 10.1038/sj.hdy.6800860. [DOI] [PubMed] [Google Scholar]
- Schlenke TA, Begun DJ. Strong selective sweep associated with a transposon insertion in Drosophila simulans. Proc Natl Acad Sci U S A. 2004;101:1626–1631. doi: 10.1073/pnas.0303793101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JT, et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer DD, Delcher AL, Salzberg SL, Pop M. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics. 2007;8:64. doi: 10.1186/1471-2105-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicer GS, Bell CD. Molecular phylogeny of the Drosophila virilis species group (Diptera: Drosophilidae) inferred from mitochondrial 12S and 16S ribosomal RNA genes. Ann Entomol Soc Am. 2002;95:156–161. [Google Scholar]
- Stefanatos R, Sanz A. Mitochondrial complex I: a central regulator of the aging process. Cell Cycle. 2011;10:1528–1532. doi: 10.4161/cc.10.10.15496. [DOI] [PubMed] [Google Scholar]
- Sweigart AL. The genetics of postmating, prezygotic reproductive isolation between Drosophila virilis and D. americana. Genetics. 2010a;184:401–410. doi: 10.1534/genetics.109.111245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweigart AL. Simple Y-autosomal incompatibilities cause hybrid male sterility in reciprocal crosses between Drosophila virilis and D. americana. Genetics. 2010b;184:779–787. doi: 10.1534/genetics.109.112896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Throckmorton LH. The virilis species group. In: Ashburner M, Novistky E, editors. The genetics and biology of Drosophila. London: Academic; 1982. pp. 227–297. [Google Scholar]
- Vieira CP, Almeida A, Dias JD, Vieira J. On the location of the gene(s) harbouring the advantageous variant that maintains the X/4 fusion of Drosophila americana. Genet Res. 2006;87:163. doi: 10.1017/S0016672306008147. [DOI] [PubMed] [Google Scholar]
- Vieira CP, Coelho PA, Vieira J. Inferences on the evolutionary history of the Drosophila americana polymorphic X/4 fusion from patterns of polymorphism at the X-linked paralytic and elav genes. Genetics. 2003;164:1459–1469. doi: 10.1093/genetics/164.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira J, Charlesworth B. X chromosome DNA variation in Drosophila virilis. Proc R Soc Lond B Biol Sci. 1999;266:1905–1912. doi: 10.1098/rspb.1999.0865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira J, McAllister BF, Charlesworth B. Evidence for selection at the fused1 locus of Drosophila americana. Genetics. 2001;158:279–290. doi: 10.1093/genetics/158.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilella AJ, Blanco-Garcia A, Hutter S, Rozas J. VariScan: analysis of evolutionary patterns from large-scale DNA sequence polymorphism data. Bioinformatics. 2005;21:2791–2793. doi: 10.1093/bioinformatics/bti403. [DOI] [PubMed] [Google Scholar]
- Wittkopp PJ, et al. Local adaptation for body color in Drosophila americana. Heredity. 2011;106:592–602. doi: 10.1038/hdy.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp PJ, et al. Intraspecific polymorphism to interspecific divergence: genetics of pigmentation in Drosophila. Science. 2009;326:540–544. doi: 10.1126/science.1176980. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}