

Summary
Changes in DNA copy number, whether confined to specific genes or affecting whole chromosomes have been identified as causes of diseases and developmental abnormalities and as sources of adaptive potential. Here, we discuss the costs and benefits of DNA copy number alterations. Changes in DNA copy number are largely detrimental. Amplifications or deletions of specific genes can elicit discrete defects. Large-scale changes in DNA copy number can also cause detrimental phenotypes that are due to the cumulative effects of copy number alterations of many genes simultaneously. On the other hand, studies in microorganisms, show that DNA copy number alterations can be beneficial, increasing survival under selective pressure. As DNA copy number alterations underlie many human diseases we will end with a discussion of gene copy number changes as therapeutic targets.
Introduction
Every species is defined by its karyotype. Maintaining this karyotype is essential for species survival, a truth perhaps best illustrated by the fact that numerous mechanisms have evolved to ensure that the species-specific karyotype is maintained. Changes in the copy number of specific genes, entire chromosomes or parts thereof can have a dramatic impact on the fitness and reproductive abilities of an organism. Trisomy 21 in humans illustrates this point. Individuals with Trisomy 21, also known as Down syndrome, exhibit a number of developmental disabilities and have a significantly reduced life-span (Yang et al., 2002). The fact that changes in copy number of small regions of the genome can have a significant impact on fitness is emphasized by genome-wide association studies of genetically complex human diseases. Small-scale often submicroscopic changes in DNA copy number are estimated to be responsible for at least 15% of human neurodevelopmental defects and are associated with psychiatric disorders, kidney and heart defects (reviewed in Girirajan et al., 2012).
Much progress has been made in our ability to identify DNA copy number alterations and to determine their biological impact. Depending on their size, DNA copy number alterations are referred to by different terms (Figure 1). Multiples of the entire genetic complement are referred to as polyploidies. Whole chromosome (34 ~230 Mbp) gains or losses are known as aneuploidies (Figure 1A, C). Changes in copy number of sub-chromosomal regions that are visible by light microscopy are referred to as partial or segmental aneuploidies. Sub-microscopic DNA copy number alterations that are between 1 kbp and 1 Mbp in length are referred to as copy number variations (CNVs; Figure 1A, B). Smaller changes in DNA copy number, ranging - 1 bp and 1 kbp in size - are called deletions or insertions depending on whether sequences are deleted or amplified, respectively (Figure 1A; Feuk et al., 2006; Lupski, 2007). The mechanisms leading to small and large-scale DNA copy number changes have been investigated in detail. We refer the reader to several excellent recent reviews that summarize our current understanding of these mechanisms (Alkan et al., 2011; Holland and Cleveland, 2012; Malhotra and Sebat, 2012; Zhang et al., 2009).
Figure 1. Defining DNA copy number alterations.
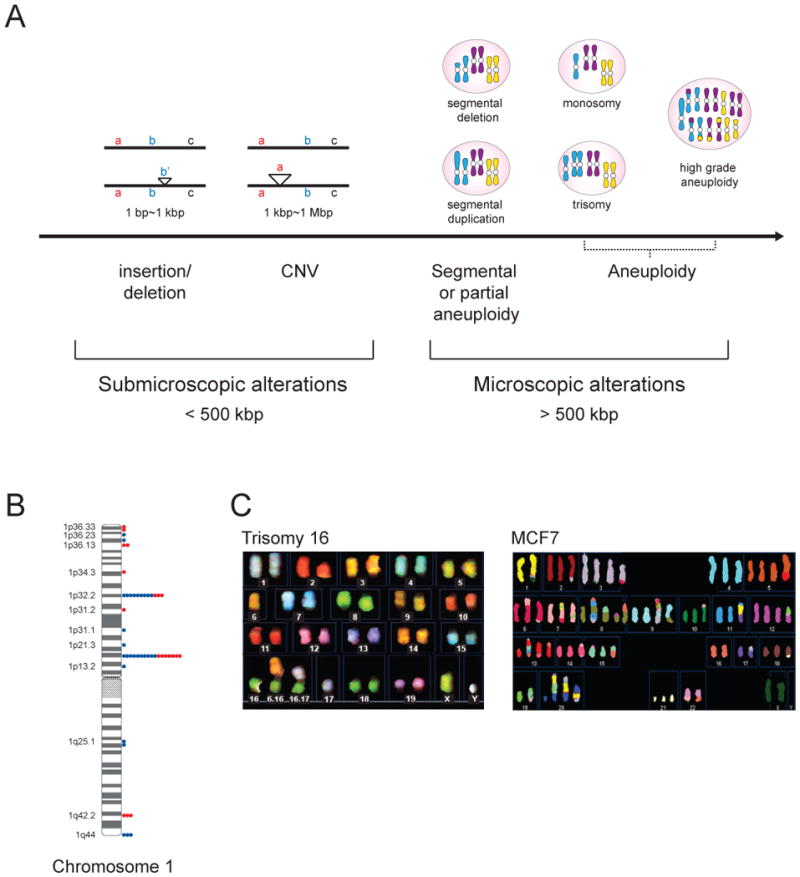
(A) DNA copy number alterations can be categorized into submicroscopic variations, which are smaller than 500 kbp and microscopy alterations, which are greater than 500 kbp. DNA copy number changes between 1 bp and 1 kbp in size are called insertions or deletions, depending on whether DNA is gained or lost, respectively. Copy number variations (CNVs) vary between 1kbp and 1Mbp in size. Examples for CNVs here are shown for duplication. Microscopically visible karyotype changes are called segmental or partial aneuploidies, when parts of chromosomes are amplified or deleted. Whole chromosomes losses or gains are called aneuploidies.
(B) CNV distribution on human chromosome 1. Dots show the number of individuals with copy gains (blue) or losses (red) among 39 unrelated, healthy control individuals. (data from (Iafrate et al., 2004).
(C) Spectral karyotyping (SKY) analysis of a trisomy 16 mouse embryonic fibroblast cell line (data from (Williams et al., 2008) and a MCF-7 breast cancer cell line (data from http://www.path.cam.ac.uk/~pawefish/BreastCellLineDescriptions/mcf7.htm).
Here we discuss the costs and benefits of small- and large-scale DNA copy number alterations. We will describe examples that illustrate how copy number alterations of individual genes can result in cellular and organismal abnormalities. We will further discuss findings that show that large-scale DNA copy number changes, such as whole chromosome gains and losses, cause general, detrimental phenotypes that result from the cumulative effects of changes in copy number of a large number of genes. The potential benefits of gene copy number alterations will also be examined, summarizing studies that show that gene copy number alterations can improve survival of microorganisms under selective pressure. Finally we will consider the possibility of targeting gene copy number changes in the treatment of human diseases.
The impact of copy number changes on gene expression
Before considering the biological impact of changes in gene copy number, it is important to address the question of whether such changes are translated into corresponding changes in gene expression or whether mechanisms are in place that ensure wild-type levels of expression irrespective of gene copy number. Such mechanisms, collectively called dosage compensation mechanisms, exist for sex chromosomes, which naturally vary in copy number between sexes (reviewed in Nguyen and Disteche, 2006). With the exception of Drosophila and some plant species (Guo and Birchler, 1994; Kim et al., 2011; Larsson et al., 2001; Miclaus et al., 2011; Stenberg and Larsson, 2011), dosage compensation mechanisms do not exist for autosomes. Gene copy number proportional expression of whole chromosomal or segmental aneuploidies has been observed in fission yeast, budding yeast, Arabidopsis, trisomic mouse embryonic fibroblasts, partially trisomic mouse tissues and human trisomies (Chikashige et al., 2007; Huettel et al., 2008; Kahlem et al., 2004; Lyle et al., 2004; Pavelka et al., 2010; Torres et al., 2007; Upender et al., 2004; Vacik et al., 2005; Williams et al., 2008; Stingele et al., 2012; Figure 2). CNVs also typically result in a corresponding change in gene expression. In humans and mice, 85–95 percent of CNVs are associated with changes in expression of the affected genes (Henrichsen et al., 2009; Stranger et al., 2007).
Figure 2. Aneuploid chromosomes are active in budding yeast.
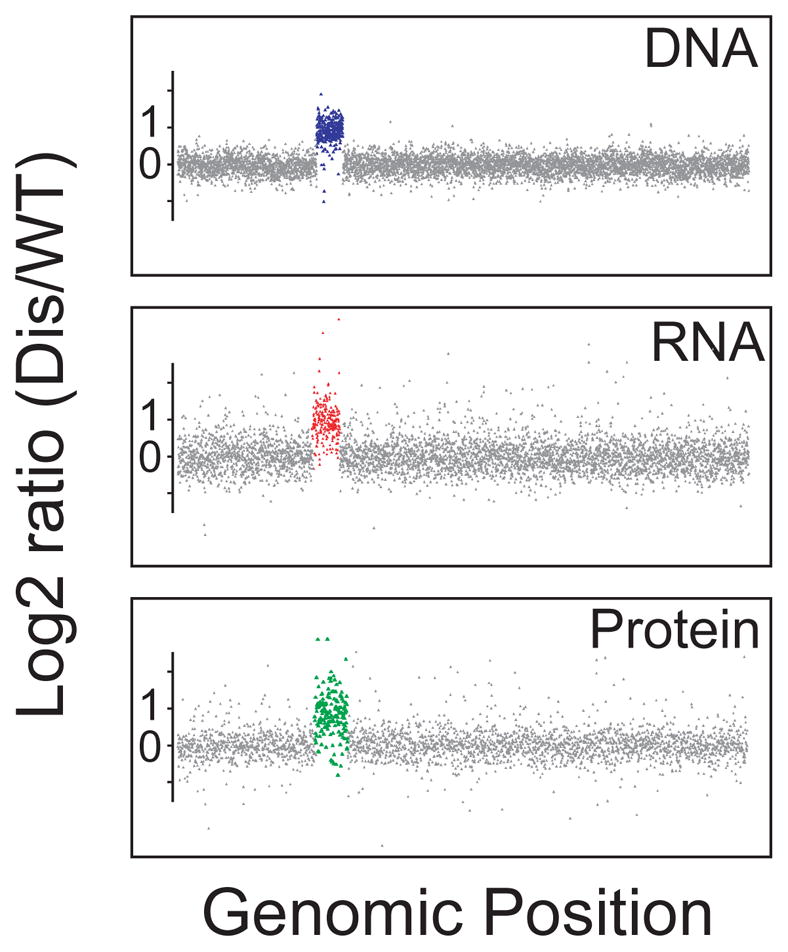
DNA, RNA and protein content of a budding yeast strain carrying an additional copy of chromosome V (data from Torres et al., 2010).
In organisms where this has been studied, increases in RNA levels result in increased protein production. Quantitative proteomic analyses in aneuploid budding yeast and human cells showed that changes in gene copy number result in changes in protein levels in the majority of cases (Pavelka et al., 2010; Stingele et al., 2012; Torres et al., 2010). The proteins that do not show this coordinate increase in protein levels with gene copy number are predominantly components of large protein complexes (Stingele et al., 2012; Torres et al., 2010), presumably because unassembled subunit of protein complexes are often unstable. Thus, gene copy number changes generally translate into changes in gene expression.
The biological impact of DNA copy number alterations
Changes in DNA copy number generally have adverse effects on fitness. The degree of adversity scales with the size of the alteration. Whole chromosome and segmental aneuploidies invariably lead to severe developmental abnormalities or death of the organism in all species analyzed to date (reviewed in Torres et al., 2008). CNVs are, for the most part, also detrimental. Sequencing of human populations revealed that CNVs are rare, occurring at a frequency of less than 1 percent (Itsara et al., 2009) and that they are under strong negative selection (Itsara et al., 2010). Insertions and deletions on the other hand are widespread in the human genome indicating that their impact on fitness is likely to be minor (Girirajan et al., 2012; Iafrate et al., 2004; Sebat et al., 2004).
How do changes in gene copy number lead to phenotypes? Changes in the copy number of individual genes can cause phenotypes. Phenotypes can also be driven by the cumulative effect of changes in copy number of a large number of genes, which individually have little or no effect. In this section we will discuss the gene-specific and general effects of changes in DNA copy number.
Gene-specific effects
It has long been known that changes in the copy number of specific genes can have a dramatic impact on organismal and cellular fitness. For example, budding yeast cells harboring an extra copy of the β-tubulin-encoding gene are inviable (Katz et al., 1990). Studies in humans suggest that as many as 15% of neurodevelopmental disorders, and other diseases are due to rare, large CNVs resulting in the imbalance of a handful of genes (for a recent review see Malhotra and Sebat, 2012). For example, duplication of PMP22 leads to Charcot-Marie-Tooth 1A neuropathy (reviewed in Hanemann and Muller, 1998). Duplication of SNCA is associated with Parkinson’s disease (Singleton et al., 2003), duplication of GSK3b with bipolar disorder (Lachman et al., 2007), and low-copy amplification of the C4 gene with lupus (Yang et al., 2007). Finally, amplifications and deletions of individual genes are major drivers of tumorigenesis. Amplification of the oncogene Myc, for example, is thought to be a driving factor in human acute myeloid leukemia (Jones et al., 2010).
Gene dosage changes of individual genes also appear to be responsible for some of the phenotypes observed in syndromes caused by chromosome-size DNA copy number changes. For example, chromosome 21 located APP which encodes a protein that when cleaved forms the main component of amyloid-β plaques, has been found duplicated in familial forms of early onset Alzheimer’s disease. This observation suggests that early onset Alzheimer’s disease, which is observed in virtually all individuals with Down Syndrome, is due to a second copy of APP (reviewed in Kingsbury et al., 2006). Many more examples have been described where imbalances in the copy number of one or a few genes elicit a discrete phenotype and we refer the reader to recent reviews that provide a comprehensive overview of the contribution of gene copy number changes to human diseases (Girirajan et al., 2012; Zhong et al., 2009).
General non-gene specific effects of large-scale DNA copy number alterations
Changes in the copy number of large regions of the genome can also cause phenotypes that are the result of cumulative effects of copy number changes in many genes, which on their own have little phenotypic consequences. Studies on whole chromosome gains in yeast and mammalian cells revealed phenotypes that are seen in many different aneuploidies (summarized in Figure 3). The severity of these phenotypes tends to scale with the degree of deviation from the euploid karyotype and thus they likely also exist in cells harboring large segmental aneuploidies, but not in cells with CNV-size DNA copy number alterations. In the following section we will describe the general phenotypes associated with large-scale DNA copy number alterations.
Figure 3. Gene-specific and general effects of aneuploidy on fitness.

See text for details.
A. Whole chromosome copy number alterations interfere with cell proliferation
Studies of S. pombe and S. cerevisiae aneuploid strains (Niwa et al., 2006; Pavelka et al., 2010; Torres et al., 2007) showed that aneuploidy impairs proliferation under standard growth conditions. Similar slowed growth was observed in mammalian cells. Trisomic mouse embryonic fibroblasts (MEFs) exhibit proliferation defects, as do cells harboring random aneuploidies caused by mutations of BubR1 or Cdc20 that lead to increased chromosome mis-segregation (Baker et al., 2004; Li et al., 2009; Thompson and Compton, 2008; Williams et al., 2008). These adverse effects on cell proliferation have severe consequences for the animal. Mice expressing mutations leading to high levels of chromosome mis-segregation die in utero or exhibit premature aging phenotypes (Baker et al., 2004; Li et al., 2009). In humans, hypomorphic mutations in BubR1 lead to mosaic-variegated aneuploidy, which is associated with growth deficiency and short stature, mental retardation, developmental defects, as well as a predisposition to cancer (Hanks et al., 2004). Several mouse models of CIN do not exhibit dramatic defects in cell proliferation and animals show little if any decline in overall fitness (reviewed in Pfau and Amon, 2012) indicating that some level of aneuploidy can be tolerated and perhaps compensated for by cells especially in the context of the organism, where proliferative potential is frequently not the rate-limiting.
How large-scale copy number changes such as chromosomal gains or losses interfere with cell proliferation is not understood. In budding yeast, fission yeast and human cells, chromosome gains cause a G1 delay (Niwa et al., 2006; Torres et al., 2007; Stingele et al., 2012). Many stresses cause a transient G1 delay, by impacting the cell cycle machinery governing the G1 – S phase transition. Gross copy number changes elicit phenotypes that are reminiscent of a stress response (see below). The observed G1 delay could thus be part of such a stress response. It is however also possible that a different genetic imbalance underlies each G1 delay in the different aneuploid strains. In mammalian cells chromosome mis-segregation and the ensuing aneuploidy causes activation of p53, which results in G1 arrest or apoptosis (Li et al., 2010; Thompson and Compton, 2010). The origins of this p53 response are controversial but could be due to various stresses associated with aneuploidy, which are discussed in the following section.
B. Whole chromosome copy number alterations cause several cellular stresses, collectively called the “aneuploidy-associated stresses”
Studies in aneuploid yeast and mouse embryonic fibroblasts (MEFs) indicate that aneuploid cells experience metabolic stress. Yeast cells and MEFs with unbalanced karyotypes produce less biomass (Li et al., 2010; Torres et al., 2007, Williams, 2008 #495). Trisomic MEFs and MEFs containing the aneuploidy-inducing CDC20AAA mutation were also found to take up more glutamine and to exhibit sensitivity to the energy stress-inducing compound AICAR (Li et al., 2010; Tang et al., 2011; Williams et al., 2008). Aneuploid cells generated by the CDC20AAA mutation also display increased glucose uptake and lactate production (Li et al., 2010). In all, these findings point toward increased energy needs of cells with large-scale karyotypic changes. Perhaps, cells waste energy by producing and then dealing with the excess proteins produced from the additional genes.
Because gains and losses of genetic information impact gene expression, large-scale changes in gene copy number have a profound impact on the cell’s protein composition. Overproduced proteins and proteins lacking their appropriate binding partners eventually mis-fold and lead to a condition known as proteotoxic stress. Thus, not surprisingly, cells with whole chromosome gains are under proteotoxic stress. Aneuploid S. cerevisiae cells are more sensitive to conditions that elicit proteotoxic stress such as elevated temperature or treatment with the protein synthesis inhibitors cycloheximide or hygromycin, and the Hsp90 inhibitor geldanamycin (Pavelka et al., 2010; Torres et al., 2007). Furthermore, aneuploid yeast cells form protein aggregates and appear challenged in their ability to fold proteins (A.B. Oromendia, S. Dodgson, and A. Amon, unpublished data). In addition, inactivation of the proteasome antagonist Ubp6 improved the fitness of several different yeast strains carrying an additional chromosome and partially attenuated the proteomic changes elicited by the aneuploid karyotype (Torres et al., 2010). Evidence for proteotoxic stress also exists in mammalian cells harboring additional chromosomes. Trisomic MEFs and human cells show increased basal levels of autophagy and the chaperone Hsp72, and are more sensitive to the Hsp90 chaperone inhibitor 17-AAG than euploid cells (Stingele et al., 2012; Tang et al., 2011). In summary, changes in gene copy number of many genes simultaneously, as occurs when entire chromosomes are lost or gained, profoundly alter the cell’s protein composition. This aneuploidy-induced change in the cell’s proteome places a burden on the cell’s protein quality control pathways and hence impacts fitness.
C. Large-scale changes in DNA copy number may elicit a stress response
Whole chromosomal aneuploidies have been shown to lead to a transcriptional response in all organisms where this has been analyzed. A gene expression signature similar to the environmental stress response (ESR) in budding yeast has been found to exist not only in aneuploid budding yeast strains (Torres et al., 2007) but also in fission yeast, Arabidopsis, mouse and human cells with whole-chromosome gains (Sheltzer et al., 2012). The ESR was first defined in budding yeast. The ESR gene expression signature is characterized by genes involved in the response to stress being up-regulated and genes associated with cell growth and proliferation being down-regulated (Gasch, 2007). The ESR is elicited by a large number of stress conditions and is associated with slow growth (Brauer et al., 2008; Gasch et al., 2000; Regenberg et al., 2006). The finding that an ESR-like gene expression pattern exists in many different whole-chromosome aneuploidies in different organisms raises the interesting possibility that aneuploidy affects similar cellular pathways and causes an anti-proliferative response in most if not all organisms.
In addition to an ESR-like transcriptional response, several studies indicate that in mammalian cells, p53 is activated in response to chromosome mis-segregation and/or aneuploidy, though the proposed mechanisms differ. The first study to implicate p53 in antagonizing the proliferation of cells that mis-segregate chromosomes or that are aneuploid, examined mouse embryos lacking the spindle assembly checkpoint component MAD2. Such embryos die by embryonic day 7.5 due to massive chromosome mis-segregation, but deletion of p53 permits such embryos to survive until embryonic day 10.5 (Burds et al., 2005; Dobles et al., 2000). In 2010, Thompson and Compton showed that the process of mis-segregating chromosomes leads to activation of p53 through the stress-kinase p38 (Thompson and Compton, 2010). Li et al. (2010) also observed p53 activation upon chromosome mis-segregation, but suggested that the accumulation of reactive oxygen species (ROS) was responsible for p53 activation (Li et al., 2010). ROS, generated by increased metabolism in aneuploid cells, activates the DNA damage checkpoint kinase ATM in a DNA damage independent manner (Li et al., 2010). ATM in turn activates p53. Interestingly, the authors of this study also found that p53 activity is correlated with aneuploidy level. A low degree of aneuploidy caused a less pronounced p53 response than high levels. This graded response to aneuploidy may explain why p53 activity was not found to be increased in trisomic MEFs, in which the degree of aneuploidy is low and cells had time to adapt to the aneuploid state (Tang et al., 2011).
The studies that observed a p53 response in aneuploid cells did not examine cells with constitutive aneuploidies, but cells that actively mis-segregate chromosomes, a condition known as chromosome instability (CIN). p53 activation may thus not be a consequence of aneuploidy per se, but could also be caused by events associated with chromosome mis-segregation. This is the conclusion that Janssen et al. (2011) arrived at. Their studies suggest that the process of mis-segregating a chromosome leads to lagging chromosomes, which suffer DNA damage during cytokinesis. This cytokinesis-inflicted DNA damage in turn causes p53 activation through the canonical DNA damage pathway (Janssen et al., 2011). It should further be noted that cytokinesis failure even in the absence of DNA damage can cause p53 activation (de Stanchina et al., 1998; Serrano et al., 1997). Consistent with the idea that p53 activation may be a consequence of chromosome mis-segregation rather than aneuploidy per se is the observation that in cells harboring whole chromosome gains without accompanying CIN, a p53 response has not been detected. Trisomic MEFs, do not mount a p53 response (Tang et al., 2011). Thus it remains to be determined whether p53 activation is a response to chromosome missegregation-induced DNA damage or cytokinesis defects or a response to the aneuploid state or all of the above. While the exact function of p53 in CIN and aneuploidy remains to be determined it is clear that mechanisms other than p53 activation also limit the proliferation of cells harboring aneuploidies or other large-scale gene copy number changes. Trisomic MEFs proliferate poorly without mounting a p53 response (Tang et al., 2011).
D. Aneuploidy and CIN cause genome instability
It has recently become clear that the presence of additional chromosomes as well as the process of gaining or losing whole chromosomes has a dramatic effect on genome stability. Budding yeast strains harboring additional chromosomes exhibit increased rates of chromosome mis-segregation, mitotic recombination, mutation and DNA damage, as well as increased sensitivity to genotoxic agents (Sheltzer et al., 2011; Zhu et al., 2012). Aneuploid fission yeast cells are also sensitive to DNA damaging agents and harbor an increased number of DNA damage foci (Sheltzer et al., 2011). The origins of these genome instability-inducing effects of whole-chromosome aneuploidies are not yet known, but have been suggested to be due to changes in the copy number of specific genes. For example, changes in the dosage of the gene encoding the kinesin Kip3 impact mitotic spindle dynamics and cause increased chromosome mis-segregation in budding yeast (Su et al., 2011). Changes in gene copy number also impact the stability of their immediate environment. Repeated DNA sequences are prone to engage in non-allelic homologous recombination especially during meiosis, which can lead to genomic rearrangements.
Two recent studies in mammalian cells showed that the process of changing the copy number of whole chromosomes, chromosome mis-segregation, also induces genomic instability, especially DNA damage. Merotelically attached chromosomes - these are chromosomes that attach to microtubules emanating from both centrosomes - remain in the center of the cell during anaphase (Janssen et al., 2011; Thompson and Compton, 2010). These lagging chromosomes are then broken during cytokinesis, which leads to translocations and other genomic rearrangements (Janssen et al., 2011).
Chromosome breaks on lagging chromosomes not only occur because of breakage of the chromosome during cytokinesis, they also form because of their inefficient replication in the subsequent cell cycle (Crasta et al., 2012). Lagging chromosomes can form micronuclei if they fail to reach the bulk of the segregated DNA prior to nuclear envelope reformation. These micronuclei then undergo defective and asynchronous DNA replication, which results in DNA damage and fragmentation and rearrangement of the chromosome in the micronucleus. This phenomenon could explain a recently discovered feature of some cancers, called chromothripsis, where individual chromosomes undergo massive breakage and rearrangement (Stephens et al., 2011). Amplifications, deletions and whole-chromosome gains or losses, are a hallmark of cancer. The genome-instability inducing effects of chromosome mis-segregation and the resulting whole chromosomal aneuploidies could fuel the dramatic karyotype changes characteristic of tumorigenesis.
E. Are some developmental defects seen in aneuploid organisms due to the general effects caused by large-scale gene copy number alterations?
In cells changes in chromosome copy number elicit a set of phenotypes that appears to be independent of the identity of the genes whose copy number is altered. Are such general phenotypes also evident at the organismal level? Most organisms with whole chromosome aneuploidies or large segmental aneuploidies are growth retarded (reviewed in Torres et al. 2008), which is most likely due to the impact of large-scale gene copy number alterations on cell proliferation (see above). Beyond the stunted growth, few commonalities are observed among different aneuploids at the organismal level. This finding indicates that most developmental aberrations observed in organisms with large DNA copy number variations are due to copy number changes of specific genes. However, it is intriguing to note that in mammals some developmental phenotypes are observed in many different trisomies. Mice and humans trisomic for any chromosome exhibit craniofacial defects such as microcephaly or cleft palate, cardiac defects and nuchal edema (Gropp et al., 1975; Hassold and Jacobs, 1984; Krushinskii et al., 1986). It may simply require a large number of dosage sensitive genes to build the organs whose development is affected in many different aneuploidies, but it is also possible that some general aspects of large-scale gene copy number changes affect pathways used in the development of these organs.
In summary, the phenotypic analyses of small and large-scale changes in DNA copy number lead to two general conclusions. First, changes in the copy number of single or a small number of genes can lead to specific phenotypes, as illustrated by human diseases such as Charcot-Marie-Tooth 1A neuropathy or APP-driven early onset Alzheimer’s Disease. Second, when the copy number of large genomic regions is altered, such as in whole chromosome aneuploidies, the origins of the phenotypes are more complex. Some phenotypes are the result of cumulative effects of changes in copy number of a large number of genes, whereas others are caused by changes in the dosage of single genes.
Why are gene copy number alterations detrimental?
Although it is possible that in some circumstances changes in specific DNA sequences (i.e. centromeres or telomeres) can have adverse effects (Futcher and Carbon, 1986; Runge and Zakian, 1989), the most likely explanation for the majority of detrimental phenotypes caused by changes in gene copy number is the gene dosage hypothesis: gains or losses of gene copies change the expression levels of the affected gene. This altered dosage impacts fitness. The comparison of the phenotypes caused by changing the copy number of autosomes and sex chromosomes illustrates this point. The consequences of losing or gaining autosomes, for which dosage compensation does not occur, are severe. Alterations in sex chromosome copy number, for which dosage compensation occurs, are mild (reviewed in Berletch et al., 2011; Prestel et al., 2010).
We can envision several ways in which an altered gene dosage can negatively impact cellular or organismal fitness:
Dramatic over or underexpression of a specific gene can cause phenotypic changes (Figure 4A; 5A). The recurrent amplifications of c-Myc or of receptor tyrosine kinases in various tumors are examples of such effects.
Changing the levels of individual genes encoding structural proteins or proteins that function in protein complexes by 50 percent can affect fitness (Kacser and Burns, 1981; Veitia, 2002) (Figure 4B, 5B).
Overexpression of individual genes that encode proteins that engage in promiscuous low-affinity interactions can result in off-target interactions thereby impairing fitness (Vavouri et al., 2009)(Figure 5C).
The detrimental phenotypes associated with gene copy number changes can also result from synergistic effects of large-scale DNA copy number alterations.
Figure 4. Effects of increasing and decreasing gene copy number.
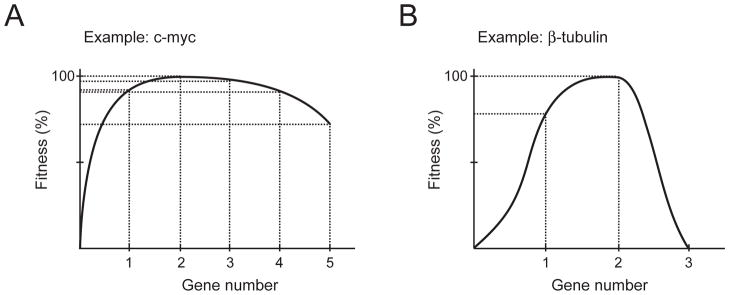
(A) Increasing or decreasing the levels of most genes by 50% has a minimal impact on fitness, but decreases or increases beyond that can affect fitness (based on Kacser and Burns, 1981).
(B) Changing the copy number of structural genes can have dramatic effects. The example of the β-tubulin-encoding gene is shown (Katz et al., 1990).
Figure 5. Consequences of changes in gene copy number.

(A) An increased dosage of a single gene, such as a rate-limiting enzyme B, can increase the output or function of a cellular pathway. Conversely, reduction of enzyme B will diminish the production of C thus decreasing pathway activity.
(B) Altered gene dosage can interfere with the function of stoichiometry-sensitive complexes, with excess of protein A or protein B inhibiting the function of C and therefore decreasing pathway activity.
(C) Overexpression of a regulatory enzyme can lead to off-target effects. For example overexpression of a protein kinase or protein phosphatase can cause deregulation of pathways that proteins usually do not function in.
(D) Changes in the copy number of many genes simultaneously can impact protein quality control mechanisms such as molecular chaperones and the ubiquitin–proteasome system (UPS). Misfolded proteins can eventually form aggregates.
Overexpression of many proteins at once can impact basic cellular functions such as energy homeostasis and protein quality control mechanisms (Figure 5D). In the absence of sufficient chaperone capacity to accommodate over-expressed proteins, other chaperone clients that failed to fold in a timely manner will be degraded or deposited as aggregates (Olzscha et al., 2011)(Figure 5D). Protein stoichiometry imbalances caused by gene copy number changes can also contribute to proteotoxicity. Many subunits of protein complexes only acquire a stable conformation by binding to other subunits of the complex (Kaizu et al., 2010; Vavouri et al., 2009). Thus, changes in dosage of genes encoding polypeptides that normally have binding partners will produce excess proteins that require the continuous assistance of chaperones, preventing chaperones from assisting other folding reactions. This could reduce the general folding capacity of the cell and thus interfere with their essential function of mediating folding of essential proteins (Olzscha et al., 2011). Indeed, the production of a mis-folded protein representing 0.1% of the total yeast proteome, leads to a significant reduction in fitness and elicits a cytoplasmic unfolded protein response (Geiler-Samerotte et al., 2010). Excess proteins also impact proteasomal degradation, as excess proteins are frequently cleared by ubiquitin-proteasomal degradation (Figure 5D). Energy stress may also exist in cells with large-scale DNA copy number alterations. The production of additional gene products and the shielding of the cell from the effects cost energy. This increased energy demand may negatively impact fitness.
Gene copy number alterations as a source of adaptive potential and their function during normal development
Gene copy number changes are not always detrimental. Experimental evolution studies demonstrate that under selection, gene copy number changes endow the organism with increased survival under the imposed selection. Perhaps the best example of such beneficial effects is the tissue-specific amplification of genes as part of normal development.
DNA copy number changes as a source of adaptive potential
In 1981, Edlund and Normark reported that amplification of the ampC locus promotes antibiotic resistance in E. coli (Edlund and Normark, 1981). Many subsequent studies have shown that gene amplifications frequently arise during adaptive evolution experiments. For example in budding yeast, growth under glucose limitation selects for cells in which the genes encoding high-affinity glucose transporters are amplified (Brown et al., 1998; Dunham et al., 2002; Kao and Sherlock, 2008) (Figure 6A). Studies employing other nutrient deprivation selections revealed similar principles; genes that facilitate the uptake and/or metabolism of the limiting factor are upregulated often through gene amplification (Gresham et al., 2008; Gresham et al., 2010). This principle is recapitulated during tumorigenesis. Amplifications of oncogenes and losses of tumor suppressor genes are an integral feature of the disease (Gordon et al., 2012).
Figure 6. Small and large-scale gene copy number alterations arise during adaptive evolution experiments.

(A) Eight S. cerevisiae strains were isolated after growth under glucose-limiting conditions for 100–500 generations and DNA content was assessed. Red and green indicate gene copy number amplification and reduction, respectively. HXT6 encodes a high-affinity hexose transporter and is amplified in evolved strains E1, 5, and 8. Data from (Dunham et al., 2002).
(B) Trisomy 3 is a transient intermediate during continuous growth at 39°C. Cells were grown for 450 generations at 39°C. Many isolates harbored an additional copy of chromosomes III. Upon return to the permissive temperature (30°C) the trisomy was quickly lost. The trisomy was also lost after continuous growth at 39°C (1000 generations) and replaced by changes in the expression of genes that confer resistance to high temperature.
Whole chromosome and segmental aneuploidies are also obtained in experimental evolution experiments, because a gene in that genomic region provides a selective advantage. For example, the pathogenic fungus C. albicans can develop resistance to the antifungal drug fluconazole through duplication of the left half of chromosome 5 (Selmecki et al., 2006). This region of chromosome 5 harbors ERG11, which encodes the biosynthetic enzyme targeted by fluconazole as well as TAC1, a transcription factor that up-regulates expression of (ABC) transporter genes (Selmecki et al., 2006). Their duplication is an important contributor to fluconazole resistance in strains harboring the segmental aneuploidy of chromosome 5. In budding yeast, whole chromosome aneuploidies have been obtained in the selection for suppressors of cytokinesis defects (Rancati et al., 2008). These aneuploidies occurred in the context of an increase in base ploidy. Polyploid cells exhibit increased chromosome instability (Storchova et al., 2006) indicating that polyploidization predisposes to aneuploidy. Polyploidy may not only facilitate the generation of aneuploidy (Storchova et al., 2006), it also attenuates the phenotypes associated with the condition (Torres et al., 2007) and may thus buffer against the adverse effects of aneuploidy allowing cells to take full advantage of potential beneficial effects associated with certain gene copy number alterations. This could explain why cancer cells, which are highly aneuploid, often show increased base ploidy.
The selective advantage gained through whole chromosome aneuploidies comes at a price. A large-scale DNA copy number alteration results in changes in the dosage of many genes, the adverse effects of which dampen the potential benefits that arise due to the duplication of gene(s) conferring a selective advantage. This was elegantly demonstrated by Yona et al. (2012). They showed that whole chromosome copy number changes are a transient occurrence in adaptive evolution experiments. They provide a quick means of adapting to a continuous selective pressure that is replaced over time by more subtle genetic changes that achieve the same goal (Yona et al., 2012). Diploid budding yeast grown continuously at an elevated temperature of 39°C developed trisomy of chromosome III. This aneuploidy was lost once the cells were returned to growth at normal temperature (30°C). Loss of the trisomy not only occurred when the selective pressure was removed, but even when cells were kept at 39°C for extended periods. After 1000 generations trisomy III was replaced by increased expression of 17–18 genes located on chromosome III and presumably of genes elsewhere in the genome that conveyed increased temperature tolerance. Interestingly, there was a significant overlap in up-regulated genes among four repetitions. These results indicate that whole chromosome aneuploidies can provide an effective means of quickly adapting to a selective pressure. This is not surprising given that chromosome loss rates are by 3 orders of magnitude higher than mutation rates (10−5 compared to 10−8). Eventually, however, the “crude” whole chromosome gain/loss solutions are replaced by ones that do not bring with them the adverse effects caused by gene dosage changes of the other genes located on the gained/lost chromosome.
Recent studies in mice indicate that that aneuploidy is also an effective way to adapt to selective pressure in multicellular organisms. The liver is a naturally aneuploid organ (Duncan et al.; Putkey et al., 2002; Weaver et al., 2007; described in detail below) providing an opportunity to determine the importance of aneuploidy in adaptation to selective pressure. Deficiency of fumarylacetoacetate hydrolase (FAH) causes chronic liver disease that is suppressed by the loss of enzymes functioning upstream of FAH such as homogentisic acid dioxygenase (HPD). Mice lacking FAH function and heterozygous for a deletion in HPD develop disease resistance through loss of the chromosome encoding the functional copy of HPD (Duncan et al., 2012) illustrating the power of aneuploidy in providing adaptive potential even in multicellular organisms.
Mutations that suppress the adverse effects of large-scale DNA copy number changes may enhance the adaptive potential of chromosome gains or losses. Aneuploidy-tolerating mutations have been found in yeast (Torres et al., 2010). Loss of p53 increases survival of cells following chromosome mis-segregation in mammalian cells (Janssen et al., 2011; Li et al., 2010; Thompson and Compton, 2010). It will be interesting to examine the contribution of different aneuploidy-tolerating mutations to disease progression in cancer where gene copy number alterations are a key feature. Indeed, p53 inactivation is a major contributor to tumorigenesis (Dai and Gu, 2010; Kruse and Gu, 2009; Toledo and Wahl, 2006).
Gene copy number alterations as part of normal development
Differentiation into specific cell types often requires increased expression of particular genes. In some instances this is achieved by amplification of specific loci. rDNA amplification occurs in amphibian oocytes, the macronucleus of Tetrahymena and budding yeast (Brown and Dawid, 1968; Claycomb and Orr-Weaver, 2005; Gall, 1968; Kobayashi et al., 1998; Nordman and Orr-Weaver, 2012; Oakes et al., 2006; Pesin and Orr-Weaver, 2008; Yao et al., 1974) and thus appears to be a common mechanism used to accommodate different protein synthesis needs in different tissues and under changing environmental conditions. While rDNA amplification is not uncommon, amplification of selective genes is rarely used to up-regulate gene expression and in the instances where this has been described, occurs in cells destined to die. In Drosophila, follicle cells amplify the chorion genes, which encode eggshell proteins (Spradling, 1981). This amplification allows follicle cells to make the eggshell. Once they have accomplished this task they become part of the eggshell.
Whole chromosome aneuploidies have been found in various tissues in mammals. In mice and humans, approximately 30 percent of neuroblasts in the embryonic brain are aneuploid (Rehen et al., 2001). The majority of these aneuploid neurons are eliminated during development, but 10% of neurons in adult brains are estimated to be aneuploid (Rehen et al., 2001; Rehen et al., 2005; Westra et al., 2008). The biological significance and the impact of this increased aneuploidy on neuronal physiology and fitness remains to be determined.
Mammalian hepatocytes acquire whole chromosome gains and losses during aging, and in response to toxic stresses and disease (Duncan et al.; Putkey et al., 2002; Weaver et al., 2007). Aneuploidy in hepatocytes is preceded by polyploidization as a result of failed cytokinesis. Polyploid cells that not only harbor additional genome copies but also additional centrosomes then undergo a mitotic division in which all centrosomes form a multipolar spindle to produce aneuploid progeny. Whether aneuploid liver cells are as fit as their euploid counter parts remains to be determined, but aneuploidy has been hypothesized to provide genetic variation so that the liver can adapt to nutritional and noxious challenges (Duncan et al., 2010). Determining the importance of aneuploidy in the function of tissues such as the brain and the liver and defining whether different cell types exhibit different sensitivities to gene copy number changes will provide critical insights into how gene copy number variations contribute to cell type specification and diseases such as cancer, in which aneuploidy is the norm yet fitness as judged by unrestricted proliferation is high.
Future directions – gene copy number alterations as a therapeutic target?
The last decade has brought to light the importance of gene copy number changes in a wide variety of human diseases. Understanding in detail of how changes in copy number of individual genes or large chromosomal regions enhance or perhaps suppress disease is a critical challenge. With an ever more detailed view of the human genome, its variations in the normal population and in disease prone families, we will be able to learn which copy number changes don’t matter and allow us to focus on those that impact function.
A particularly exciting future direction is the pursuit of gene copy number changes in developing therapies. The realization that CNVs contribute to difficult to treat diseases, such as neurological and psychiatric disorders brings with it the possibility of targeting the gene products amplified/deleted in particular disorders. For example duplication of AUTS4, a gene encoding the GABAA receptor subunit, has been associated with autism spectrum disorder (Zhang et al., 2009). Autistic individual with this CNV may benefit from compounds that downregulate GABAA receptor function. CNVs causing disruptions of MYT1L, CTNND2 and ASTN2 have been seen in patients with Schizophrenia. Crohn’s disease, a bowl inflammatory disease, is linked to copy number reduction of HBD2. Compounds that increase the function of the remaining copy or inhibit the function of negative regulators of the pathways that these genes function in may have a significant therapeutic index.
Cancer is the prime example in which gene amplifications and deletions have been shown to drive disease (Gordon et al., 2012). Therapies where overexpressed or amplified oncogenic drivers are targeted have been developed. The gene encoding EGFR is amplified in non-small-cell lung cancer. Small molecules such as gefitinib or erlotinib have been applied to inhibit EGFR with success (Carling, 2004; Paez et al., 2004). ERBB2, which encodes the EGFR receptor HER2, is amplified in ~30% of primary breast cancers (Slamon et al., 1987). Trastuzumab, an anti-HER2 antibody, has been used in the therapy of HER2 amplified breast cancers with great success (Baselga et al., 1998). These successes raise the exciting possibility that targeting amplified disease drivers provides opportunities for therapy in cancer, psychiatric disorders and autoimmune diseases, where effective treatments are scarce.
Whether large-scale gene copy number changes as are the cause of Down syndrome or as occur in cancer can be targeted in therapy remains to be determined. In Down syndrome, gene dosage changes of many different genes contribute to the associated phenotypes, making the development of therapeutics a challenge. The identification of individual genes responsible for specific phenotypes could enable the development of therapeutics that target specific aspects of the condition. For example, individuals with Down syndrome could benefit from therapies that lower APP protein levels, to prevent early onset Alzheimer’s disease; the development of which has, unfortunately, failed so far.
In cancer, the situation is even more complex. In this disease, the contribution of gene dosage changes of many genes is augmented by the variability of an ever-changing genetic make-up. Many cancers do however harbor specific aneuploidies that could be targeted in therapy. For example, trisomy 8 is frequently observed in patients with AML and associated with poor survival when present together with other genetic aberrations (Wolman et al., 2002). Drugs that target cells with amplified chromosome 8 may aid in the treatment of AML. Genomic instability in cancers also leads to loss of many genomic regions. These genetic lesions could provide additional therapeutic targets (Nijhawan et al., 2012). The general stress phenotypes associated with aneuploidy could also be explored in cancer treatment. The advantage of such compounds is that they would show efficacy against a broad spectrum of cancers. Compounds that preferentially inhibit the proliferation of aneuploid cell lines have been shown to exist and appear to exaggerate the general stress phenotypes associated with whole chromosome copy number changes (Tang et al., 2011). These compounds included AICAR, an agonist of the stress activated AMP kinase and the Hsp90 inhibitor 17-AAG (Tang et al., 2011). Thus targeting the general stresses associated with aneuploidy could be developed as cancer drug targets. It is worth noting that Hsp90 inhibitors such as 17-AAG, displaye anti-tumor efficacy in HER2/ErbB2-positive breast cancer and are currently in phase II and III clinical trails. The proteasome inhibitor bortezomib is used in the treatment of multiple myeloma (Richardson et al., 2005). Other inhibitors of the ubiquitin-proteasome system such as highly specific inhibitors of the proteasome-associated deubiquitinating enzyme Usp14 (Lee et al., 2010) could also show efficacy against aneuploid cells and thus could be used in the treatment of aneuploid cancers. We note that, compounds that target aneuploid cells may be especially effective when combined with chemotherapeutics that increase chromosome mis-segregation such as Taxol.
Changes in gene copy number, large and small in scale, contribute to population diversity and are significant contributors to disease. Understanding their cost and benefits will provide critical insights into evolution and diseases.
Acknowledgments
We thank D. Pellman, M. Dunham, F. Solomon, S. Pfau, J. Sheltzer, J. Rock, and K. Knouse for their critical reading of this manuscript. Work in the Amon lab was supported by NIH grant GM56800. A. Amon is also an Investigator of the Howard Hughes Medical Institute. Y.-C. Tang is supported by a Human Frontier Science Program Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. 2011;12:363–376. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36:744–749. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- Baselga J, Norton L, Albanell J, Kim YM, Mendelsohn J. Recombinant humanized anti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res. 1998;58:2825–2831. [PubMed] [Google Scholar]
- Berletch JB, Yang F, Xu J, Carrel L, Disteche CM. Genes that escape from X inactivation. Hum Genet. 2011;130:237–245. doi: 10.1007/s00439-011-1011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauer MJ, Huttenhower C, Airoldi EM, Rosenstein R, Matese JC, Gresham D, Boer VM, Troyanskaya OG, Botstein D. Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol Biol Cell. 2008;19:352–367. doi: 10.1091/mbc.E07-08-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CJ, Todd KM, Rosenzweig RF. Multiple duplications of yeast hexose transport genes in response to selection in a glucose-limited environment. Mol Biol Evol. 1998;15:931–942. doi: 10.1093/oxfordjournals.molbev.a026009. [DOI] [PubMed] [Google Scholar]
- Brown DD, Dawid IB. Specific gene amplification in oocytes. Oocyte nuclei contain extrachromosomal replicas of the genes for ribosomal RNA. Science. 1968;160:272–280. doi: 10.1126/science.160.3825.272. [DOI] [PubMed] [Google Scholar]
- Burds AA, Lutum AS, Sorger PK. Generating chromosome instability through the simultaneous deletion of Mad2 and p53. Proc Natl Acad Sci U S A. 2005;102:11296–11301. doi: 10.1073/pnas.0505053102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling D. The AMP-activated protein kinase cascade--a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Chikashige Y, Haraguchi T, Hiraoka Y. Another way to move chromosomes. Chromosoma. 2007;116:497–505. doi: 10.1007/s00412-007-0114-8. [DOI] [PubMed] [Google Scholar]
- Claycomb JM, Orr-Weaver TL. Developmental gene amplification: insights into DNA replication and gene expression. Trends Genet. 2005;21:149–162. doi: 10.1016/j.tig.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53–58. doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med. 2010;16:528–536. doi: 10.1016/j.molmed.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Stanchina E, McCurrach ME, Zindy F, Shieh SY, Ferbeyre G, Samuelson AV, Prives C, Roussel MF, Sherr CJ, Lowe SW. E1A signaling to p53 involves the p19(ARF) tumor suppressor. Genes Dev. 1998;12:2434–2442. doi: 10.1101/gad.12.15.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobles M, Liberal V, Scott ML, Benezra R, Sorger PK. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 2000;101:635–645. doi: 10.1016/s0092-8674(00)80875-2. [DOI] [PubMed] [Google Scholar]
- Duncan AW, Hanlon Newell AE, Bi W, Finegold MJ, Olson SB, Beaudet AL, Grompe M. Aneuploidy as a mechanism for stress-induced liver adaptation. J Clin Invest. 2012;122:3307–3315. doi: 10.1172/JCI64026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan AW, Taylor MH, Hickey RD, Hanlon Newell AE, Lenzi ML, Olson SB, Finegold MJ, Grompe M. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 2010;467:707–710. doi: 10.1038/nature09414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham MJ, Badrane H, Ferea T, Adams J, Brown PO, Rosenzweig F, Botstein D. Characteristic genome rearrangements in experimental evolution of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2002;99:16144–16149. doi: 10.1073/pnas.242624799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlund T, Normark S. Recombination between short DNA homologies causes tandem duplication. Nature. 1981;292:269–271. doi: 10.1038/292269a0. [DOI] [PubMed] [Google Scholar]
- Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- Futcher B, Carbon J. Toxic effects of excess cloned centromeres. Mol Cell Biol. 1986;6:2213–2222. doi: 10.1128/mcb.6.6.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall JG. Differential synthesis of the genes for ribosomal RNA during amphibian oogenesis. Proc Natl Acad Sci U S A. 1968;60:553–560. doi: 10.1073/pnas.60.2.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP. Comparative genomics of the environmental stress response in ascomycete fungi. Yeast. 2007;24:961–976. doi: 10.1002/yea.1512. [DOI] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiler-Samerotte KA, Dion MF, Budnik BA, Wang SM, Hartl DL, Drummond DA. Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc Natl Acad Sci U S A. 2010;108:680–685. doi: 10.1073/pnas.1017570108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Campbell CD, Eichler EE. Human copy number variation and complex genetic disease. Annu Rev Genet. 2012;45:203–226. doi: 10.1146/annurev-genet-102209-163544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet. 2012;13:189–203. doi: 10.1038/nrg3123. [DOI] [PubMed] [Google Scholar]
- Gresham D, Desai MM, Tucker CM, Jenq HT, Pai DA, Ward A, DeSevo CG, Botstein D, Dunham MJ. The repertoire and dynamics of evolutionary adaptations to controlled nutrient-limited environments in yeast. PLoS Genet. 2008;4:e1000303. doi: 10.1371/journal.pgen.1000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresham D, Usaite R, Germann SM, Lisby M, Botstein D, Regenberg B. Adaptation to diverse nitrogen-limited environments by deletion or extrachromosomal element formation of the GAP1 locus. Proc Natl Acad Sci U S A. 2010;107:18551–18556. doi: 10.1073/pnas.1014023107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gropp A, Kolbus U, Giers D. Systematic approach to the study of trisomy in the mouse. II. Cytogenet Cell Genet. 1975;14:42–62. doi: 10.1159/000130318. [DOI] [PubMed] [Google Scholar]
- Guo M, Birchler JA. Trans-acting dosage effects on the expression of model gene systems in maize aneuploids. Science. 1994;266:1999–2002. doi: 10.1126/science.266.5193.1999. [DOI] [PubMed] [Google Scholar]
- Hanemann CO, Muller HW. Pathogenesis of Charcot-Marie-Tooth 1A (CMT1A) neuropathy. Trends Neurosci. 1998;21:282–286. doi: 10.1016/s0166-2236(97)01222-8. [DOI] [PubMed] [Google Scholar]
- Hanks S, Coleman K, Reid S, Plaja A, Firth H, Fitzpatrick D, Kidd A, Mehes K, Nash R, Robin N, et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet. 2004;36:1159–1161. doi: 10.1038/ng1449. [DOI] [PubMed] [Google Scholar]
- Hassold TJ, Jacobs PA. Trisomy in man. Annu Rev Genet. 1984;18:69–97. doi: 10.1146/annurev.ge.18.120184.000441. [DOI] [PubMed] [Google Scholar]
- Henrichsen CN, Vinckenbosch N, Zollner S, Chaignat E, Pradervand S, Schutz F, Ruedi M, Kaessmann H, Reymond A. Segmental copy number variation shapes tissue transcriptomes. Nat Genet. 2009;41:424–429. doi: 10.1038/ng.345. [DOI] [PubMed] [Google Scholar]
- Holland AJ, Cleveland DW. Losing balance: the origin and impact of aneuploidy in cancer. EMBO Rep. 2012;13:501–514. doi: 10.1038/embor.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettel B, Kreil DP, Matzke M, Matzke AJ. Effects of aneuploidy on genome structure, expression, and interphase organization in Arabidopsis thaliana. PLoS Genet. 2008;4:e1000226. doi: 10.1371/journal.pgen.1000226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- Itsara A, Cooper GM, Baker C, Girirajan S, Li J, Absher D, Krauss RM, Myers RM, Ridker PM, Chasman DI, et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itsara A, Wu H, Smith JD, Nickerson DA, Romieu I, London SJ, Eichler EE. De novo rates and selection of large copy number variation. Genome Res. 2010;20:1469–1481. doi: 10.1101/gr.107680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science. 2011;333:1895–1898. doi: 10.1126/science.1210214. [DOI] [PubMed] [Google Scholar]
- Jones L, Wei G, Sevcikova S, Phan V, Jain S, Shieh A, Wong JC, Li M, Dubansky J, Maunakea ML, et al. Gain of MYC underlies recurrent trisomy of the MYC chromosome in acute promyelocytic leukemia. J Exp Med. 2010;207:2581–2594. doi: 10.1084/jem.20091071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacser H, Burns JA. The molecular basis of dominance. Genetics. 1981;97:639–666. doi: 10.1093/genetics/97.3-4.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlem P, Sultan M, Herwig R, Steinfath M, Balzereit D, Eppens B, Saran NG, Pletcher MT, South ST, Stetten G, et al. Transcript level alterations reflect gene dosage effects across multiple tissues in a mouse model of down syndrome. Genome Res. 2004;14:1258–1267. doi: 10.1101/gr.1951304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaizu K, Moriya H, Kitano H. Fragilities caused by dosage imbalance in regulation of the budding yeast cell cycle. PLoS Genet. 2010;6:e1000919. doi: 10.1371/journal.pgen.1000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao KC, Sherlock G. Molecular characterization of clonal interference during adaptive evolution in asexual populations of Saccharomyces cerevisiae. Nat Genet. 2008;40:1499–1504. doi: 10.1038/ng.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz W, Weinstein B, Solomon F. Regulation of tubulin levels and microtubule assembly in Saccharomyces cerevisiae: consequences of altered tubulin gene copy number. Mol Cell Biol. 1990;10:5286–5294. doi: 10.1128/mcb.10.10.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JC, Nordman J, Xie F, Kashevsky H, Eng T, Li S, MacAlpine DM, Orr-Weaver TL. Integrative analysis of gene amplification in Drosophila follicle cells: parameters of origin activation and repression. Genes Dev. 2011;25:1384–1398. doi: 10.1101/gad.2043111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsbury MA, Yung YC, Peterson SE, Westra JW, Chun J. Aneuploidy in the normal and diseased brain. Cell Mol Life Sci. 2006;63:2626–2641. doi: 10.1007/s00018-006-6169-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Heck DJ, Nomura M, Horiuchi T. Expansion and contraction of ribosomal DNA repeats in Saccharomyces cerevisiae: requirement of replication fork blocking (Fob1) protein and the role of RNA polymerase I. Genes Dev. 1998;12:3821–3830. doi: 10.1101/gad.12.24.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krushinskii LV, Dyban AP, Baranov VS, Poletaeva II, Romanova LG. Behavior of mice with robertsonian translocations of chromosomes. Genetika. 1986;22:434–441. [Google Scholar]
- Lachman HM, Pedrosa E, Petruolo OA, Cockerham M, Papolos A, Novak T, Papolos DF, Stopkova P. Increase in GSK3beta gene copy number variation in bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:259–265. doi: 10.1002/ajmg.b.30498. [DOI] [PubMed] [Google Scholar]
- Larsson J, Chen JD, Rasheva V, Rasmuson-Lestander A, Pirrotta V. Painting of fourth, a chromosome-specific protein in Drosophila. Proc Natl Acad Sci U S A. 2001;98:6273–6278. doi: 10.1073/pnas.111581298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Fang X, Baker DJ, Guo L, Gao X, Wei Z, Han S, van Deursen JM, Zhang P. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc Natl Acad Sci U S A. 2010 doi: 10.1073/pnas.1005960107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Fang X, Wei Z, York JP, Zhang P. Loss of spindle assembly checkpoint-mediated inhibition of Cdc20 promotes tumorigenesis in mice. J Cell Biol. 2009;185:983–994. doi: 10.1083/jcb.200904020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR. Genomic rearrangements and sporadic disease. Nat Genet. 2007;39:S43–47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- Lyle R, Gehrig C, Neergaard-Henrichsen C, Deutsch S, Antonarakis SE. Gene expression from the aneuploid chromosome in a trisomy mouse model of down syndrome. Genome Res. 2004;14:1268–1274. doi: 10.1101/gr.2090904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223–1241. doi: 10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miclaus M, Xu JH, Messing J. Differential gene expression and epiregulation of alpha zein gene copies in maize haplotypes. PLoS Genet. 2011;7:e1002131. doi: 10.1371/journal.pgen.1002131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DK, Disteche CM. Dosage compensation of the active X chromosome in mammals. Nat Genet. 2006;38:47–53. doi: 10.1038/ng1705. [DOI] [PubMed] [Google Scholar]
- Nijhawan D, Zack TI, Ren Y, Strickland MR, Lamothe R, Schumacher SE, Tsherniak A, Besche HC, Rosenbluh J, Shehata S, et al. Cancer vulnerabilities unveiled by genomic loss. Cell. 2012;150:842–854. doi: 10.1016/j.cell.2012.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa O, Tange Y, Kurabayashi A. Growth arrest and chromosome instability in aneuploid yeast. Yeast. 2006;23:937–950. doi: 10.1002/yea.1411. [DOI] [PubMed] [Google Scholar]
- Nordman J, Orr-Weaver TL. Regulation of DNA replication during development. Development. 2012;139:455–464. doi: 10.1242/dev.061838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes ML, Johzuka K, Vu L, Eliason K, Nomura M. Expression of rRNA genes and nucleolus formation at ectopic chromosomal sites in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 2006;26:6223–6238. doi: 10.1128/MCB.02324-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH, Tartaglia GG, Vendruscolo M, Hayer-Hartl M, Hartl FU, Vabulas RM. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell. 2011;144:67–78. doi: 10.1016/j.cell.2010.11.050. [DOI] [PubMed] [Google Scholar]
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, Sanderson BW, Hattem GL, Li R. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature. 2010;468:321–325. doi: 10.1038/nature09529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesin JA, Orr-Weaver TL. Regulation of APC/C activators in mitosis and meiosis. Annu Rev Cell Dev Biol. 2008;24:475–499. doi: 10.1146/annurev.cellbio.041408.115949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfau SJ, Amon A. Chromosomal instability and aneuploidy in cancer: from yeast to man. EMBO Rep. 2012;13:515–527. doi: 10.1038/embor.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestel M, Feller C, Becker PB. Dosage compensation and the global re-balancing of aneuploid genomes. Genome Biol. 2010;11:216. doi: 10.1186/gb-2010-11-8-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putkey FR, Cramer T, Morphew MK, Silk AD, Johnson RS, McIntosh JR, Cleveland DW. Unstable kinetochore-microtubule capture and chromosomal instability following deletion of CENP-E. Dev Cell. 2002;3:351–365. doi: 10.1016/s1534-5807(02)00255-1. [DOI] [PubMed] [Google Scholar]
- Rancati G, Pavelka N, Fleharty B, Noll A, Trimble R, Walton K, Perera A, Staehling-Hampton K, Seidel CW, Li R. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell. 2008;135:879–893. doi: 10.1016/j.cell.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regenberg B, Grotkjaer T, Winther O, Fausboll A, Akesson M, Bro C, Hansen LK, Brunak S, Nielsen J. Growth-rate regulated genes have profound impact on interpretation of transcriptome profiling in Saccharomyces cerevisiae. Genome Biol. 2006;7:R107. doi: 10.1186/gb-2006-7-11-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehen SK, McConnell MJ, Kaushal D, Kingsbury MA, Yang AH, Chun J. Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc Natl Acad Sci U S A. 2001;98:13361–13366. doi: 10.1073/pnas.231487398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehen SK, Yung YC, McCreight MP, Kaushal D, Yang AH, Almeida BS, Kingsbury MA, Cabral KM, McConnell MJ, Anliker B, et al. Constitutional aneuploidy in the normal human brain. J Neurosci. 2005;25:2176–2180. doi: 10.1523/JNEUROSCI.4560-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352:2487–2498. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- Runge KW, Zakian VA. Introduction of extra telomeric DNA sequences into Saccharomyces cerevisiae results in telomere elongation. Mol Cell Biol. 1989;9:1488–1497. doi: 10.1128/mcb.9.4.1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- Selmecki A, Forche A, Berman J. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science. 2006;313:367–370. doi: 10.1126/science.1128242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Sheltzer JM, Blank HM, Pfau SJ, Tange Y, George BM, Humpton TJ, Brito IL, Hiraoka Y, Niwa O, Amon A. Aneuploidy drives genomic instability in yeast. Science. 2011;333:1026–1030. doi: 10.1126/science.1206412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM, Torres EM, Dunham MJ, Amon A. Transcriptional consequences of aneuploidy. Proc Natl Acad Sci U S A. 2012 doi: 10.1073/pnas.1209227109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- Spradling AC. The organization and amplification of two chromosomal domains containing Drosophila chorion genes. Cell. 1981;27:193–201. doi: 10.1016/0092-8674(81)90373-1. [DOI] [PubMed] [Google Scholar]
- Stenberg P, Larsson J. Buffering and the evolution of chromosome-wide gene regulation. Chromosoma. 2011;120:213–225. doi: 10.1007/s00412-011-0319-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele S, Stoehr G, Peplowska K, Cox J, Mann M, Storchova Z. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol Syst Biol. 2012;8:608. doi: 10.1038/msb.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storchova Z, Breneman A, Cande J, Dunn J, Burbank K, O’Toole E, Pellman D. Genome-wide genetic analysis of polyploidy in yeast. Nature. 2006;443:541–547. doi: 10.1038/nature05178. [DOI] [PubMed] [Google Scholar]
- Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, Redon R, Bird CP, de Grassi A, Lee C, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Qiu W, Gupta ML, Jr, Pereira-Leal JB, Reck-Peterson SL, Pellman D. Mechanisms underlying the dual-mode regulation of microtubule dynamics by Kip3/kinesin-8. Mol Cell. 2011;43:751–763. doi: 10.1016/j.molcel.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YC, Williams BR, Siegel JJ, Amon A. Identification of aneuploidy-selective antiproliferation compounds. Cell. 2011;144:499–512. doi: 10.1016/j.cell.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Compton DA. Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol. 2008;180:665–672. doi: 10.1083/jcb.200712029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Compton DA. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol. 2010;188:369–381. doi: 10.1083/jcb.200905057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6:909–923. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, Gygi SP, Dunham MJ, Amon A. Identification of aneuploidy-tolerating mutations. Cell. 2010;143:71–83. doi: 10.1016/j.cell.2010.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916–924. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- Torres EM, Williams BR, Amon A. Aneuploidy: cells losing their balance. Genetics. 2008;179:737–746. doi: 10.1534/genetics.108.090878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upender MB, Habermann JK, McShane LM, Korn EL, Barrett JC, Difilippantonio MJ, Ried T. Chromosome transfer induced aneuploidy results in complex dysregulation of the cellular transcriptome in immortalized and cancer cells. Cancer Res. 2004;64:6941–6949. doi: 10.1158/0008-5472.CAN-04-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacik T, Ort M, Gregorova S, Strnad P, Blatny R, Conte N, Bradley A, Bures J, Forejt J. Segmental trisomy of chromosome 17: a mouse model of human aneuploidy syndromes. Proc Natl Acad Sci U S A. 2005;102:4500–4505. doi: 10.1073/pnas.0500802102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vavouri T, Semple JI, Garcia-Verdugo R, Lehner B. Intrinsic protein disorder and interaction promiscuity are widely associated with dosage sensitivity. Cell. 2009;138:198–208. doi: 10.1016/j.cell.2009.04.029. [DOI] [PubMed] [Google Scholar]
- Veitia RA. Exploring the etiology of haploinsufficiency. Bioessays. 2002;24:175–184. doi: 10.1002/bies.10023. [DOI] [PubMed] [Google Scholar]
- Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Westra JW, Peterson SE, Yung YC, Mutoh T, Barral S, Chun J. Aneuploid mosaicism in the developing and adult cerebellar cortex. J Comp Neurol. 2008;507:1944–1951. doi: 10.1002/cne.21648. [DOI] [PubMed] [Google Scholar]
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322:703–709. doi: 10.1126/science.1160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolman SR, Gundacker H, Appelbaum FR, Slovak ML. Impact of trisomy 8 (+8) on clinical presentation, treatment response, and survival in acute myeloid leukemia: a Southwest Oncology Group study. Blood. 2002;100:29–35. doi: 10.1182/blood.v100.1.29. [DOI] [PubMed] [Google Scholar]
- Yang Q, Rasmussen SA, Friedman JM. Mortality associated with Down’s syndrome in the USA from 1983 to 1997: a population-based study. Lancet. 2002;359:1019–1025. doi: 10.1016/s0140-6736(02)08092-3. [DOI] [PubMed] [Google Scholar]
- Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, Zhou B, Hebert M, Jones KN, Shu Y, Kitzmiller K, et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet. 2007;80:1037–1054. doi: 10.1086/518257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao MC, Kimmel AR, Gorovsky MA. A small number of cistrons for ribosomal RNA in the germinal nucleus of a eukaryote, Tetrahymena pyriformis. Proc Natl Acad Sci U S A. 1974;71:3082–3086. doi: 10.1073/pnas.71.8.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Gu W, Hurles ME, Lupski JR. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet. 2009;10:451–481. doi: 10.1146/annurev.genom.9.081307.164217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11:468–476. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Pavelka N, Bradford WD, Rancati G, Li R. Karyotypic determinants of chromosome instability in aneuploid budding yeast. PLoS Genet. 2012;8:e1002719. doi: 10.1371/journal.pgen.1002719. [DOI] [PMC free article] [PubMed] [Google Scholar]