

The right ventricle (RV) of the heart, which pumps blood through a low pressure, low resistance “lesser” circulation, is now receiving more attention by researchers and clinicians, after many years of benign neglect.[1–7] Some of the many reasons for neglecting the RV assumed that (1) In the context of global cardiac performance, the RV was not very important;[2] and (2) The cellular and molecular mechanisms of RV failure (RVF) were not different from those responsible for LV failure. Perhaps the biggest knowledge gap originated from the lack of experimental studies modeling the development of RVF, and from attempts to extrapolate mechanisms of chronic RVF from studies originally designed to study acute RVF. These likely explain why “pressure overload” is the most frequent—and sometimes exclusively—quoted mechanism of RVF. Indeed, patients with chronic, progressive pulmonary vascular disease most frequently die of RV failure.[8,9] However, as we enlarge our knowledge of in the pathobiology of RVF, we should also begin to consider RVF as a “progressive,” but not necessarily “chronic,” phenomenon.
THE CASE FOR INVESTIGATING RV FUNCTION AND DYSFUNCTION
We propose that the outcome of the patient with pulmonary arterial hypertension (PAH) is not fundamentally determined by pathophysiological characteristics germane to the lung disease (e.g., vasoconstriction and pulmonary vascular remodeling), but rather by the response of the RV to increased afterload and to additional mechanistically important factors imposed on the RV such as neuroendocrine system activation,[3,10] and, perhaps, factors released from a “sick-lung circulation”[11] (Fig. 1). In the management of patients with severe PAH, we still face (as a barrier) the fact that prevention of RV failure is not a realized treatment goal. For the past 15 years, clinical studies have utilized vasodilator drugs expecting to significantly reduce pulmonary arterial pressure, halt or delay the progression of the lung vascular disease, reduce RV afterload, and prevent the development of RVF. However, this goal is not often accomplished in the clinical setting, as a recent meta-analysis of PAH clinical trials reported a weighted mean reduction in mean pulmonary arterial pressure of 3 mmHg after treatment.[2] Moreover, it has been demonstrated that even after many years of prostacyclin therapy (at least with epoprostenol treatment), pathological lung vascular remodeling does not stop.[12] Lastly, a recent cardiac MRI study from the Netherlands’ pulmonary hypertension center reported that the survival of medically-treated patients with PAH was not determined by a vasodilator-induced decrease of the pulmonary vascular resistance, but rather by an improved RV ejection fraction (RVEF).[13] Thus, if current vasodilator drugs have failed to reverse the vascular disease, should we not start thinking about RV-targeted therapies while we wait for new PAH lung-specific therapies to get in the pipeline?
Figure 1.
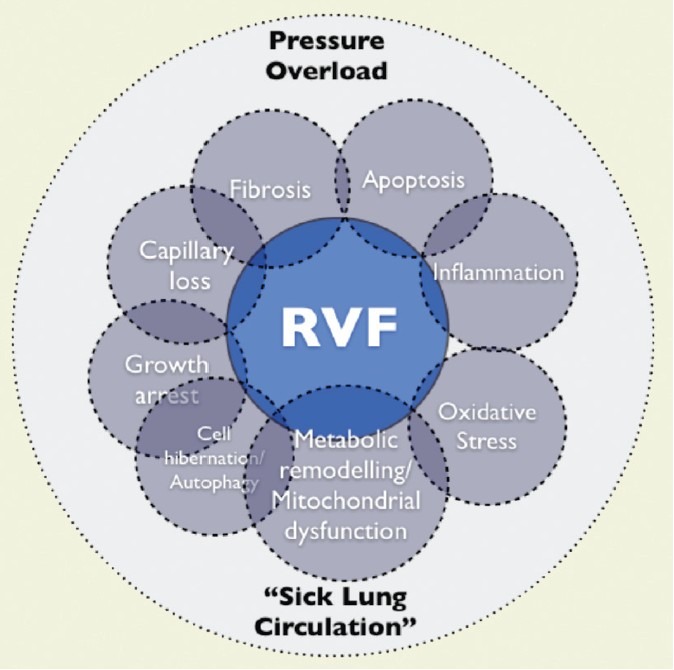
The right ventricle between “a rock and a hard place”, challenged by the pressure overload and the sick lung circulation. The grey circles symbolize cellular and molecular mechanisms. RVF: right ventricular failure.[3]
Altogether, current data highlights unmet needs in the current concepts of the pathobiology of RVF: (1) We lack detailed cellular and molecular mechanistic elements to explain RV failure (“RV failure program”); and (2) we do not fully understand the pathobiology behind the transition from compensated RV hypertrophy to RV failure. Possibly the most compelling argument in support of RV failure investigations is based on the postulate that RV failure is reversible: the end-stage RVF of PAH patients fully recovers after lung transplantation, usually within weeks. Unfortunately, we do not have any mechanistic studies to help explain how the RV recovers during the post-transplant period.
MECHANISMS CONTRIBUTING TO RV FAILURE
The prevailing paradigm holds that increased RV afterload is sufficient as an explanation of RV failure, but there are several problems with this view:
Under most circumstances, the RV adequately adapts to chronic pressure overload (in contrast to acute pressure overload). On average, at least 55% of incident patients with PAH live (with chronic progressive pressure overload) for three years.
Even in the prepulmonary hypertension “drug era” (prior to 1991) there were long-term PAH patient survivors,[14] most prominently patients diagnosed with aminorex- (appetite suppressant) induced PAH.[15]
There are patients with severe PAH that remain highly functional (NYHA Functional Class I) for many years without developing RV failure.[12]
Patients with Eisenmenger physiology live prepulm.hypert.drugera number of years with strikingly elevated pulmonary arterial pressure but do not develop failure until later in life.[16]
Experimental studies show that after pulmonary artery banding (PAB), the RV develops adaptive hypertrophy which is more resistant to pressure overload and does not necessarily develop failure.[17]
In the aggregate, there seems to be solid evidence to suggest that chronic, progressive pressure overload might be the initial component responsible for triggering maladaptive changes in the RV; however, it is not necessarily the only one. Other factors such as ischemia, inflammation, oxidative damage, epigenetics, and abnormal cardiac energetics could equally contribute to the development of RVF (Figure 1).
Right ventricular ischemia
Patients with severe PAH can develop chest pain which may be due to RV ischemia. Wolferen et al.[18] have shown changes in the right coronary artery blood flow pattern in patients with PAH, while Gómez, Sandoval, and collaborators employed stress myocardial scintigraphy using technetium sestamibi to demonstrate RV ischemia associated with an RV end-diastolic pressure elevation.[6] Unfortunately, human histological studies of the RV from patients with chronic severe PAH are unavailable, and it has never been demonstrated that the ratio of RV muscle-to-capillary density, in patients with severe PAH, is altered as it has been reported in LV failure or in experimental RVF. Hein et al. studied LV tissue biopsies obtained at the time of heart surgery for aortic stenosis valve replacement, and described capillary rarefaction and discuss their finding of autophagy. In order to explain RV ischemia in the setting of PAH, investigators have pointed toward increased RV-wall stress[19,20] and myocardial fibrosis, but it is presently unclear whether impairment, RV wall stress, RV fibrosis-or all these factors in combination—contribute to RV myocardial ischemia.
In the SU5416/ chronic hypoxia (Su/Hx) model of severe PAH and RV failure,[10,17] we have demonstrated a dramatic reduction of the number of perfused myocardial capillaries using in vivo tomato-lectin endothelial cell (EC) labeling (Fig. 2B). Histologically, we have also showed diffuse myocardial fibrosis.[17] . Capillary rarefaction has also been reported in the monocrotaline (MCT) model of PAH.[21] Surprisingly, the capillary density in the RV from rats with pulmonary artery banding (mechanically induced RV hypertrophy) is only mildly decreased.[17] We asked whether the Su/Hx model of PAH presents with additional functional alterations of the remaining RV capillaries. Thus, we used electron microscopy to show damaged EC in cardiac capillaries (Fig. 2C). By immunohistochemistry, we found regional loss of proteins associated with normal EC function (unpublished data).
Figure 2.
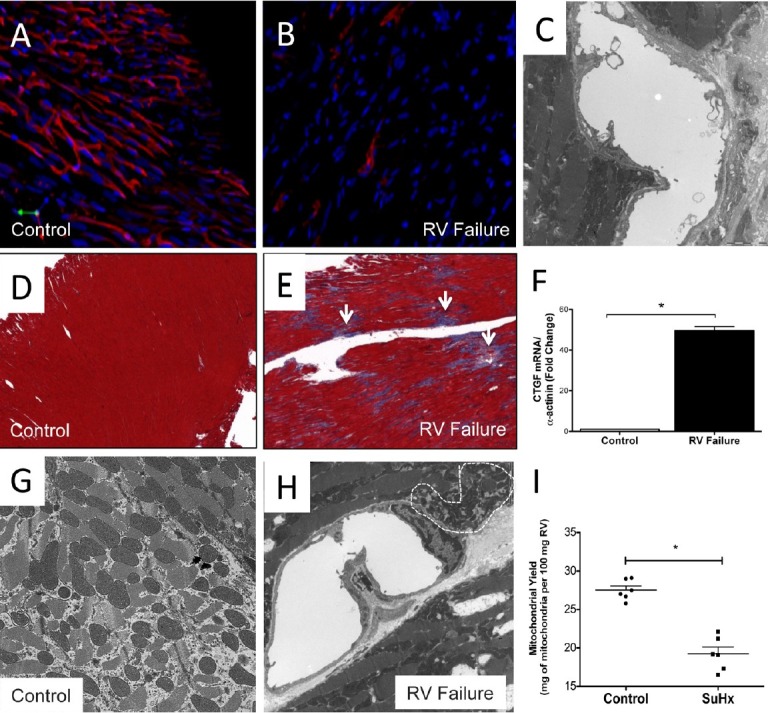
“Right ventricle; changes of the microcirculation (A-C). Confocal microscopy of in vivo labeled right heart muscle capillaries; immunohistochemistry, tomato lectin stain = red (A,B). One single capillary lumen, electron microscopy, in vivo fixation. The image shows destruction of the endothelial cell with bleb formation (C). Trichrome stain (D,E), expression of connective tissue growth factor (CTGF) gene in the failing (Su/Hx) right ventricle (F). Electron microscopy, normal right ventricle mitochondrial morphology (G), small, dense mitochondria in the Su/Hx failing right venticle (H). Reduction of mitochondria (mitochondrial yield) in the Su/Hx failing right ventricle.
Based on our results, we propose three mechanisms that could account for the capillary rarefaction observed in experimental RVF: 1) EC apoptosis, and/or 2) endothelial cell mesenchymal transformation, as proposed by Zeissberg et al.,[22] and 3) insufficient angiogenesis in a context of rapid myocardial hypertrophy. EC death could be partially explained by a decreased expression of the angiogenic protein vascular endothelial growth factor (VEGF), as it has been observed in the failing RV from Su/Hx animals.[17] This hypothesis is largely based on the data generated by Eli Keshet's group, which have shown that genetically engineered, reversible reduction of VEGF expression in the mouse myocardium results in capillary rarefaction, suggesting that VEGF signaling is necessary for the maintenance of myocardial capillaries.[23] Handoko et al. have also reported decreased VEGF and RV-capillary rarefaction in rats treated with MCT.[21,24] Taken together, it appears that the work from several laboratories supports the notion that there are microcirculatory problems in the failing RV. However, whether impaired microcirculation function is sufficient to explain the rest of maladaptive changes observed in the failing RV remains to be investigated.
Inflammation
Inflammatory cells have been localized in the RV from rats with monocrotaline-induced PAH;[25] however, data relating to human RV failure are lacking and no systematic study has been undertaken to examine the role of inflammatory cells or inflammatory mediators in RV failure. A recent experimental study of LV failure in mice generated a novel concept of a toll-like receptor 9 (TLR-9)-dependent failure component. In this model of chronic severe LV pressure overload, mitochondrial DNA from damaged heart cells generated a cardiac inflammatory response via TLR 9.[26]
Oxidative stress
Oxidative cell damage and oxidant-dependent transcriptional control occur and participate in inflammatory organ and tissue responses. Little is known about the role of oxidant stress in RV failure with the exception of the important study of Yet et al.[27] These authors exposed hemeoxygenase 1 KO mice to five weeks of chronic hypoxia and found that the RV, but not the LV, was damaged and dilated. Thus, it appears that the HO-1 function is required for a successful adaptation of the RV to pressure overload. The expression of HO-1 in the RV tissue from the Su/Hx pulmonary hypertensive rats is reduced, perhaps reflecting an impaired response to oxidant stress.[17]
MicroRNA
Several recent publications have demonstrated microRNA dependent regulation of gene expression in association with pulmonary artery cell growth[28–30] and plasma microRNA expression patterns after acute myocardial infarcts or in patients with heart failure[31,32] (Table 1). In mouse models, miRNAs play a role in the development of heart failure and the literature on cardiac microRNAs is still expanding.[33–35] The transition of the RV from hypertrophy to failure has been investigated in the rat Su/Hx model and we reported a decrease in miR 21,34c, 133a, and 139-3p in Su/Hx-induced RV failure when compared to PAB-induced RV hypertrophy.[36] More recently, Reddy et al. reported RV-specific microRNA expression changes in a murine model.[31] Unfortunately, the mechanisms by which miRNAs could affect gene and protein expression during RVF are incompletely understood.
Table 1.
microRNA in PAH

Mitochondrial energy metabolism
Abnormal cardiac metabolism has long been recognized as a problem in chronic left heart failure.[37,38] Similarly, data derived from experimental models of PH suggest that the failing RV is also characterized by a certain degree of “metabolic remodeling.” In the SU/Hx rat model, the RV exhibits increased expression of glycolysis-related genes,[36] whereas dysfunctional RV hypertrophy in the monocrotaline rat model of PH is associated with increased glycolysis enzymatic rates.[39] Unfortunately, a complete characterization of RV “metabolic remodeling” in human PAH is still lacking. Positron emission tomography studies have shown increased accumulation of the glucose analog 18F-2-Deoxy-2-Fluoro-D-Glucose (18-FDG) in the RV of PAH patients.[40,41] However, 18-FDG uptake studies have multiple limitations and the physiological interpretation is not straightforward (i.e., 18-FDG uptake studies do not directly measure glycolysis). RV oxygen consumption can also be measured by [11C]-acetate PET scanning.[42] However, such studies are limited to a few specialized centers and are difficult to perform.[8] Based on experimental models, there is now evidence to suggest that the machinery required for fatty acid metabolism in the failing RV is compromised on multiple levels (fatty acid transport, oxidation and upstream transcriptional regulation).[43] Furthermore, there is evidence to suggest important alterations of mitochondrial structure and function. The expression of multiple critical transcription factors involved in the regulation of mitochondrial biogenesis, including the mitochondrial transcription factor A (TFAm) and the peroxisome proliferator-activated receptor (PPAR)-γ coactivator-1α (PGC-1α), is significantly downregulated in the failing RV.[44] In accordance, RVF seems to exhibit a net loss in the mitochondrial number per gram of RV tissue[10] (Fig. 2I). The remaining mitochondria demonstrate abnormal ultrastructure on electron microscopy (Fig. 2H), as well as decreased oxidative capacity.[10] Akin to chronic left heart failure, the data support that dysfunctional RV hypertrophy, in the setting of severe PAH, is characterized by a metabolic switch from aerobic to anaerobic metabolism. Whether this switch is an adaptive or maladaptive mechanism remains elusive. However, multiple studies have shown that the rate of fatty acid oxidation is preserved or increased in physiological/adaptive left ventricular hypertrophy, and that it decreases during the progression of heart failure.[45] Similar results have been reported for the nondysfunctional, mechanically-induced RV hypertrophy from rats with pulmonary artery banding, which exhibit high rates of fatty acid oxidation.[46] In addition, pure mechanical pressure overload is insufficient to change the expression of PGC-1α, PPAR-α or the expression of the estrogen-related receptor-α[43] which, along with PGC-1α, are the main transcription factors that coordinate fatty acid oxidation in the heart.[47] Thus, it appears that decreased fatty acid oxidation may contribute to the development of RV failure, along with fibrosis, capillary dysfunction and, oxidative stress (Fig. 1). It is clear that pressure overload alone is not sufficient to explain RVF,[17] or the metabolic switch.[43] Whether capillary dysfunction (or the consequent tissue hypoxia) is sufficient to explain the metabolic remodeling remains to be investigated. Whereas the role of metabolic modulators for the treatment of RVF in PAH is of potential interest, their efficiency could be limited by underlying mitochondrial abnormalities. If the mitochondrial machinery is not working adequately, it would not matter which substrate is being oxidized. Finally, we have begun to investigate the mechanisms whereby carvedilol improves RV function in Su/Hx RVF model. Some of the changes in the RV gene expression pattern as they relate to mitochondrial function are listed in Table 2. Interestingly, treatment of normal rats with carvedilol is sufficient to induce gene expression changes in the heart, and carvedilol has been shown to be cardio-protective in cultured cardiomyocytes.
Table 2.
Gene expression changes during right ventricular failure and after treatment with carvedilol
SUMMARY AND CONCLUSION
Right heart failure is characterized by morphological and functional changes. We wish to advance the concept that the RV afterload by itself is insufficient as an explanation of the mechanism of RV failure and that RVF prevention and preservation of RV function are important treatment goals for patients with severe PAH. We also take the position that RVF is potentially reversible. This has been shown experimentally in the Su/Hx rat model of severe RVF, where carvedilol treatment has reversed RVF without reducing the RV afterload.[10] This “proof of principle” investigation has stimulated the search for mechanisms and molecular targets of RVF reversibility (Table 2). Mitochondrial energy metabolism is compromised during RVF and β-adrenergic receptor blockade[43] and perhaps diet or excersie may improve cardiac mitochondrial function.[48,49] Both the positive and negative impact of neuroendocrine factors[3,50] on a right ventricle “under pressure” deserve detailed studies.[51]
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Bristow MR, Zisman LS, Lowes BD, Abraham WT, Badesch DB, Groves BM, et al. The pressure-overloaded right ventricle in pulmonary hypertension. Chest, 1998;114(1 Suppl):101S–6S. doi: 10.1378/chest.114.1_supplement.101s. [DOI] [PubMed] [Google Scholar]
- 2.Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, et al. Right Ventricular Function and Failure: Report of a National Heart, Lung, and Blood Institute Working Group on Cellular and Molecular Mechanisms of Right Heart Failure. Circulation. 2006;114:1883–91. doi: 10.1161/CIRCULATIONAHA.106.632208. [DOI] [PubMed] [Google Scholar]
- 3.Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest. 2009;135:794–804. doi: 10.1378/chest.08-0492. [DOI] [PubMed] [Google Scholar]
- 4.Haddad F, Doyle R, Murphy DJ, Hunt SA. Right ventricular function in cardiovascular disease, part II: pathophysiology, clinical importance, and management of right ventricular failure. Circulation. 2008;117:1717–31. doi: 10.1161/CIRCULATIONAHA.107.653584. [DOI] [PubMed] [Google Scholar]
- 5.Haddad F, Hunt SA, Rosenthal DN, Murphy DJ. Right ventricular function in cardiovascular disease, part I: Anatomy, physiology, aging, and functional assessment of the right ventricle. Circulation. 2008;117:1436–48. doi: 10.1161/CIRCULATIONAHA.107.653576. [DOI] [PubMed] [Google Scholar]
- 6.Gómez A, Bialostozky D, Zajarias A, Santos E, Palomar A, Martínez ML, et al. Right ventricular ischemia in patients with primary pulmonary hypertension. J Am Coll Cardiol. 2001;38:1137–42. doi: 10.1016/s0735-1097(01)01496-6. [DOI] [PubMed] [Google Scholar]
- 7.Meyer P, Desai RV, Mujib M, Feller MA, Adamopoulos C, Banach M, et al. Right ventricular ejection fraction <20% is an independent predictor of mortality but not of hospitalization in older systolic heart failure patients. Int J Cardiol. 2012;155:120–5. doi: 10.1016/j.ijcard.2011.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vonk Noordegraaf A, Galie N. The role of the right ventricle in pulmonary arterial hypertension. Eur Respir Rev. 2011;20:243–53. doi: 10.1183/09059180.00006511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voelkel NF, et al. Right Ventricle in Pulmonary Hypertension. Compr Physiol. 2011;1:595–610. doi: 10.1002/cphy.c090008. [DOI] [PubMed] [Google Scholar]
- 10.Bogaard HJ, Natarajan R, Mizuno S, Abbate A, Chang PJ, Chau VQ, et al. Adrenergic Receptor Blockade Reverses Right Heart Remodeling and Dysfunction in Pulmonary Hypertensive Rats. Am J Respir Crit Care Med. 2010;182:652–60. doi: 10.1164/rccm.201003-0335OC. [DOI] [PubMed] [Google Scholar]
- 11.Voelkel NF. The Wide Spectrum of Pulmonary Vascular Diseases: “The Sick Lung Circulation”. In: Voelkel NF, editor. Pulmonary Hypertension The Presence and Future. USA: People's Medical Publishing House; 2011. [Google Scholar]
- 12.Rich S, Pogoriler J, Husain AN, Toth PT, Gomberg-Maitland M, Archer SL. Long-term effects of epoprostenol on the pulmonary vasculature in idiopathic pulmonary arterial hypertension. Chest. 2010;138:1234–9. doi: 10.1378/chest.09-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van de Veerdonk MC, Kind T, Marcus JT, Mauritz GJ, Heymans MW, Bogaard HJ, et al. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol. 2011;58:2511–9. doi: 10.1016/j.jacc.2011.06.068. [DOI] [PubMed] [Google Scholar]
- 14.Voelkel NF, Reeves JT. Primary Pulmonary Hypertension. In: Moser KM, editor. Pulmonary Vascular Diseases. New York: Marcel Dekker; 1979. pp. 573–6. [Google Scholar]
- 15.Lang I, Kneussl M, Frank H, Mlczoch J. [Long-term follow-up of pulmonary hypertension of unknown etiology] Pneumologie. 1990;44:913–4. [PubMed] [Google Scholar]
- 16.Hopkins WE, Ochoa LL, Richardson GW, Trulock EP. Comparison of the hemodynamics and survival of adults with severe primary pulmonary hypertension or Eisenmenger syndrome. J Heart Lung Transplant. 1996;15:100–5. [PubMed] [Google Scholar]
- 17.Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, et al. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation. 2009;120:1951–60. doi: 10.1161/CIRCULATIONAHA.109.883843. [DOI] [PubMed] [Google Scholar]
- 18.van Wolferen SA, Marcus JT, Westerhof N, Spreeuwenberg MD, Marques KM, Bronzwaer JG, et al. Right coronary artery flow impairment in patients with pulmonary hypertension. Eur Heart J. 2008;29:120–7. doi: 10.1093/eurheartj/ehm567. [DOI] [PubMed] [Google Scholar]
- 19.Hein S, Arnon E, Kostin S, Schönburg M, Elsässer A, Polyakova V, et al. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation. 2003;107:984–91. doi: 10.1161/01.cir.0000051865.66123.b7. [DOI] [PubMed] [Google Scholar]
- 20.Elsässer A, Decker E, Kostin S, Hein S, Skwara W, Müller KD, et al. A self-perpetuating vicious cycle of tissue damage in human hibernating myocardium. Mol Cell Biochem. 2000;213:17–28. doi: 10.1023/a:1007182617215. [DOI] [PubMed] [Google Scholar]
- 21.Handoko ML, de Man FS, Happé CM, Schalij I, Musters RJ, Westerhof N, et al. Opposite effects of training in rats with stable and progressive pulmonary hypertension. Circulation. 2009;120:42–9. doi: 10.1161/CIRCULATIONAHA.108.829713. [DOI] [PubMed] [Google Scholar]
- 22.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–61. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 23.May D, Gilon D, Djonov V, Itin A, Lazarus A, Gordon O, et al. Transgenic system for conditional induction and rescue of chronic myocardial hibernation provides insights into genomic programs of hibernation. Proc Natl Acad Sci U S A. 2008;105:282–7. doi: 10.1073/pnas.0707778105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Partovian C, Adnot S, Eddahibi S, Teiger E, Levame M, Dreyfus P, et al. Heart and lung VEGF mRNA expression in rats with monocrotaline- or hypoxia-induced pulmonary hypertension. Am J Physiol. 1998;275:H1948–56. doi: 10.1152/ajpheart.1998.275.6.H1948. [DOI] [PubMed] [Google Scholar]
- 25.Watts JA, Zagorski J, Gellar MA, Stevinson BG, Kline JA. Cardiac inflammation contributes to right ventricular dysfunction following experimental pulmonary embolism in rats. J Mol Cell Cardiol. 2006;41:296–307. doi: 10.1016/j.yjmcc.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 26.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–5. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yet SF, Perrella MA, Layne MD, Hsieh CM, Maemura K, Kobzik L, et al. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J Clin Invest. 1999;103:R23–9. doi: 10.1172/JCI6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drake KM, Zygmunt D, Mavrakis L, Harbor P, Wang L, Comhair SA, et al. Altered microRNA Processing in Heritable Pulmonary Arterial Hypertension: an Important Role for Smad-8. Am J Respir Crit Care Med. 2011;184:1400–8. doi: 10.1164/rccm.201106-1130OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pullamsetti SS, Doebele C, Fischer A, Savai R, Kojonazarov B, Dahal BK, et al. Inhibition of microRNA-17 improves lung and heart function in experimental pulmonary hypertension. Am J Respir Crit Care Med. 2012;185:409–19. doi: 10.1164/rccm.201106-1093OC. [DOI] [PubMed] [Google Scholar]
- 30.Courboulin A, Paulin R, Giguère NJ, Saksouk N, Perreault T, Meloche J, et al. Role for miR-204 in human pulmonary arterial hypertension. J Exp Med. 2011;208:535–48. doi: 10.1084/jem.20101812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reddy S, Zhao M, Hu DQ, Fajardo G, Hu S, Ghosh Z, et al. Dynamic microRNA Expression During the Transition from Right Ventricular Hypertrophy to Failure. Physiol Genomics. 2012;44:562–75. doi: 10.1152/physiolgenomics.00163.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu J, Zhao J, Evan G, Xiao C, Cheng Y, Xiao J. Circulating microRNAs: Novel biomarkers for cardiovascular diseases. J Mol Med (Berl) 2011;90:865–75. doi: 10.1007/s00109-011-0840-5. [DOI] [PubMed] [Google Scholar]
- 33.Gladka MM, da Costa Martins PA, De Windt LJ. Small changes can make a big difference - microRNA regulation of cardiac hypertrophy. J Mol Cell Cardiol. 2012;52:74–82. doi: 10.1016/j.yjmcc.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 34.Wang K, Lin ZQ, Long B, Li JH, Zhou J, Li PF. Cardiac Hypertrophy Is Positively Regulated by MicroRNA miR-23a. J Biol Chem. 2012;287:589–99. doi: 10.1074/jbc.M111.266940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montgomery RL, Hullinger TG, Semus HM, Dickinson BA, Seto AG, Lynch JM, et al. Therapeutic Inhibition of miR-208a Improves Cardiac Function and Survival During Heart Failure. Circulation. 2011;124:1537–47. doi: 10.1161/CIRCULATIONAHA.111.030932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drake JI, Bogaard HJ, Mizuno S, Clifton B, Xie B, Gao Y, et al. Molecular signature of a right heart failure program in chronic severe pulmonary hypertension. Am J Respir Cell Mol Biol. 2011;45:1239–47. doi: 10.1165/rcmb.2010-0412OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007;356:1140–51. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 38.Taegtmeyer H. Cardiac metabolism as a target for the treatment of heart failure. Circulation. 2004;110:894–6. doi: 10.1161/01.CIR.0000139340.88769.D5. [DOI] [PubMed] [Google Scholar]
- 39.Piao L, Fang YH, Cadete VJ, Wietholt C, Urboniene D, Toth PT, et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: Resuscitating the hibernating right ventricle. J Mol Med (Berl) 2010;88:47–60. doi: 10.1007/s00109-009-0524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Can MM, Kaymaz C, Tanboga IH, Tokgoz HC, Canpolat N, Turkyilmaz E, et al. Increased right ventricular glucose metabolism in patients with pulmonary arterial hypertension. Clin Nucl Med. 2011;36:743–8. doi: 10.1097/RLU.0b013e3182177389. [DOI] [PubMed] [Google Scholar]
- 41.Oikawa M, Kagaya Y, Otani H, Sakuma M, Demachi J, Suzuki J, et al. Increased [18F]fluorodeoxyglucose accumulation in right ventricular free wall in patients with pulmonary hypertension and the effect of epoprostenol. J Am Coll Cardiol. 2005;45:1849–55. doi: 10.1016/j.jacc.2005.02.065. [DOI] [PubMed] [Google Scholar]
- 42.Wong YY, Ruiter G, Lubberink M, Raijmakers PG, Knaapen P, Marcus JT, et al. Right ventricular failure in idiopathic pulmonary arterial hypertension is associated with inefficient myocardial oxygen utilization. Circ Heart Fail. 2011;4:700–6. doi: 10.1161/CIRCHEARTFAILURE.111.962381. [DOI] [PubMed] [Google Scholar]
- 43.Gomez-Arroyo JG, et al. Adrenergic Receptor Blocker Improves Metabolic Remodeling in Experimental Right Ventricular Failure Due to Pulmonary Hypertension. Am J Respir Crit Care Med. 2011;183:A2520. [Google Scholar]
- 44.Gomez-Arroyo J. Metabolic Remodeling in Right Heart Failure is Associated with Abnormal Mitochondrial Biogenesis. Am J Respir Crit Care Med. 2012;185:A3455. [Google Scholar]
- 45.Abel ED, T Doenst. Mitochondrial adaptations to physiological vs.pathological cardiac hypertrophy. Cardiovasc Res. 2011;90:234–42. doi: 10.1093/cvr/cvr015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fang YH, Piao L, Hong Z, Toth PT, Marsboom G, Bache-Wiig P, et al. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: exploiting Randle's cycle. J Mol Med (Berl) 2012;90:31–43. doi: 10.1007/s00109-011-0804-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Finck BN, Kelly DP. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–22. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Benit P, Rustin P. Changing the diet to make more mitochondria and protect the heart. Circ Res. 2012;110:1047–8. doi: 10.1161/RES.0b013e318255408a. [DOI] [PubMed] [Google Scholar]
- 49.Krebs P, Fan W, Chen YH, Tobita K, Downes MR, Wood MR, et al. Lethal mitochondrial cardiomyopathy in a hypomorphic Med30 mouse mutant is ameliorated by ketogenic diet. Proc Natl Acad Sci U S A. 2011;108:19678–82. doi: 10.1073/pnas.1117835108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Donaldson C, Eder S, Baker C, Aronovitz MJ, Weiss AD, Hall-Porter M, et al. Estrogen attenuates left ventricular and cardiomyocyte hypertrophy by an estrogen receptor-dependent pathway that increases calcineurin degradation. Circ Res. 2009;104:265–75. doi: 10.1161/CIRCRESAHA.108.190397. 11p following 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghosh G, Subramanian IV, Adhikari N, Zhang X, Joshi HP, Basi D, et al. Hypoxia-induced microRNA-424 expression in human endothelial cells regulates HIF-alpha isoforms and promotes angiogenesis. J Clin Invest. 2010;120:4141–54. doi: 10.1172/JCI42980. [DOI] [PMC free article] [PubMed] [Google Scholar]