

Abstract
Pulmonary arterial hypertension (PAH) is a syndrome in which pulmonary vascular cross sectional area and compliance are reduced by vasoconstriction, vascular remodeling, and inflammation. Vascular remodeling results in part from increased proliferation and impaired apoptosis of vascular cells. The resulting increase in afterload promotes right ventricular hypertrophy (RVH) and RV failure. Recently identified mitochondrial-metabolic abnormalities in PAH, notably pyruvate dehydrogenase kinase-mediated inhibition of pyruvate dehydrogenase (PDH), result in aerobic glycolysis in both the lung vasculature and RV. This glycolytic shift has diagnostic importance since it is detectable early in experimental PAH by increased lung and RV uptake of 18F-fluorodeoxyglucose on positron emission tomography. The metabolic shift also has pathophysiologic and therapeutic relevance. In RV myocytes, the glycolytic switch reduces contractility while in the vasculature it renders cells hyperproliferative and apoptosis-resistant. Reactivation of PDH can be achieved directly by PDK inhibition (using dichloroacetate), or indirectly via activating the Randle cycle, using inhibitors of fatty acid oxidation (FAO), trimetazidine and ranolazine. In experimental PAH and RVH, PDK inhibition increases glucose oxidation, enhances RV function, regresses pulmonary vascular disease by reducing proliferation and enhancing apoptosis, and restores cardiac repolarization. FAO inhibition increases RV glucose oxidation and RV function in experimental RVH. The trigger for metabolic remodeling in the RV and lung differ. In the RV, metabolic remodeling is likely triggered by ischemia (due to microvascular rarefaction and/or reduced coronary perfusion pressure). In the vasculature, metabolic changes result from redox-mediated activation of transcription factors, including hypoxia-inducible factor 1α, as a consequence of epigenetic silencing of SOD2 and/or changes in mitochondrial fission/fusion. Randomized controlled trials are required to assess whether the benefits of enhancing glucose oxidation are realized in patients with PAH.
Keywords: aerobic glycolysis, fatty acid oxidation, pyruvate dehydrogenase kinase, right ventricular ischemia, the Randle cycle
The fetal right ventricle (RV) is a thick-walled chamber that ejects blood at relatively high pressure into a high resistance, fetal pulmonary vascular bed with thick walled pulmonary arteries. After birth, structural remodeling, marked by reduced muscularity of small pulmonary arteries, combines with active vasodilatation to lower pulmonary vascular resistance (PVR). This reduction in afterload, and associated changes in the fetal gene package, alters RV metabolism and reduces RV hypertrophy (RVH). Chronic pressure overload, as occurs in diseases such as pulmonary hypertension (PH),[1] stimulates RVH. In left ventricular hypertrophy (LVH), reactivation of the fetal gene package, mediated by activation of proto-oncogenes (e.g., c-Myc), reverts expression of key enzymes to their fetal forms. Thus in LVH one sees re-emergence of β myosin heavy chain isoform and skeletal muscle α-actin in the heart.[2] Whether similar changes occur is RVH is unclear.
The RVH associated with group 1 PH (pulmonary arterial hypertension, PAH) theoretically is a compensatory mechanism to maintain cardiac output (CO) and attenuate increases in wall stress. However, RVH is rarely fully compensatory, and sustained RV pressure overload often culminates in RV ischemia and RV failure (RVF). RV ischemia may reflect rarefaction of the RV microcirculation, hypoperfusion of the right coronary artery (RCA) due to a reduced perfusion pressure and/or a supply/demand mismatch. The evidence for RV ischemia in RVH includes animal studies demonstrating reduced RCA coronary perfusion pressure,[3,4] human studies showing small leaks of troponin,[5,6] reduced RV perfusion flow reserve,[7] and increased RV uptake of 18F-fluorodeoxyglucose on positron emission tomography (FDG-PET) in PAH patients,[8] and rodents with PAH (Fig. 1).
Figure 1.
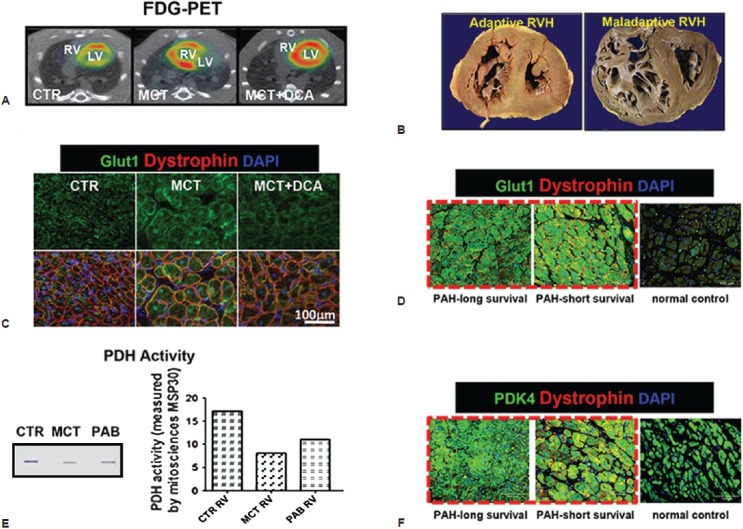
Increased glycolysis in the RV in experimental RVH in PAH patients. (A) Increased RV FDG-PET in MCT models is reduced by dichloroacetate (DCA). (B) Increased Glut1 expression in RV myocyte membranes in a monocrotaline (MCT) model is reduced by DCA. (C) RV PDH activity is reduced in MCT and PAB, especially in MCT model. (D) The cross sections of RVs from patients with adaptive versus maladaptive RVH. RV chambers are enlarged in both patients however adaptive RVH is concentric with less dilatation and fibrosis. (E and F) Immunostaining shows up-regulation of Glut1 and PDK4 expression in RV myocytes is less profound in the PAH patient with adaptive RVH. The figure is partially adapted from references 13 and 21, with permission.
ADAPTIVE AND MALADAPTIVE RIGHT VENTRICULAR HYPERTROPHY
Although it is the pulmonary vascular disease in PAH that initiates the RVH, ultimately it is the decline in RV function that determines prognosis in PAH. Impaired RV ejection fraction (RVEF) predicts clinical worsening in PAH,[9,10] even more accurately than does elevated pulmonary vascular resistance (PVR).[9] Reduction in RVEF and/or late gadolinium enhancement (LGE) at the RV-LV septal hinge points on magnetic resonance imaging (MRI) predicts clinical worsening in PAH.[10] Development of RVF is an ominous sign in PAH,[11] and PAH patients admitted to an intensive care unit who receive catecholamines to treat RVF have a 46% in-hospital mortality rate.[12] Hopefully a better understanding of metabolic derangements in the pulmonary vasculature and RV in PAH will offer new therapeutic targets to enhance RV function.
There is individual variation in the susceptibility to RVF with some PAH patients rapidly decompensating while others remain stable, despite similar RVH and PA pressures.[13] There is also differential predilection to RVF between subtypes of WHO Group I PH. RV dysfunction is more prevalent in those with scleroderma,[14,15] than those with idiopathic PAH or Eisenmenger's syndrome.[16] These differences in the RV response to pressure overload can be divided into adaptive and maladaptive phenotypes. The adaptive attributes include a concentric pattern of RVH with minimal dilatation or fibrosis and relatively preserved RVEF; conversely, the maladaptive phenotype includes RV dilatation, hypokinesis and fibrosis. Recently we reported the case of a woman with Group 1 PH for over 20 years who retained near normal RV function and remained Functional Class II, before rapidly succumbing to colon cancer (adaptive RVH in Fig. 1D). Her lung histology revealed severe plexogenic arteriopathy suggesting that her excellent functional class related to her concentrically hypertrophied, nondilated RV. On molecular interrogation, her RV was less glycolytic with lower expression of both pyruvate dehydrogenase kinase, PDK4, and the fetal isoform of the glucose transporter 1 (Glut1) than the RV of a demographically similar patient who had rapidly deteriorated and died of RV failure (Fig. 1E).[17] Rat PAH models can also be categorized as adaptive or maladaptive. RVH in rats post pulmonary artery banding (PAB) is better tolerated (longer survival, better exercise tolerance) than RVH induced by injection of endothelial toxins, including monocrotaline (MCT) and the vascular endothelial growth factor receptor (VEGF-R) antagonist, Sugen 5416, plus chronic hypoxia (CH-SU).[18] That greater prevalence of RVF in the MCT and CH-SU models suggests a possible role for endothelial damage/dysfunction in the RV microcirculation in the pathogenesis of RVF.
Emerging evidence suggests that the RV's metabolic response is one of the factors that determines whether the RVH is adaptive or maladaptive. Two abnormalities that appear common to maladaptive RVH are ischemia,[19] and aerobic glycolysis.[8,13,20] Glycolysis is a common response to ischemia and when sufficiently severe it impairs energetics, reducing RV function and creating a vicious cycle of ischemia, compromised energetics, reduced cardiac output, and further ischemia. It has been proposed that the hypokinetic RV in PAH is viable, hibernating myocardium and that the reduced RVEF reflects ischemia-initiated, metabolically mediated RV dysfunction.[20] Favoring the argument that the RV in PAH is hibernating, severe RV dysfunction reverses after successful single-lung transplantation. In one series there was an increase in RVEF from pretransplant values of 28 ± 8% to 60 ± 11%, within three months of lung transplantation for PH.[21]
ISCHEMIA OF THE RIGHT VENTRICLE
RV ischemia in RVH has been thought to reflect decreased systolic perfusion of the right coronary artery (RCA) caused by the reduced perfusion pressure that occurs when RV systolic pressure (RVSP) approaches aortic pressure.[22,23] This form of ischemia has been observed and associated with RV dysfunction in acute, severe PAB models,[3] and in humans with PAH.[22] However, impaired RV function only occurs if RCA perfusion pressure falls below 50 mmHg.[24] An alternative or additional cause of ischemia in RVF is capillary rarefaction.[4] In comparing PAB versus MCT RVH models in which RVSP and RVH were similar, PAB rats have less reduction in RV free wall thickening and CO (Fig. 2). This greater RV dysfunction in MCT PAH cannot be explained by more severe reduction in coronary perfusion pressure. In previously unpublished data presented at the Aspen conference we show a more severe decrease in RV microvascular density in the MCT versus PAB RVH (Fig. 3), consistent with the work of Bogaard et al.[4] Likewise, mortality is higher and RV contractility lower in scleroderma-associated PAH vs. idiopathic PAH,[25] despite somewhat milder PH in the scleroderma cohort.[14] If RVF was initiated primarily by a drop in coronary perfusion pressure then current strategies, like infusion of phenylephrine to increase the aortic-RV pressure gradient, as suggested by Vlahakes et al., might be rational.[3] However, if there is significant microvascular disease in the RV (i.e., capillary rarefaction, as suggested by Bogaard et al.[4] ), it is more logical to treat RVF by promoting angiogenesis or pharmacologically changing metabolism to “do more with less.”
Figure 2.
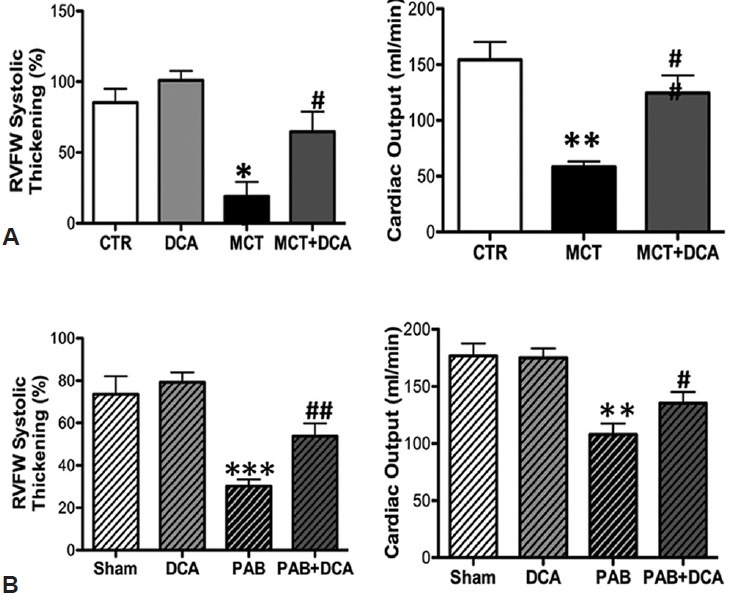
PDK inhibition by DCA improves RV function in experimental RVH. (A and B) Dichloroacetate increases RV free wall (RVFW) systolic thickening, measured by echocardiography, and CO (measured using thermodilution technique) in both MCT and PAB. The figure is adapted from reference 21, with permission.
Figure 3.
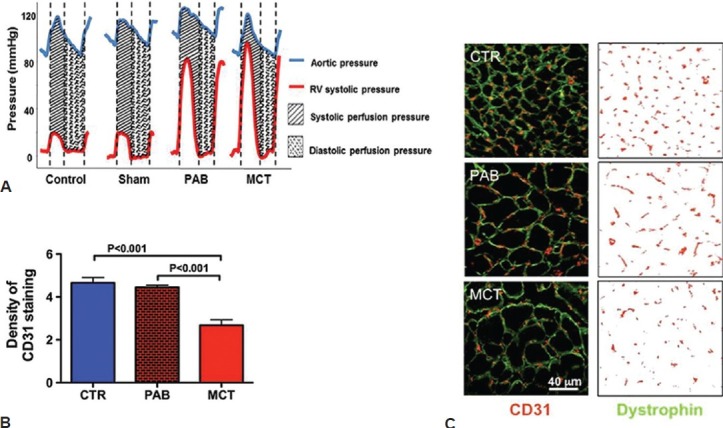
Possible mechanisms of RV Ischemia in rats. (A) Catheterization data shows simultaneous RV pressure and aortic pressure (AoP) in various rat RVH models. Despite similar coronary perfusion pressure (defined as the Δ Pressure Ao-RV) there is worse RV function and exercise capacity in MCT versus PAB (not shown). This argues against epicardial coronary perfusion pressure being the major cause of ischemia or impaired RV function in these models. (B and C) Evidence for capillary rarefaction in RVH: Representative images and mean data show a reduced density of RV capillaries (stained with CD31) in maladaptive RVH induced by MCT.
METABOLISM IN THE RIGHT VENTRICLE IN PULMONARY ARTERIAL HYPERTENSION
In contrast to the normal RV, which can vary its substrate utilization from fatty acids to glucose as needed, RVH is associated with a persistent reliance on glucose metabolism.[26] In experimental RVH, there is increased RV glycolysis, evidenced by increased uptake of FDG-PET (Fig. 1A) and by direct measurement of metabolism in an RV working heart model.[8,20] Glycolysis generates pyruvate and if this pyruvate cannot be used by mitochondrial pyruvate dehydrogenase (PDH) to fuel oxidative phosphorylation, only two ATP molecules/glucose are obtained (vs. 32 ATP during glucose oxidation (GO)) and lactate is produced. One cause of impaired GO in RVH is increased PDK activity/expression. There are 4 PDK isoforms and they inhibit pyruvate dehydrogenase (PDH) by phosphorylating its E1-α subunit. This inhibits formation of acetyl CoA and slows Krebs’ cycle,[27] accounting for the reduced O2- consumption/g tissue in RVH and reduced energy production. This PDK-mediated metabolic switch is associated with impaired cardiac mechanical function (impaired RV contractility and decreased cardiac output) and electrical remodeling (prolongation of the RV action potential and QT interval prolongation on the surface EKG).[20] In the RV it appears that PDK4 and to a lesser extent PDK2 are the key regulators of PDH (unpublished data). PDH is inhibited more in maladaptive MCT RVH than in adaptive PAB RVH (Fig. 1C).[13] The PDK inhibitor, dichloroacetate decreases PDH phosphorylation and improves RV contractility (Fig. 2) and restores RV repolarization in rodent models of RVH.[20]
Fatty acid oxidation (FAO) is the major source of ATP production in the adult heart whereas glucose metabolism is considered a secondary source (generating 10-40% of energy production).[28] There is a reciprocal relationship between the two major oxidative metabolic pathways such that inhibiting FAO increases the GO.[29] This phenomenon called the Randle cycle,[30] reflects, in part, the production of citrate during FAO (Fig. 4). Citrate inhibits phosphofructokinase, causing accumulation of glucose-6-phosphate. This inhibits hexokinase and decreases the production of pyruvate. Another important mechanism underlying the Randle cycle is the inhibition of PDH by the acetyl CoA generated from FAO (Fig. 4). Since FAO uses 12% more oxygen than GO to generate a given amount of ATP, we explored the possibility that increasing GO by inhibiting FAO might be beneficial in RVH.[31] Direct metabolic measurements in an RV working heart model, using the dual isotope technique, showed increased FAO and reduced GO/glycolysis ratio in PAB versus sham animals (Fig. 5A and 5C).[32] We also used the seahorse extracellular flux analyzer to study the metabolism of enzymatically dispersed RV myocytes and confirmed that when palmitate was the only substrate in the perfusate, increased FAO was seen in PAB myocytes (Fig. 5D). When using glucose as the only substrate, RV myocyte metabolism was reduced, consistent with impaired GO in PAB RVH. In vivo it appears that the net effect of the increase in FAO and suppression of GO in RVH is a metabolic shift to glycolysis. To explore whether FAO in PAB RVH was beneficial or harmful we used two putative FAO inhibitors, trimetazidine and ranolazine (both of which are used in humans).[32] Trimetazidine inhibits the long-chain isoform of the final enzyme in β-FAO, 3-ketoacyl coenzyme A (CoA) thiolase.[33] This mechanism is important to trimetazidine's ability to increase GO in patients with ischemic heart disease.[33] In patients with idiopathic dilated cardiomyopathy, PET studies show that, using palmitate and acetate tracers, trimetazidine decreases FAO and causes a compensatory increase in GO.[34] Ranolazine is approved for refractory angina in the USA.[35] Ranolazine has been shown to inhibit FAO and activate PDH in perfused, normoxic rat hearts.[36] In ischemic models, ranolazine improves cardiac work by activating PDH and stimulating GO.[37]
Figure 4.
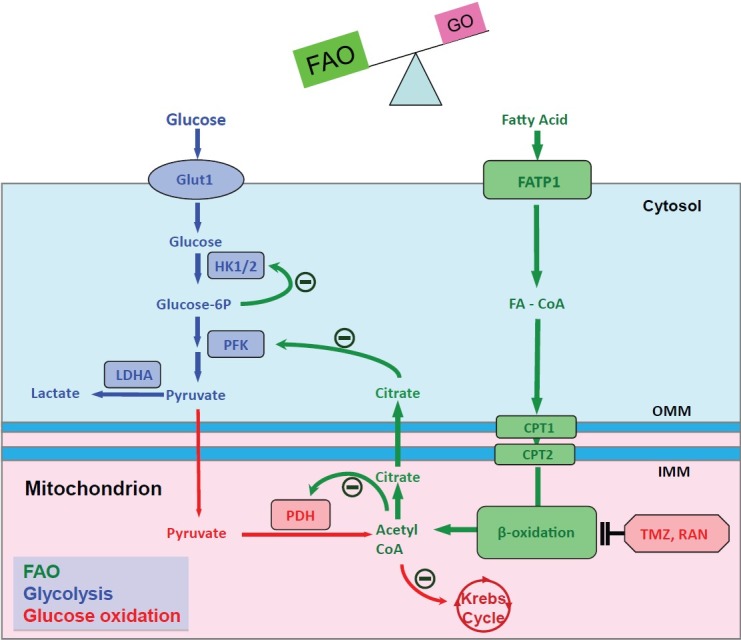
The Randle cycle in RVH. The reciprocal relationship between GO and FAO is referred to as Randle's cycle. The production of citrate and acetyl CoA from β-oxidation inhibits HK1/2, PFK and PDH, thereby inhibiting glucose oxidation. In PAB-induced RVH, increased FAO inhibits GO. Conversely, FAO inhibitors (TMZ and Ran) increase PDH activity and restore GO. FAO: fatty acid oxidation; GO: glucose oxidation; Glut1: glucose transporter 1; HK1/2: hexokinase1/2; PFK: phosphofructokinase; LDHA: lactate dehydrogenase A; PDH: pyruvate dehydrogenase; FATP1: fatty acid transport protein 1; FA-CoA: fatty acyl-CoA; OMM and IMM: outer and inner mitochondrial membrane; CPT1 and CPT2: carnitine palmitoyltransferase I and II; TMZ: trimetazidine; RAN: ranolazine. The diagram is adapted from reference 34, with permission.
Figure 5.
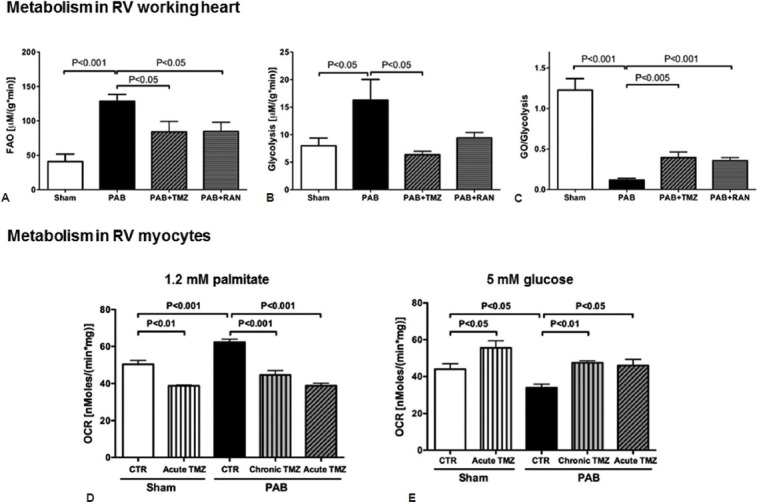
The metabolic changes in RV in PAB-induced RVH. (A-C) Metabolic measurement in the PAB RV working heart model, using the dual isotope technique, show that FAO and glycolysis are increased whereas GO is decreased. The inhibitors of b-oxidation, trimetazidine and ranolazine decrease FAO and increase GO. (D and E) Metabolic measurement in RV myocytes, using the Seahorse Extracellular Flux Analyzer, show that FAO is increased in RV myocytes in PAB model when 1.2 mM palmitate is the only substrate. GO is reduced when 5 mM glucose is the only substrate. Both acute treatment of trimetazidine (two hours incubation) and chronic treatment of trimetazidine (0.7 g/L in drinking water for 8 weeks) reduce FAO and improves GO. The figure is adapted from reference 34, with permission.
In RVH, trimetazidine and ranolazine inhibited FAO and did not prolong the QTc interval. The long-term use of trimetazidine and ranolazine In vivo in rats with PAB increased treadmill exercise capacity and CO, suggesting that the increased FAO in PAB is maladaptive. The benefit of the FAO inhibition reflected activation of the Randle cycle, since FAO inhibitors increased GO and elevated RV ATP concentrations.[32] Like the PDK inhibitors, the FAO inhibitors also cause some regression of RVH. The RV's reliance on glycolysis is evident in the increased expression of Glut1 and hexokinase 1 (HKI) in MCT and PAB models.[20,32] As expected, RV glycogen increases in the presence of suppressed GO and glycogen levels fall with peripheral FAO inhibition.[32] FAO inhibition also reduces the expression of glycolytic genes. As would be expected with increased FAO, RV expression of carnitine palmitoyltransferase 1, a key regulatory enzyme, is elevated in PAB-induced RVH and decreases with chronic inhibition of FAO.[32] Although we consider the PAB model one of adaptive RVH, relative to MCT or CH-SU models (i.e., mortality is low and resting CO is well maintained), there is clearly inadequate bioenergetic reserve to support exercise, without lactic academia.[32] Thus, severe PAB results in borderline RV reserve, similar to that seen clinically in patients with severe pulmonic stenosis. The beneficial effects of inhibiting FAO, despite persistence of the mechanical obstruction to RV outflow, may have clinical implications for PAH and for patients with pulmonic stenosis. Guarnieri and Muscari reported similar beneficial effects of trimetazidine in MCT-induced RVH.[38] They found that trimetazidine increases O2-consumption and enhances cardiac mitochondrial function, while reducing the formation of oxygen free radicals.[39] Research is required to determine whether combining PDK inhibitors and FAO inhibitors would have benefit in RVH.
METABOLISM IN THE PULMONARY VASCULATURE IN PULMONARY ARTERIAL HYPERTENSION
The parallel glycolytic shift in the lung and RV in PAH offer therapeutic opportunities to simultaneously treat the RV and pulmonary vasculature. While in the RV metabolic remodeling is likely triggered by ischemia, the basis for aerobic glycolysis in the pulmonary vasculature is different (although the role of PDH inhibition is common to both). In the vasculature, metabolic changes appear to be triggered by changes in redox state that activate transcription factors, such as HIF-1α. Examples of redox changes in PAH include epigenetic silencing of SOD2 and/or changes in mitochondrial fission/fusion. Once oxidative metabolism is inhibited, a vicious cycle maintains the redox abnormalities and favors persistent glycolysis. As mentioned previously, there are similarities between cancer and PAH, including a shared reliance on aerobic glycolysis, as originally noted by Dr. Otto Warburg.[40] Warburg suggested that a shift in glucose metabolism from oxidative phosphorylation to glycolysis was central to the cause/maintenance of cancers and occurred despite abundant oxygen.[40] Although originally controversial, recent oncology,[42,42] and PAH investigations,[43–45] suggest that PAH and cancer do share an aerobic glycolysis phenotype. As in lung cancer,[46] the mitochondria in the PASMC in PAH are fragmented.[47] Mitochondria in PAH are deficient in complex I expression and superoxide dismutase 2 (SOD2) and their membrane potential is hyperpolarized.[48] These changes in the mitochondria (which are the PASMC's oxygen sensor,[49] ) create a "pseudohypoxic" redox milieu in which ambient PO 2 and the production of the reactive oxygen species (that serve as short-lived signaling molecules) are dissociated. The consequence of this redox disorder is normoxic activation of HIF-1α, both in experimental,[48] and human PAH.[48,50,51] This pathological HIF-1α activation contributes to the aerobic glycolysis. The up-regulation of glycolysis in the lung vasculature can be detected by lung 18F-FDG PET scans in humans,[45] and rats.[52] The HIF-1α-positive, glycolytic cells in experimental PAH are hyperproliferative,[52] suggesting a similar link between metabolism and proliferation as is seen in cancer.[46,53]
HIF-1α activation. Several other lines of evidence implicate HIF-1α in the pathogenesis of PAH. Mutations of the von Hippel-Lindau tumor suppressor gene (which also leads to normoxic activation of HIF-1α) causes patients to develop mild pulmonary hypertension and polycythemia (a syndrome called Chuvash pulmonary hypertension).[54] There is also a rodent model of Chuvash disease created in mice that are homozygous for the R200W von Hippel-Lindau (VHL) tumor suppressor protein mutation at codon 200 (R200W). These mice develop PH and pulmonary fibrosis, independent of polycythemia. However in this murine model it appears HIF-2α may be more important than HIF-1α.[55] Moreover, PAH develops spontaneously in Fawn-hooded rats (FHR), a strain in which normoxic HIF-1α activation results from impairment of mitochondrial network formation and redox signaling.[56] Conversely, HIF-1α haploinsufficiency is protective against chronic hypoxic pulmonary hypertension in mice.[57,58] Recently we have shown that the HIF-1α activation persists in human PAH PASMC and FHR PASMC in culture despite abundant PO2. This persistent and heritable HIF-1α activation suggests a heritable mechanism, and indeed an epigenetic basis for this observation has recently been noted.[44]
HIF-1α is redox regulated. For example, hydrogen peroxide inhibits HIF-1α activation.[59] HIF-1α activation in PAH likely results from a series of interconnected mitochondrial-metabolic abnormalities that regulate hydrogen peroxide production. One such abnormality is seen in FHR where DNA methyltransferase activation in the lung mediates epigenetic silencing of superoxide dismutase 2, SOD2, the mitochondrial enzyme responsible for generation of hydrogen peroxide.[44]
Epigenetic silencing of SOD2. Epigenetic regulation of genes involves modifications that alter gene expression without alteration of gene sequence.[60] Epigenetic down regulation of the mitochondrial SOD2 occurs in the lungs and PAs (but not systemic arteries) of FHR. The SOD2 gene expression is reduced by ~50% due to transcriptional repression by methylation of two key CpG islands in the gene.[44] This methylation interferes with gene transcription and reduces hydrogen peroxide production, leading to a redox-mediated HIF-1α activation that promotes a glycolytic metabolic phenotype, which promotes PASMC proliferation. In an interesting proof of concept experiment, we showed that suppressing SOD2 expression in normal PASMC (using siRNA) is sufficient to activate HIF-1α during normoxia.[44] DNA methyltransferase inhibition (using 5-azacytidine) demethylates the SOD2 gene, restoring SOD2 expression and inhibiting PASMC proliferation.[44] For reasons that remain unclear, the dysregulation of DNA methyltransferases in experimental PAH is restricted to the lung, perhaps accounting for some of the tissue specificity of the syndrome.[44] Interestingly humans with PAH have the same HIF-1α activation and SOD2 deficiency,[48,61] although it is not proven that this is epigenetically mediated. A similar epigenetic inhibition of SOD2 has been implicated in the hyperproliferative phenotype of various cancer cells.[62,63]
PDK and PDH. As in the RV, PDK activation occurs in the lungs in PAH. HIF-1α increases expression of certain PDK isoforms (e.g., PDK1) via interaction with its hypoxia response element.[64] The transcriptional up-regulation of PDK by HIF-1α inhibits mitochondrial PDH creating a glycolytic state. Under aerobic conditions the mitochondrial PDH complex catalyzes the rate-limiting step in the oxidation of glucose to acetyl CoA. Acetyl CoA fuels Krebs′ cycle thereby providing the electron transport chain with electron donors driving oxidative metabolism. Rapid regulation of PDH is achieved by reversible phosphorylation of the α-subunit of the E1 (pyruvate decarboxylase) component of the complex. The phosphorylated form of PDH is inactive. Loss-of-function mutations in the PDH complex,[65] or pathological activation of PDK, as occurs in cancer,[66] or PAH,[43] favor excessive cell proliferation and impair apoptosis.[47,48] PDK presents a specific target that is tractable to the relatively nontoxic, small molecule xenobiotic, dichloroacetate (DCA). DCA has an appealing profile because it corrects the pathologic inhibition of PDH and GO in both the lung (inhibiting proliferation and inducing apoptosis) and RV (enhancing energetics and contractility), while having little effect on normal vascular or cardiac cells (in which PDK isoforms are relatively inactive).[48,67–69]
Mitochondrial Fission. We recently discovered an additional consequence of HIF-1α activation in the lung vasculature, namely increased mitochondrial fragmentation. The morphology of the mitochondrial network in PASMC is tightly regulated by the opposing effects of fission and fusion, which are mediated by various GTPase. Tight coordination between mitochondrial division and mitosis, particularly in hyperproliferation, ensures equal distribution of mitochondria organelles to daughter cells. Increased fission and mitosis are coordinated by their shared dependence on the cyclin B1-CDK1. Cyclin B1-CDK1 activates mitosis and also phosphorylates (and activates) dynamin-related protein-1 (DRP-1), the key mediator of mitochondrial fission.[47] The expression and activity of both DRP-1 and cyclin B1-CDK1 are increased in human PAH. Inhibition of mitochondrial fission, using the small molecular inhibitor of DRP1 (mdivi-1) arrests PAH PASMC in the G2/M phase of the cell cycle. This indicates that mitochondrial fission is a mitotic checkpoint. In human and experimental PAH, excessive activation of the GTPase DRP-1 promotes excessive fission. HIF-1α promotes fission by activating DRP-1. HIF-1α activation can be stimulated by administration of cobalt, and within two hours this fragments the mitochondrial network through a DRP-1-dependent mechanism. In vivo, administration of cobalt leads to a form of PAH.[47] In this cobalt model, inhibition of DRP-1 slows PASMC proliferation and regresses experimental PAH. Interestingly, similar abnormalities (DRP-1 mediated mitochondrial fission and hyperproliferation) also occur in lung cancer.[47] It is uncertain what the metabolic consequences of fission and fusion are; however this is a rich area for future research. We suspect, based on preliminary studies, that the consequence of fission and fusion are contextual, varying amongst cell types and temporally. For example, the brief fission of mitosis is distinct from prolonged fission, as occurs with cell death.
FAO in the vasculature. Sutendra recently showed that mice deficient in malonyl-coenzyme A (CoA) decarboxylase (MCD) are protected from hypoxic pulmonary hypertension.[70] The lack of MCD inhibits fatty acid oxidation, which promotes GO and prevents the glycolytic shift in metabolism that otherwise occurs in the vascular media during chronic hypoxia in wild-type mice.[70] Metabolism in the MCD knockout mouse is similar to that achieved in rats with RVH treated with the FAO inhibitors ranolazine and trimetazidine.
In conclusion, mitochondrial metabolism is dysregulated in PAH, both in the lung and the RV with common features being PDH inhibition and aerobic glycolysis. These pathways are tractable to therapeutic intervention and merit further study.
Footnotes
Source of Support: This work is supported in part by the Harold Hines Jr. Chair in Medicine and NIH.RO1.HL071115 (Stephen L. Archer), 1RC1HL099462-01 (Stephen L. Archer), and the American Heart Association (AHA).
Conflict of Interest: None declared.
REFERENCES
- 1.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 2.Izumo S, Nadal-Ginard B, Mahdavi V. Protooncogene induction and reprogramming of cardiac gene expression produced by pressure overload. Proc Natl Acad Sci USA. 1988;85:339–43. doi: 10.1073/pnas.85.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vlahakes GJ, Turley K, Hoffman JI. The pathophysiology of failure in acute right ventricular hypertension: Hemodynamic and biochemical correlations. Circulation. 1981;63:87–95. doi: 10.1161/01.cir.63.1.87. [DOI] [PubMed] [Google Scholar]
- 4.Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, et al. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation. 2009;120:1951–60. doi: 10.1161/CIRCULATIONAHA.109.883843. [DOI] [PubMed] [Google Scholar]
- 5.Torbicki A, Kurzyna M, Kuca P, Fijalkowska A, Sikora J, Florczyk M, et al. Detectable serum cardiac troponin t as a marker of poor prognosis among patients with chronic precapillary pulmonary hypertension. Circulation. 2003;108:844–8. doi: 10.1161/01.CIR.0000084544.54513.E2. [DOI] [PubMed] [Google Scholar]
- 6.Heresi GA, Tang WH, Aytekin M, Hammel J, Hazen SL, Dweik RA. Sensitive cardiac troponin I predicts poor outcomes in pulmonary arterial hypertension. Eur Respir J. 2012;39:939–44. doi: 10.1183/09031936.00067011. [DOI] [PubMed] [Google Scholar]
- 7.Vogel-Claussen J, Skrok J, Shehata ML, Singh S, Sibley CT, Boyce DM, et al. Right and left ventricular myocardial perfusion reserves correlate with right ventricular function and pulmonary hemodynamics in patients with pulmonary arterial hypertension. Radiology. 2011;258:119–27. doi: 10.1148/radiol.10100725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oikawa M, Kagaya Y, Otani H, Sakuma M, Demachi J, Suzuki J, et al. Increased [18F] fluorodeoxyglucose accumulation in right ventricular free wall in patients with pulmonary hypertension and the effect of epoprostenol. J Am Coll Cardiol. 2005;45:1849–55. doi: 10.1016/j.jacc.2005.02.065. [DOI] [PubMed] [Google Scholar]
- 9.van de Veerdonk MC, Kind T, Marcus JT, Mauritz GJ, Heymans MW, Bogaard HJ, et al. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol. 2011;58:2511–9. doi: 10.1016/j.jacc.2011.06.068. [DOI] [PubMed] [Google Scholar]
- 10.Freed BH, Gomberg-Maitland M, Chandra S, Mor-Avi V, Rich S, Archer SL, et al. Late gadolinium enhancement cardiovascular magnetic resonance predicts clinical worsening in patients with pulmonary hypertension. J Cardiovasc Magn Reson. 2012;14:11. doi: 10.1186/1532-429X-14-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sztrymf B, Souza R, Bertoletti L, Jais X, Sitbon O, Price LC, et al. Prognostic factors of acute heart failure in patients with pulmonary arterial hypertension. Eur Respir J. 2010;35:1286–93. doi: 10.1183/09031936.00070209. [DOI] [PubMed] [Google Scholar]
- 12.Campo A, Mathai SC, Le Pavec J, Zaiman AL, Hummers LK, Boyce D, et al. Outcomes of hospitalisation for right heart failure in pulmonary arterial hypertension. Eur Respir J. 2011;38:359–67. doi: 10.1183/09031936.00148310. [DOI] [PubMed] [Google Scholar]
- 13.Rich S, Pogoriler J, Husain AN, Toth PT, Gomberg-Maitland M, Archer SL. Long-term effects of epoprostenol on the pulmonary vasculature in idiopathic pulmonary arterial hypertension. Chest. 2010;138:1234–9. doi: 10.1378/chest.09-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawut SM, Taichman DB, Archer-Chicko CL, Palevsky HI, Kimmel SE. Hemodynamics and survival in patients with pulmonary arterial hypertension related to systemic sclerosis. Chest. 2003;123:344–50. doi: 10.1378/chest.123.2.344. [DOI] [PubMed] [Google Scholar]
- 15.Kuhn KP, Byrne DW, Arbogast PG, Doyle TP, Loyd JE, Robbins IM. Outcome in 91 consecutive patients with pulmonary arterial hypertension receiving epoprostenol. Am J Respir Crit Care Med. 2003;167:580–6. doi: 10.1164/rccm.200204-333OC. [DOI] [PubMed] [Google Scholar]
- 16.Hopkins WE, Ochoa LL, Richardson GW, Trulock EP. Comparison of the hemodynamics and survival of adults with severe primary pulmonary hypertension or eisenmenger syndrome. J Heart Lung Transplant. 1996;15(1 Pt 1):100–5. [PubMed] [Google Scholar]
- 17.Rich S, Pogoriler J, Husain AN, Toth PT, Gomberg-Maitland M, Archer SL. Long-term effects of epoprostenol on the pulmonary vasculature in idiopathic pulmonary arterial hypertension. Chest. 2010;138:1234–9. doi: 10.1378/chest.09-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oka M, Homma N, Taraseviciene-Stewart L, Morris KG, Kraskauskas D, Burns N, et al. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res. 2007;100:923–9. doi: 10.1161/01.RES.0000261658.12024.18. [DOI] [PubMed] [Google Scholar]
- 19.Kurzyna M, Zylkowska J, Fijalkowska A, Florczyk M, Wieteska M, Kacprzak A, et al. Characteristics and prognosis of patients with decompensated right ventricular failure during the course of pulmonary hypertension. Kardiol Pol. 2008;66:1033–9. [PubMed] [Google Scholar]
- 20.Piao L, Fang YH, Cadete VJ, Wietholt C, Urboniene D, Toth PT, et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: Resuscitating the hibernating right ventricle. J Mol Med. 2010;88:47–60. doi: 10.1007/s00109-009-0524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ritchie M, Waggoner AD, Davila-Roman VG, Barzilai B, Trulock EP, Eisenberg PR. Echocardiographic characterization of the improvement in right ventricular function in patients with severe pulmonary hypertension after single-lung transplantation. J Am Coll Cardiol. 1993;22:1170–4. doi: 10.1016/0735-1097(93)90433-2. [DOI] [PubMed] [Google Scholar]
- 22.van Wolferen SA, Marcus JT, Westerhof N, Spreeuwenberg MD, Marques KM, Bronzwaer JG, et al. Right coronary artery flow impairment in patients with pulmonary hypertension. Eur Heart J. 2008;29:120–7. doi: 10.1093/eurheartj/ehm567. [DOI] [PubMed] [Google Scholar]
- 23.Vlahakes GJ, Baer RW, Uhlig PN, Verrier ED, Bristow JD, Hoffmann JI. Adrenergic influence in the coronary circulation of conscious dogs during maximal vasodilation with adenosine. Circ Res. 1982;51:371–84. doi: 10.1161/01.res.51.3.371. [DOI] [PubMed] [Google Scholar]
- 24.Bian X, Williams AG, Jr, Gwirtz PA, Downey HF. Right coronary autoregulation in conscious, chronically instrumented dogs. Am J Physiol. 1998;275:H169–75. doi: 10.1152/ajpheart.1998.275.1.H169. [DOI] [PubMed] [Google Scholar]
- 25.Overbeek MJ, Lankhaar JW, Westerhof N, Voskuyl AE, Boonstra A, Bronzwaer JG, et al. Right ventricular contractility in systemic sclerosis-associated and idiopathic pulmonary arterial hypertension. Eur Respir J. 2008;31:1160–6. doi: 10.1183/09031936.00135407. [DOI] [PubMed] [Google Scholar]
- 26.Sharma S, Taegtmeyer H, Adrogue J, Razeghi P, Sen S, Ngumbela K, et al. Dynamic changes of gene expression in hypoxia-induced right ventricular hypertrophy. Am J Physiol Heart Circ Physiol. 2004;286:H1185–92. doi: 10.1152/ajpheart.00916.2003. [DOI] [PubMed] [Google Scholar]
- 27.Sugden MC, Langdown ML, Harris RA, Holness MJ. Expression and regulation of pyruvate dehydrogenase kinase isoforms in the developing rat heart and in adulthood: Role of thyroid hormone status and lipid supply. Biochem J. 2000;352:731–8. [PMC free article] [PubMed] [Google Scholar]
- 28.Stanley WC, Lopaschuk GD, Hall JL, McCormack JG. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions.Potential for pharmacological interventions. Cardiovasc Res. 1997;33:243–57. doi: 10.1016/s0008-6363(96)00245-3. [DOI] [PubMed] [Google Scholar]
- 29.Randle PJ, Priestman DA, Mistry SC, Halsall A. Glucose fatty acid interactions and the regulation of glucose disposal. J Cell Biochem. 1994;55(Suppl):1–11. doi: 10.1002/jcb.240550002. [DOI] [PubMed] [Google Scholar]
- 30.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle.Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–9. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 31.Abozguia K, Clarke K, Lee L, Frenneaux M. Modification of myocardial substrate use as a therapy for heart failure. Nat Clin Pract Cardiovasc Med. 2006;3:490–8. doi: 10.1038/ncpcardio0583. [DOI] [PubMed] [Google Scholar]
- 32.Fang YH, Piao L, Hong Z, Toth PT, Marsboom G, Bache-Wiig P, et al. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: Exploiting randle's cycle. J Mol Med (Berl) 2012;90:31–43. doi: 10.1007/s00109-011-0804-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kantor PF, Lucien A, Kozak R, Lopaschuk GD. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme a thiolase. Circ Res. 2000;86:580–8. doi: 10.1161/01.res.86.5.580. [DOI] [PubMed] [Google Scholar]
- 34.Tuunanen H, Engblom E, Naum A, Nagren K, Scheinin M, Hesse B, et al. Trimetazidine, a metabolic modulator, has cardiac and extracardiac benefits in idiopathic dilated cardiomyopathy. Circulation. 2008;118:1250–8. doi: 10.1161/CIRCULATIONAHA.108.778019. [DOI] [PubMed] [Google Scholar]
- 35.Wilson SR, Scirica BM, Braunwald E, Murphy SA, Karwatowska-Prokopczuk E, Buros JL, et al. Efficacy of ranolazine in patients with chronic angina observations from the randomized, double-blind, placebo-controlled merlin-timi (metabolic efficiency with ranolazine for less ischemia in non-st-segment elevation acute coronary syndromes) 36 trial. J Am Coll Cardiol. 2009;53:1510–6. doi: 10.1016/j.jacc.2009.01.037. [DOI] [PubMed] [Google Scholar]
- 36.Clarke B, Wyatt KM, McCormack JG. Ranolazine increases active pyruvate dehydrogenase in perfused normoxic rat hearts: Evidence for an indirect mechanism. J Mol Cell Cardiol. 1996;28:341–50. doi: 10.1006/jmcc.1996.0032. [DOI] [PubMed] [Google Scholar]
- 37.McCormack JG, Barr RL, Wolff AA, Lopaschuk GD. Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation. 1996;93:135–42. doi: 10.1161/01.cir.93.1.135. [DOI] [PubMed] [Google Scholar]
- 38.Guarnieri C, Muscari C. Beneficial effects of trimetazidine on mitochondrial function and superoxide production in the cardiac muscle. Cardiovasc Drugs Ther. 1990:814–5. doi: 10.1007/BF00051282. [DOI] [PubMed] [Google Scholar]
- 39.Guarnieri C, Muscari C. Beneficial effects of trimetazidine on mitochondrial function and superoxide production in the cardiac muscle of monocrotaline-treated rats. Biochem Pharmacol. 1988;37:4685–8. doi: 10.1016/0006-2952(88)90338-3. [DOI] [PubMed] [Google Scholar]
- 40.Warburg O. Berline: Julius Springer; 1926. Uber den stoffwechsel der tumoren. [Google Scholar]
- 41.Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2:31ra4. doi: 10.1126/scitranslmed.3000677. [DOI] [PubMed] [Google Scholar]
- 42.Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer. 2008;99:989–94. doi: 10.1038/sj.bjc.6604554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: A mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol. 2008;294:H570–8. doi: 10.1152/ajpheart.01324.2007. [DOI] [PubMed] [Google Scholar]
- 44.Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation. 2010;121:2661–71. doi: 10.1161/CIRCULATIONAHA.109.916098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci USA. 2007;104:1342–7. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, et al. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012;26:2175–86. doi: 10.1096/fj.11-196543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res. 2012;110:1484–97. doi: 10.1161/CIRCRESAHA.111.263848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thébaud B, Bonnet SN, et al. An abnormal mitochondrial-inducible factor-1alpha-Kv channel pathway disrupts oxygen-sensing and triggers pulmonary arterial hypertension (pah) in fawn-hooded rats: Similarities to human pah. Circulation. 2006;113:2630–41. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 49.Weir EK, Lopez-Barneo J, Buckler KJ, Archer SL. Acute oxygen-sensing mechanisms. N Engl J Med. 2005;353:2042–55. doi: 10.1056/NEJMra050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tuder RM, Chacon M, Alger L, Wang J, Taraseviciene-Stewart L, Kasahara Y, et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. J Pathol. 2001;195:367–74. doi: 10.1002/path.953. [DOI] [PubMed] [Google Scholar]
- 51.Fijalkowska I, Xu W, Comhair SA, Janocha AJ, Mavrakis LA, Krishnamachary B, et al. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol. 2010;176:1130–8. doi: 10.2353/ajpath.2010.090832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marsboom G, Wietholt C, Haney CR, Toth PT, Ryan JJ, Morrow E, et al. Lung 18F-fluorodeoxyglucose pet for diagnosis and monitoring of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;185:670–9. doi: 10.1164/rccm.201108-1562OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-k+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 54.Gladwin MT. Polycythemia, HIF-1alpha and pulmonary hypertension in chuvash. Haematologica. 2006;91:722. [PubMed] [Google Scholar]
- 55.Hickey MM, Richardson T, Wang T, Mosqueira M, Arguiri E, Yu H, et al. The von hippel-lindau chuvash mutation promotes pulmonary hypertension and fibrosis in mice. J Clin Invest. 2010;120:827–39. doi: 10.1172/JCI36362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith TG, Brooks JT, Balanos GM, Lappin TR, Layton DM, Leedham DL, et al. Mutation of von hippel-lindau tumour suppressor and human cardiopulmonary physiology. PLoS Med. 2006;3:e290. doi: 10.1371/journal.pmed.0030290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shimoda LA, Manalo DJ, Sham JS, Semenza GL, Sylvester JT. Partial HIF-1alpha deficiency impairs pulmonary arterial myocyte electrophysiological responses to hypoxia. Am J Physiol Lung Cell Mol Physiol. 2001;281:L202–8. doi: 10.1152/ajplung.2001.281.1.L202. [DOI] [PubMed] [Google Scholar]
- 58.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest. 1999;103:691–6. doi: 10.1172/JCI5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang LE, Arany Z, Livingston DM, Bunn HF. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J Biol Chem. 1996;271:32253–9. doi: 10.1074/jbc.271.50.32253. [DOI] [PubMed] [Google Scholar]
- 60.Wolffe AP, Matzke MA. Epigenetics: Regulation through repression. Science. 1999;286:481–6. doi: 10.1126/science.286.5439.481. [DOI] [PubMed] [Google Scholar]
- 61.Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med. 2004;169:764–9. doi: 10.1164/rccm.200301-147OC. [DOI] [PubMed] [Google Scholar]
- 62.Hitchler MJ, Oberley LW, Domann FE. Epigenetic silencing of SOD2 by histone modifications in human breast cancer cells. Free Radic Biol Med. 2008;45:1573–80. doi: 10.1016/j.freeradbiomed.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hitchler MJ, Wikainapakul K, Yu L, Powers K, Attatippaholkun W, Domann FE. Epigenetic regulation of manganese superoxide dismutase expression in human breast cancer cells. Epigenetics. 2006;1:163–71. doi: 10.4161/epi.1.4.3401. [DOI] [PubMed] [Google Scholar]
- 64.Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–69. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 65.Patel KP, O’Brien TW, Subramony SH, Shuster J, Stacpoole PW. The spectrum of pyruvate dehydrogenase complex deficiency: Clinical, biochemical and genetic features in 371 patients. Mol Genet Metab. 2012;106:385–94. doi: 10.1016/j.ymgme.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2:31ra4. doi: 10.1126/scitranslmed.3000677. [DOI] [PubMed] [Google Scholar]
- 67.Guignabert C, Tu L, Izikki M, Dewachter L, Zadigue P, Humbert M, et al. Dichloroacetate treatment partially regresses established pulmonary hypertension in mice with SM22alpha-targeted overexpression of the serotonin transporter. Faseb J. 2009;23:4135–47. doi: 10.1096/fj.09-131664. [DOI] [PubMed] [Google Scholar]
- 68.McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, et al. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. 2004;95:830–40. doi: 10.1161/01.RES.0000145360.16770.9f. [DOI] [PubMed] [Google Scholar]
- 69.Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, et al. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: Role of increased expression and activity of voltage-gated potassium channels. Circulation. 2002;105:244–50. doi: 10.1161/hc0202.101974. [DOI] [PubMed] [Google Scholar]
- 70.Sutendra G, Bonnet S, Rochefort G, Haromy A, Folmes KD, Lopaschuk GD, et al. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med. 2010;2:44ra58. doi: 10.1126/scitranslmed.3001327. [DOI] [PubMed] [Google Scholar]