

Abstract
Pulmonary hypertension is a prevalent complication of chronic obstructive pulmonary disease (COPD) that is associated with poor prognosis. Although pulmonary hypertension is usually diagnosed in patients with advanced disease, changes in pulmonary vessels are already apparent at early disease stages, and in smokers without airflow obstruction. Changes in pulmonary vessels include intimal hyperplasia, resulting from proliferating mesenchymal cells, and elastic and collagen deposition as well as endothelial dysfunction. Dysregulation of endothelium-derived mediators and growth factors and inflammatory mechanisms underlie the endothelial dysfunction and vessel remodeling. Circumstantial and experimental evidence suggests that cigarette smoke products can initiate pulmonary vascular changes in COPD and that, at advanced disease stages, hypoxia may amplify the effects of cigarette smoke on pulmonary arteries. Bone marrow-derived progenitor cells may contribute to vessel repair and to vessel remodeling, a process that appears to be facilitated by transforming growth factor-β.
Keywords: endothelium, vascular remodeling, cigarette smoke, progenitor cells
Chronic obstructive pulmonary disease (COPD) is a highly prevalent disorder and a major cause of mortality worldwide. It is characterized by progressive airflow limitation, which is associated with a chronic inflammatory process in the airways and lung parenchyma, in response to noxious particles or gases, particularly cigarette smoke. Small airways disease and parenchymal destruction are the main structural abnormalities, although changes in pulmonary vessels also represent an important component of the disease. Alterations in vessel structure may progress and result in pulmonary hypertension, a complication that may develop in > 50% of patients with an advanced disease.[1] The presence of pulmonary hypertension is associated with shorter survival[2] and more frequent exacerbation episodes.[3] In the present review, we will address the current knowledge on the underlying mechanisms for the development of pulmonary hypertension in COPD.
VASCULAR CHANGES
Patients with COPD present conspicuous changes in pulmonary vessels (vascular remodeling) that affect predominantly small size vessels (muscular arteries and arterioles). Intimal hyperplasia is the most prominent feature in pulmonary muscular arteries. It is apparent in vessels of different sizes, although it is more pronounced in small arteries with a diameter of < 500 μm.[4,5] In addition, there is muscularization of the arterioles, which also show intimal enlargement. Changes in the tunica media are less conspicuous and the majority of studies have failed to show any striking differences in the thickness of the muscular layer in COPD patients as compared with controls.[4–7]
Intimal hyperplasia is produced by proliferation of cells that express smooth muscle α-actin and other mesenchymal markers like vimentin.[8] In addition, there is deposition of elastic and collagen fibers[8] (Fig. 1). Pulmonary vascular remodeling is apparent in patients with different degrees of COPD severity, and the presence of pulmonary hypertension does not seem to be associated with greater derangement of the vessel structure.[7] Furthermore, intimal hyperplasia[8] and muscularization of small pulmonary arteries[9] of similar magnitude than in COPD also occur in heavy smokers with normal lung function. In fact, studies in experimental models of COPD indicate that pulmonary vascular changes antecede the development of emphysema.[9,10]
Figure 1.
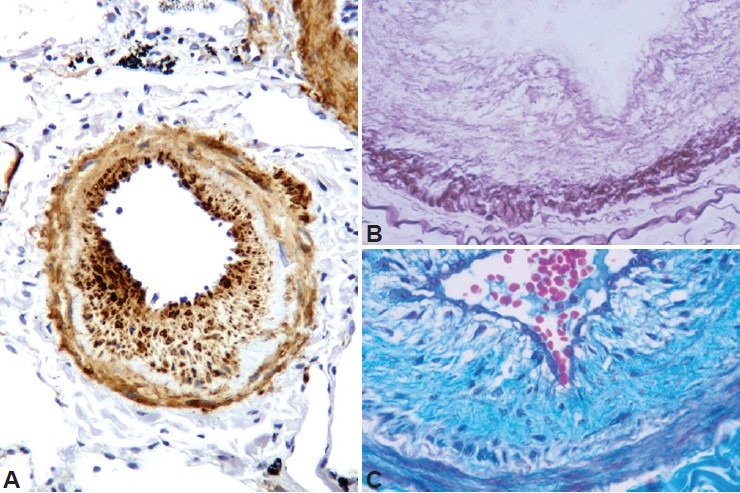
Characteristics of remodeling of pulmonary arteries in COPD. (A) Pulmonary muscular artery with an enlarged intima showing abundant cells with positive immunostaining to α-smooth muscle actin antibody. (B) Section of a hyperplasic intima of a muscular artery stained with orcein. Note the abundant deposition of elastic fibers. (C) Collagen fibers stained blue with Masson's trichrome stain.
Patients with COPD and smokers without airflow obstruction show an increased number of inflammatory cells infiltrating the adventitia of pulmonary muscular arteries, largely constituted by activated T lymphocytes with a predominance of the CD8+ T cell subset.[11,12] The number of neutrophils, macrophages, and B lymphocytes are minimal and do not differ from control nonsmokers.
At the inspection with electron microscopy, the endothelial surface of pulmonary arteries of patients with COPD may show loss of cell junctions and areas of endothelial denudation (Fig. 2).
Figure 2.
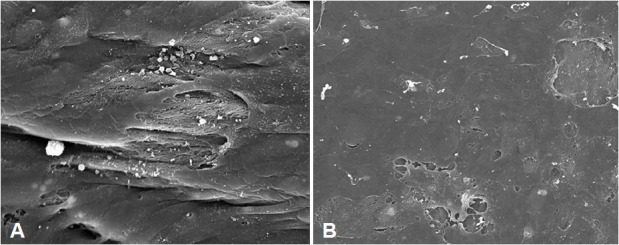
Scanning electron microscopy of the endothelial surface of pulmonary arteries. Patients with COPD show frequent detachments between endothelial cells (A) and areas of denuded endothelium (B).
RELEVANT MEDIATORS
In COPD, changes in vessel structure are accompanied by changes in endothelial function and in the expression of angiogenic and growth factors. Reduced endothelium-dependent relaxation, denoting endothelial dysfunction of pulmonary arteries, has been shown in patients with end-stage COPD who underwent lung transplantation[13] as well as in patients with mild-to-moderate COPD.[6] Endothelial dysfunction is associated with reduced expression of endothelial nitric oxide synthase (eNOS)[14] and prostacyclin synthase (PGI2-S)[15] changes that favor vessel contraction and cell proliferation. Pulmonary arteries of patients with COPD also show increased expression of vascular endothelial growth factor (VEGF)[16] and the receptor-II of transforming growth factor-beta (TGF-βRII).[17]
In a recent study, Seimetz et al.[9] showed in lungs of patients with advanced COPD downregulation of eNOS mRNA and protein, whereas inducible NOS (iNOS) was upregulated. Similar findings were observed in lungs of smokers without COPD and in mice exposed to cigarette smoke.[9] Upregulation of iNOS may favor vascular remodeling either through activation of an inflammatory mechanism or the differentiation of bone marrow-derived progenitor cells.[9] Overall, downregulation of endothelium-derived antiproliferative agents associated with increased growth factor activity may concur and contribute to cell proliferation and remodeling in pulmonary arteries in COPD.
EFFECTS OF CIGARETTE SMOKE
Different mechanisms may contribute to the development of endothelial dysfunction and vascular remodeling in pulmonary vessels, including hypoxia and inflammation. However, in recent years, attention has been focused on the potential effects of cigarette smoke products as initiating factors of the pulmonary vascular derangement in COPD.[18] Circumstantial evidence supports this contention, since most changes observed in pulmonary arteries of COPD patients have also been noticed in pulmonary arteries of smokers without COPD. Studies from our laboratory have shown that “healthy” smokers without airflow obstruction show intimal hyperplasia,[8] reduced expression of eNOS,[14] increased expression of VEGF,[16] and inflammatory cell infiltrate[11] in pulmonary arteries at a similar level than in patients with COPD, which is clearly different from that of nonsmokers. Furthermore, a recent study evaluating genes potentially involved in pulmonary vascular changes has shown more similarities in lung gene expression between “healthy” smokers and patients with mild-to-moderate COPD than between the later and patients with end-stage COPD.[19] This has prompted the hypothesis that cigarette-smoke products play a key role in initiating vascular changes that may eventually progress to pulmonary hypertension in COPD.[18,20]
Experimental studies have provided support to this hypothesis. Su et al.[21] showed that in human pulmonary artery endothelial cells (HPAEC) exposed to cigarette smoke extract, eNOS activity, and protein content decreased in a time- and dose-dependent manner. Furthermore, prostacyclin synthase (PGI 2S) mRNA and protein content in HPAEC also decreased when cells were exposed to cigarette smoke extract and acrolein, a potent αβ unsaturated aldehyde found in cigarette smoke.[15]
Guinea pigs[10,22] and mice[9] chronically exposed to cigarette smoke develop changes in pulmonary vessels that resemble those seen in COPD patients. Interestingly, studies in experimental models of COPD have revealed that remodeling (muscularization of small intrapulmonary vessels) may antecede the development of pulmonary emphysema.[9,10,22] This finding agrees with the observation of pulmonary vascular changes in “healthy” smokers or in patients with mild COPD and provides support to the hypothesis that pulmonary vascular remodeling in COPD might be initiated by the effects of cigarette smoke products on pulmonary vessels.
Guinea pigs exposed to cigarette smoke show increased numbers of inflammatory cells (neutrophils, macrophages, and eosinophils) in the adventitia of intrapulmonary vessels,[23] suggesting that vascular changes in exposed animals might have, at least in part, an inflammatory mechanism. In this experimental model, cigarette smoke produces endothelial dysfunction in pulmonary arteries but not in systemic arteries, indicating that vascular impairment induced by cigarette smoke develops earlier in pulmonary vessels, presumably as a consequence of greater lung concentration of smoke products.[22]
Guinea pigs exposed to cigarette smoke develop pulmonary hypertension and right ventricular hypertrophy at levels similar to those seen in animals exposed to hypoxia.[24] Interestingly, animals exposed to cigarette smoke and subsequently to hypoxia show greater increase in pulmonary artery pressure and more prominent remodeling in small pulmonary vessels that when exposed to cigarette smoke or hypoxia alone.[24] This suggests synergism between the effects of cigarette smoke and hypoxia, which might explain the well-established association between hypoxemia and pulmonary hypertension in COPD. Conceivably, patients with pulmonary vascular dysfunction and remodeling originated by the effects of cigarette smoke products are more susceptible to the effects of hypoxia. The latter, which usually develops in patients with advanced disease, may further amplify pulmonary vascular changes induced by cigarette smoke.
PROGENITOR CELLS
Intimal hyperplasia of pulmonary arteries in COPD is produced by the proliferation of cells expressing common mesenchymal and smooth muscle cell (SMC) markers (Fig. 1) and lack some markers characteristic of mature SMC such as desmin.[8] The expression pattern of intermediate filaments may discriminate between a synthetic phenotype of SMC and the contractile phenotype observed in mature cells.[25] Accordingly, vimentin-positive, desmin-negative cells may represent a subpopulation of mesenchymal cells (poorly differentiated SMCs, myofibroblasts) that may possess synthetic capacity and take part in an ongoing process of vascular remodeling.
The origin of these poorly differentiated cells in the intima of pulmonary arteries is not fully understood. Hypothetically, several possibilities can be considered: (1) dedifferentiation of mature SMC in the muscular layer and migration toward the intima,[26] (2) transdifferentiation from endothelial cells through an endothelial-to-mesenchymal-transition process, (3) differentiation from resident precursor cells, and (4) recruitment and differentiation from circulating bone marrow-derived progenitor cells.
The bone marrow has the capacity to synthesize and mobilize progenitor cells that can regenerate several tissues, among them the so-called endothelial progenitor cells (EPCs) that may contribute to vascular re-endothelization and vasculogenesis.[27] The characterization and functional properties of EPCs remains controversial.[28] Using antibodies against CD133, a marker of progenitor cells derived from the bone marrow, we have identified cells showing positive immunoreactivity to this antigen in denuded areas of the endothelium and within the intimal layer of pulmonary arteries from patients with COPD.[29] Interestingly, the number of progenitor cells present in the vessel wall was associated with both the contractile response to hypoxic stimulus, which is associated with endothelial function,[30] and the degree of remodeling of pulmonary arteries, thus suggesting that these cells could be involved not only in endothelial repair but also in the remodeling of pulmonary vessels.[29] Therefore, bone marrow-derived progenitor vascular cells might exert a dual, opposite effect, contributing to vascular repair through differentiation into endothelial cells or to vessel remodeling through differentiation into SMC. In fact, labeled CD133+ cells coincubated with human isolated PA could migrate from the vessel lumen to the endothelial surface, where they differentiated into endothelial cells, or deeper into the intima, where they differentiated into SMC[31] (Fig. 3).
Figure 3.
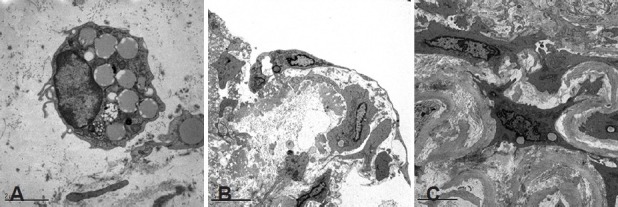
Transmission electron microscopy images of (A) CD133 cells labeled with Dil-acLDL-l that were injected into the lumen of a pulmonary artery. (B) After 4 days incubation, CD133 labeled cells, identified by large cytoplasmic vesicles, differentiated into endothelial cells at the lumen surface. (C) CD133 cells infiltrating the intima acquired a smooth muscle cell-like phenotype.
Little is known about the mechanisms by which bone marrow-derived circulating EPCs can migrate and differentiate into SMCs in the intima of pulmonary arteries and contribute to vessel remodeling. Progenitor cells lying on the luminal surface of pulmonary arteries may come into contact with endothelial cells or they may be chemo-attracted by SMCs and migrate into the intima.[31] Dνez et al.[32] showed that pulmonary SMCs exert greater stimulus on EPCs migration than endothelial cells, suggesting that in pathological conditions this phenomenon could alter the normal process of endothelium repair and, thus, promote pulmonary vascular remodeling. Smooth muscle cells can release several stimulatory cytokines, including TGFί1, which can actively induce EPCs to migrate toward the intima.
Smooth muscle cells could also originate by transdifferentiation from endothelial cells, a process known as endothelial-to-mesenchymal-transition (EnMT). This process plays an important role in vascular development and in some vascular disorders.[33] The EnMT process requires the activation of related transcription factors like Snail, Slug, Zeb1, and Zeb 2,[34] which in endothelial cells switch the gene expression profile to upregulation of mesenchymal genes along with the downregulation of typical endothelial gene markers such as VE-cadherin and CD31. In our laboratory, we have shown that coculture of CD133+ cells with SMCs increases the expression of mesenchymal cell markers [smooth muscle (SM) α-actin and SM22-α and myocardin] and decreases the expression of the endothelial cell marker CD31.[32] Furthermore, in the same conditions, we have observed a concomitant increase in the gene expression of EnMT-related transcription factors (Slug, Snail, Zeb1), indicating that mesenchymal phenotype acquisition occurred through an EnMT-like process. Interestingly, inhibition of TGFβ receptor I (TGFβRI) downregulated Snail gene expression, blocked the EnMT, and facilitated the differentiation of EPCs to the endothelial cell lineage. Furthermore, TGFβRI inhibition decreased migration of EPCs stimulated by SMCs without affecting their functionality and adhesion capacity.[32] These findings suggest that bone marrow-derived progenitor cells may differentiate into mesenchymal cells in hyperplasic intima through an EnMT-like process and that TGFβ-1 plays an important role in the fate of these progenitor cells.
A better understanding of mechanisms regulating the differentiation of progenitor cells into endothelial cells or SMCs might provide the clue for modulating pulmonary vascular remodeling and, hence, the development of pulmonary hypertension in COPD.
Footnotes
Source of Support: This work has been partially funded by a research grant from the Instituto de Salud Carlos III (PS09/00536).
Conflict of Interest: None declared.
REFERENCES
- 1.Thabut G, Dauriat G, Stern JB, Logeart D, Lew A, Marrash-Chahla R, et al. Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest. 2005;127:1531–6. doi: 10.1378/chest.127.5.1531. [DOI] [PubMed] [Google Scholar]
- 2.Oswald-Mammosser M, Weitzenblum E, Quoix E, Moser G, Chaouat A, Charpentier C, et al. Prognostic factors in COPD patients receiving long-term oxygen therapy.Importance of pulmonary artery pressure. Chest. 1995;107:1193–8. doi: 10.1378/chest.107.5.1193. [DOI] [PubMed] [Google Scholar]
- 3.Kessler R, Faller M, Fourgaut G, Mennecier B, Weitzenblum E. Predictive factors of hospitalization for acute exacerbation in a series of 64 patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;159:158–64. doi: 10.1164/ajrccm.159.1.9803117. [DOI] [PubMed] [Google Scholar]
- 4.Barberà JA, Riverola A, Roca J, Ramirez J, Waqner PD, Ros D, et al. Pulmonary vascular abnormalities and ventilation-perfusion relationships in mild chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1994;149:423–9. doi: 10.1164/ajrccm.149.2.8306040. [DOI] [PubMed] [Google Scholar]
- 5.Magee F, Wright JL, Wiggs BR, Paré PD, Hogg JC. Pulmonary vascular structure and function in chronic obstructive pulmonary disease. Thorax. 1988;43:183–9. doi: 10.1136/thx.43.3.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peinado VI, Barberà JA, Ramirez J, Gomez FP, Roca J, Jover L, et al. Endothelial dysfunction in pulmonary arteries of patients with mild COPD. Am J Physiol. 1998;274:L908–13. doi: 10.1152/ajplung.1998.274.6.L908. [DOI] [PubMed] [Google Scholar]
- 7.Wright JL, Petty T, Thurlbeck WM. Analysis of the structure of the muscular pulmonary arteries in patients with pulmonary hypertension and COPD: National Institutes of Health nocturnal oxygen therapy trial. Lung. 1992;170:109–24. doi: 10.1007/BF00175982. [DOI] [PubMed] [Google Scholar]
- 8.Santos S, Peinado VI, Ramirez J, Melgosa T, Roca J, Rodriguez-Roisin R, et al. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur Respir J. 2002;19:632–8. doi: 10.1183/09031936.02.00245902. [DOI] [PubMed] [Google Scholar]
- 9.Seimetz M, Parajuli N, Pichl A, Veit F, Kwapiszewska G, Weisel FC, et al. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell. 2011;147:293–305. doi: 10.1016/j.cell.2011.08.035. [DOI] [PubMed] [Google Scholar]
- 10.Wright JL, Churg A. Effect of long-term cigarette smoke exposure on pulmonary vascular structure and function in the guinea pig. Exp Lung Res. 1991;17:997–1009. doi: 10.3109/01902149109064331. [DOI] [PubMed] [Google Scholar]
- 11.Peinado VI, Barberà JA, Abate P, Ramírez J, Roca J, Santos S, et al. Inflammatory reaction in pulmonary muscular arteries of patients with mild chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;159:1605–11. doi: 10.1164/ajrccm.159.5.9807059. [DOI] [PubMed] [Google Scholar]
- 12.Saetta M, Baraldo S, Corbino L, Turato G, Braccioni F, Rea F, et al. CD8+ve cells in the lungs of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160:711–7. doi: 10.1164/ajrccm.160.2.9812020. [DOI] [PubMed] [Google Scholar]
- 13.Dinh-Xuan AT, Higenbottam TW, Clelland CA, Pepke-Zaba J, Cremona G, Butt AY, et al. Impairment of endothelium-dependent pulmonary-artery relaxation in chronic obstructive pulmonary disease. N Engl J Med. 1991;324:1539–47. doi: 10.1056/NEJM199105303242203. [DOI] [PubMed] [Google Scholar]
- 14.Barberà JA, Peinado VI, Santos S, Ramirez J, Roca J, Rodriguez-Roisin R. Reduced expression of endothelial nitric oxide synthase in pulmonary arteries of smokers. Am J Respir Crit Care Med. 2001;164:709–13. doi: 10.1164/ajrccm.164.4.2101023. [DOI] [PubMed] [Google Scholar]
- 15.Nana-Sinkam SP, Lee JD, Sotto-Santiago S, Stearman RS, Keith RL, Choudhury Q, et al. Prostacyclin prevents pulmonary endothelial cell apoptosis induced by cigarette smoke. Am J Respir Crit Care Med. 2007;175:676–85. doi: 10.1164/rccm.200605-724OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santos S, Peinado VI, Ramirez J, Morales-Blanhir J, Bastos R, Roca J, et al. Enhanced expression of vascular endothelial growth factor in pulmonary arteries of smokers and patients with moderate chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2003;167:1250–6. doi: 10.1164/rccm.200210-1233OC. [DOI] [PubMed] [Google Scholar]
- 17.Beghe B, Bazzan E, Baraldo S, Calabrese F, Rea F, Loy M, et al. Transforming growth factor-{beta} type II receptor in pulmonary arteries of patients with very severe COPD. Eur Respir J. 2006;28:556–62. doi: 10.1183/09031936.06.00077105. [DOI] [PubMed] [Google Scholar]
- 18.Peinado VI, Pizarro S, Barbera JA. Pulmonary vascular involvement in COPD. Chest. 2008;134:808–14. doi: 10.1378/chest.08-0820. [DOI] [PubMed] [Google Scholar]
- 19.Llinas L, Peinado VI, Ramon Goni J, Rabinovich R, Pizarro S, Rodriguez-Roisin R, et al. Similar gene expression profiles in smokers and patients with moderate COPD. Pulm Pharmacol Ther. 2011;24:32–41. doi: 10.1016/j.pupt.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 20.Barberà JA, Peinado VI, Santos S. Pulmonary hypertension in chronic obstructive pulmonary disease. Eur Respir J. 2003;21:892–905. doi: 10.1183/09031936.03.00115402. [DOI] [PubMed] [Google Scholar]
- 21.Su Y, Han W, Giraldo C, Li YD, Block ER. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am J Respir Cell Mol Biol. 1998;19:819–25. doi: 10.1165/ajrcmb.19.5.3091. [DOI] [PubMed] [Google Scholar]
- 22.Ferrer E, Peinado VI, Diez M, Carrasco JL, Musri MM, Martínez A, et al. Effects of cigarette smoke on endothelial function of pulmonary arteries in the guinea pig. Respir Res. 2009;10:76. doi: 10.1186/1465-9921-10-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dominguez-Fandos D, Peinado VI, Puig-Pey R, Ferrer E, Musri MM, Ramírez J, et al. Pulmonary Inflammatory Reaction and Structural Changes Induced by Cigarette Smoke Exposure in the Guinea Pig. COPD. 2012 doi: 10.3109/15412555.2012.691999. [DOI] [PubMed] [Google Scholar]
- 24.Ferrer E, Peinado VI, Castaneda J, Prieto-Lloret J, Olea E, et al. Effects of cigarette smoke and hypoxia on pulmonary circulation in the guinea pig. Eur Respir J. 2011;38:617–27. doi: 10.1183/09031936.00105110. [DOI] [PubMed] [Google Scholar]
- 25.van der Loop FT, Gabbiani G, Kohnen G, Ramaekers FC, van Eys GJ. Differentiation of smooth muscle cells in human blood vessels as defined by smoothelin, a novel marker for the contractile phenotype. Arterioscler Thromb Vasc Biol. 1997;17:665–71. doi: 10.1161/01.atv.17.4.665. [DOI] [PubMed] [Google Scholar]
- 26.Rabinovitch M. Elastase and the pathobiology of unexplained pulmonary hypertension. Chest. 1998;114:213S–24S. doi: 10.1378/chest.114.3_supplement.213s. [DOI] [PubMed] [Google Scholar]
- 27.Leone AM, Valgimigli M, Giannico MB, Zaccone V, Perfetti M, D’Amario D, et al. From bone marrow to the arterial wall: The ongoing tale of endothelial progenitor cells. Eur Heart J. 2009;30:890–9. doi: 10.1093/eurheartj/ehp078. [DOI] [PubMed] [Google Scholar]
- 28.Yoder MC, Ingram DA. Endothelial progenitor cell: Ongoing controversy for defining these cells and their role in neoangiogenesis in the murine system. Curr Opin Hematol. 2009;16:269–73. doi: 10.1097/MOH.0b013e32832bbcab. [DOI] [PubMed] [Google Scholar]
- 29.Peinado VI, Ramirez J, Roca J, Rodriguez-Roisin R, Barbera JA. Identification of vascular progenitor cells in pulmonary arteries of patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2006;34:257–63. doi: 10.1165/rcmb.2005-0255OC. [DOI] [PubMed] [Google Scholar]
- 30.Peinado VI, Santos S, Ramirez J, Roca J, Rodriguez-Roisin R, Barberà JA. Response to hypoxia of pulmonary arteries in COPD: An in vitro study. Eur Respir J. 2002;20:332–38. doi: 10.1183/09031936.02.00282002. [DOI] [PubMed] [Google Scholar]
- 31.Diez M, Barbera JA, Ferrer E, Fernández-Lloris R, Pizarro S, Roca J, et al. Plasticity of CD133+cells: Role in pulmonary vascular remodeling. Cardiovasc Res. 2007;76:517–27. doi: 10.1016/j.cardiores.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 32.Diez M, Musri MM, Ferrer E, Barbera JA, Peinado VI. Endothelial progenitor cells undergo an endothelial-to-mesenchymal transition-like process mediated by TGFbetaRI. Cardiovasc Res. 2010;88:502–11. doi: 10.1093/cvr/cvq236. [DOI] [PubMed] [Google Scholar]
- 33.Arciniegas E, Frid MG, Douglas IS, Stenmark KR. Perspectives on endothelial-to-mesenchymal transition: Potential contribution to vascular remodeling in chronic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1–8. doi: 10.1152/ajplung.00378.2006. [DOI] [PubMed] [Google Scholar]
- 34.Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3:155–66. doi: 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]