

Abstract
Current and past clinical trials in pulmonary hypertension, while valuable, are limited by the absence of mechanistic aims, by dissatisfaction with endpoints and the inability to share data. Clinical studies in pulmonary hypertension might be enhanced by a consortium approach that utilizes the expertise of academic medicine, the treatment initiatives of the pharmaceutical industry and study design from funding agencies interested in biological mechanisms. A meeting of interested parties, the Pulmonary Hypertension Academic Research Consortium (PHARC), was held from 30 April to 1 May 2012 in Bethesda, Maryland. Members at the conference were from the USA Federal Drug Administration (FDA); pharmaceutical industry (Pfizer, Novartis, Bayer and Gilead); USA National Institutes of Health (NHLBI); the Pulmonary Vascular Research Institute (PVRI), a non-governmental organization (NGO); and research and clinical members of pulmonary hypertension programs of international scope. A recommendation to develop a clinical trials consortium was the product of the working group on academic standards in clinical trials. The working group concluded that clinical trials hold immense promise to move the field of pulmonary hypertension forward if the trials are designed by a consortium with input from multiple groups. This would result in study design, conduct and analysis determined by consortium members with a high degree of independent function. The components of a well-balanced consortium that give it scientific effectiveness are: (1) the consortium can work with multiple companies simultaneously; (2) sponsors with special interests, such as testing biological mechanisms, can add investigations to a study at lower cost than with present granting strategies; (3) data handling including archiving, analysis and future sharing would be improved; (4) ancillary studies supported by the collection and dissemination of tissues and fluids would generate a broader approach to discovery than is now possible; and (5) development of improved endpoints in consultation with regulatory agencies, industry and academia would be possible.
Keywords: clinical trial, consortium, Federal Drug Administration, international, National Institutes of Health, Non-Governmental Organization, pulmonary hypertension, Pulmonary Vascular Research Institute, pharmaceutical, industry
Experimental design, data analysis, and publication are the ingredients, product, and currency of scientific research. Because sponsorship of clinical studies varies from industry to foundations to government, issues such as the rights to design the experiment and control data also vary. Many other potential issues of control may limit the quality of a clinical trial. It is difficult, therefore, to propose rules that would apply to all studies. This is why, a consortium approach is a powerful solution to the mixture of interests, goals and needs that differ among stakeholders. In a consortium, each stakeholder has input, but decisions regarding final design and implementation are made by the consortium. In addition, certain methodologies such as data management are agreed on upfront and therefore cannot be breached. It is clear that analysis of data must be scientifically based, without bias, for highest impact, quality and durability of the published observations. In addition, multiple opportunities to revisit data need to be built into a data management agreement allowing for new questions as the field progresses. In the past, sequestration or loss of data because of failure to ensure a proper repository has severely hampered progress. This problem has occurred with government funded research as well as industry funded work. Systems can be agreed to and implemented for the proper collection, evaluation, archiving and sharing of data. These agreements result from dialog and decision making within the study group, not imposed from outside.
High scientific and academic standards are essential in the design, conduct, interpretation and publication of clinical trials.[1] It is mandatory to ensure that all the patients meet all the entry criteria, that the exclusion criteria are not infringed, that the investigations are carried out to the highest standards, that the data are analyzed in an unbiased fashion and that the resulting publication gives an accurate account of the outcome of the trial, including negative data. Secondary analysis should be built into the process. Beyond these fundamental precepts, basic issues that relate to the quality and impact of clinical trials are discussed below.[2,3] These considerations should be built into any consortium agreement that moves forward with clinical studies. Some of the unmet needs and opportunities in pulmonary hypertension (PH) clinical research are bulleted in Figure 1.
Figure 1.

A summary of perceived limitations of current clinical trials in pulmonary hypertension. Approaches to improvement in trial design are built in the consortium approach developed in this paper.
The opportunity for pharmaceutical company resource sharing has been well summarized by Munos and Chin.[4] In their commentary on how to revive breakthrough innovation in the pharmaceutical industry, they note that the volume of unused scientific information generated and sequestered by each company is enormous and that a sharing plan might allow innovation by other companies that could use some of this knowledge and would benefit both institutions. In fact, an early experiment is occurring with a collaboration among Eli Lilly, Merck and Pfizer, called the Asian Cancer Research Group. Companies are sharing pharmacogenomic data on over 2000 tissue samples that will also be shared with academia and made public over time.[5]
What follows is an example for a structured approach to the development of a pulmonary hypertension clinical trial consortium (PHCTC) that addresses the goals of research enumerated above. The overall concept of the relationships and new partnerships within a consortium are shown in Figure 2. Some of this proposed structure has been adapted from the National Institutes of Health (NIH) funded ARDSnet, a consortium of academic centers in the USA that creates, implements and analyzes national trials in the acute respiratory distress syndrome (ARDS).[6]
Figure 2.
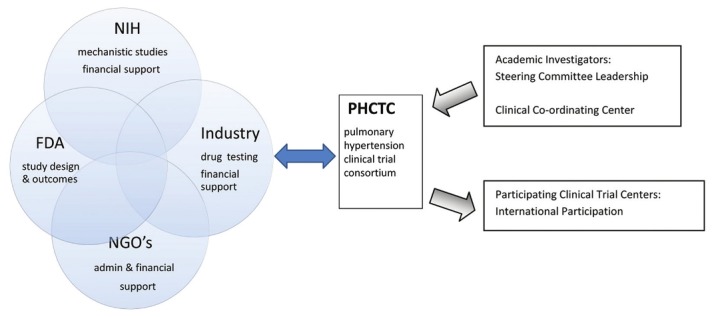
The PHCTC derives input from four separate organizations with overlapping interests, all of whom have a stake in the purpose, design, implementation and outcomes of a clinical trial. The FDA gives input and receives opinion about endpoints and design. NGOs offer administrative, financial and scientific input. The NIH/ medical research council (MRC) or other agencies can fund directly or by offering biological or epidemiological initiatives embedded in the clinical trials. The PHCTC is composed of members of academic pulmonary hypertension centers and international sites are chosen by the PHCTC.
OVERALL STRUCTURE AND FUNCTIONS
A PHCTC would be a consortium of clinical sites selected by meeting certain research standards to participate in multicenter clinical trials. The PHCTC would be composed of leadership from North America, Great Britain and Europe. It would be essential to combine expertise across national boundaries for this to succeed. The core group could be assembled by assessment and agreement of a group brought together by a committee of governmental, industrial, non-governmental organization (NGO) and academic participants. Clinical sites are usually from PH Centers, each having a principal investigator whose main academic function is study and treatment of patients with PH. It is clear that the number of clinical centers ultimately needed to implement a trial will be between 50 and 100 for large trials, so that adjunct participating centers will be needed. The PHCTC's responsibility is to get a system in place that will ensure that the highest research standards are met and provide guidance and facilitate communication among sponsors and research sites. PHCTC funding would be supported by the pharmaceutical or durable goods vendor and co-funded by public and NGO sources to add to scientific inquiry in the studies. Each site's contract specifies a deliverable, a minimum number of patients enrolled in clinical trials annually.
Mission
The mission of the PHCTC is to create and conduct studies to improve and test important clinical outcomes such as survival, function, physical capacity and measures of cardiac and vascular function of patients with PH. Integrated studies of mediators, genomics and tissue responses should be expected as part of any research plan. Protocols with only surrogate outcome endpoints (e.g., 6-min walk alone) and those only examining mechanism or pathophysiology (e.g., biomarker studies) would not likely be considered sufficient as primary studies. Such topics are ancillary studies and typically funded through alternate mechanisms including K, RO1, industry and institutional funds. The PHCTC also acts to assist industry in developing protocols with mechanistic experimental features. An example of a funded ancillary relationship is between the NIH (NHLBI) which is funding 20 RO3 basic science grants using materials generated by the Pulmonary Hypertension Breakthrough Initiative (PHBI), funded by an NGO, and the Cardiovascular Medical Research and Education Fund (CMREF).[7,8]
Depending on the funding sources, applications for studies could be initiated by members of the PHCTC, industry, or by Request for Applications (RFA) from a governmental agency or NGO. These clinical trial proposals will be used to develop collaborative science to enhance clinical trials. Centers must demonstrate they are in compliance with all regulations including those pertaining to nondiscrimination, minorities and all Federal standards, including institutional review board (IRB).
Membership
Clinical sites in leadership are selected for inclusion in PHCTC through a competitive process conducted every five years. The mechanism for choosing is not developed and would depend on the nature of the funding, but could include input from governmental agencies that contribute, NGOs and industry. Criteria for site selection are personnel qualifications, institutional resources and environment, demonstrated ability to successfully conduct clinical research in the population of interest and related populations and the nature of a clinical trial proposal submitted.
The Steering Committee
A Steering Committee is composed of each PHCTC principal investigator (or a surrogate). All PHCTC decisions are made by the Steering Committee using typical rules of order followed by a simple majority vote. The Steering Committee has a nonvoting member appointed by each funding sponsor. The steering committee meets by teleconference monthly and in person at least once annually. An executive committee, consisting of the chairs of the subcommittees and all active protocol chairs, the Steering Committee chair and a representative of the sponsor meet by teleconference monthly to set the agenda for Steering Committee calls and meet ad hoc to manage problems needing broad input. All Steering Committee meetings are structured around subcommittee reports. A possible structural model is shown in Figure 3.
Figure 3.

Possible internal organization of the PHCTC. This is a plan similar to that used by the ARDSnet. A clinical coordinating committee is likely to be a staffed site giving logistic support to the PHCTC and reporting to the Steering Committee. Details within text.
The Steering Committee's major responsibility is to evaluate, select and modify (if appropriate) the protocol(s) that will be conducted by the group. Given the lag time between site selection and protocol selection, additional protocols may be presented that are the result of interim therapeutic developments. All centers must participate in all protocols, i.e., there is no “opting out.” The protocol selection process can be competitive.
Clinical Coordinating Center
A Clinical Coordinating Center (CCC) would be selected to provide logistical support to investigators, which includes organization of meetings and teleconferences of the Steering Committee, its subcommittees and clinical site coordinators and production and maintenance of a website for both the public and investigators. The CCC personnel include nurses experienced in patient care and clinical investigation, a pulmonary critical care physician, biostatisticians and secretarial personnel. The CCC would maintain a 24-hour a day call referral center dedicated to answering questions about patient eligibility and problems with conduct of the protocols. The CCC may assist the funding agency in negotiations for subagreements with vendors, for example, clinical trial material distribution, materials for laboratory functions, laboratory equipment, etc. The CCC would be expected to work closely with the funding body. In the absence of a funded, highly focused CCC, members of the PHCTC will assume responsibility for the functions of a CCC listed above. The CCC may be a virtual center, but it is likely that the key personnel need to be funded and a principle investigator (PI) be available on site with the main administrative person.
The design of data collection forms and electronic programs, anonymity and secure maintenance of all data during studies and its archiving and public access after studies are completed are also the responsibility of the CCC. The PHCTC will use online electronic data entry to expedite data capture, accuracy and analysis and to facilitate quality control through the publication of monthly performance reports. The CCC audits case report form data according to a prespecified plan dependent upon study characteristics, for example, the USA Federal Drug Administration (FDA) registration study. The CCC biostatisticians conduct power calculations, design the a priori data analysis plan and conduct all biostatistical analyses including interim analyses for the data and safety monitoring board (DSMB). For DSMB interim analyses, the CCC provides ad hoc requested analyses. The CCC also produces a final study report. Clinical site investigators are blinded to all clinical trial results until all data are audited and the database is locked and final analysis is conducted. The CCC also conducts exploratory analyses of prior trials for purposes of planning future studies and is available to assist non-network investigators who wish to evaluate public access data from previous trials.
The PHCTC designates a center to be responsible for management of biological samples. Biological samples including blood, urine and DNA are collected by sites in accordance with each protocol. Processed samples collected at each site are batched and shipped to a sponsored storage facility for subsequent access by PHCTC investigators and non-PHCTC investigators. Genomic and proteomic material is extracted and stored at an appropriate facility by subcontract.
Increasing scientific efficiency and power
The PHCTC makes a particular effort for efficiency of studies by employing a design feature of “factorial design trials” whenever scientifically valid. This process allows the simultaneous testing of two strategies in four cells. Other strategies such as adaptive design trials can be considered.[2] Protocols may include FDA approved and unapproved drugs, devices, or simply care protocols. Support and enthusiasm of PH patient and family associations such as the Pulmonary Hypertension Association (www.phassociation.org) can stimulate interest and funding for such endeavors.
STANDING SUBCOMMITTEES
Publications Committee
The Publications Committee is a subcommittee of the Steering Committee. The Publications Committee reviews all PHCTC publications and presentations of PHCTC data and produces a set of presentation slides for the results of all major studies. All projects using PHCTC data or samples authored by PHCTC investigators require manuscript review/approval by the Publications Committee in the time between study completion and when the data may become public domain. Publications in which protocol data are presented from single PHCTC centers are not allowed. A center can take the lead on an ancillary paper, but must go through the PHCTC publications process. These agreements ensure high-quality publications and transparent communications.
Authorship rules and agreements would need to be addressed early in the creation of the PHCTC. One system would list the PHCTC as the author of all “main study” publications.[9] That is, all main publications would come from the group, without a named lead author. A listing of writing committee members and investigators would be included as an appendix. The list of writing committee members should reflect the sequence of responsibility for the text, with the main writing author first on the list. This system is intended to prevent disagreements over the number and order of authors and acknowledges the reality that dozens if not hundreds of people are key to the completion of the study. The ultimate authorship rules would be decided by the Steering Committee. The writing committee is permitted three months to produce the final manuscript from main studies. If this time window is exceeded, the Publications Committee chair may re-assign the writing duties. Other systems can work, but the authorship issue needs to be on the first agenda of the PHCTC Steering Committee.
Investigator-initiated manuscripts and presentations
Examples of investigator-initiated publications or presentations include those generated by probing the database of a primary PHCTC study or any subset; those generated by using clinical material (e.g., blood or urine samples, radiographs, DNA) to perform unique assays or determinations; and those which utilize any PHCTC resource. Publications during the “proprietary period” must have one (or can have more) Steering Committee member of the PHCTC as a coauthor. Named authors will/may assume the primary position on the masthead. In all cases where PHCTC data, samples, funds, or other resources are utilized, the PHCTC will be credited as an author using the terminology “for the Pulmonary Hypertension Clinical Trials Consortium” and membership will be included in the manuscript. Appropriate contract members must also be included. The proprietary period is three years from the closure of a clinical study.
Public domain data
The PHCTC has no authority over non-PHCTC authors when they use public domain data or samples; however, the Publications Committee will provide nonbinding review and comment on any such manuscript at the author's request. It is likely that not all data will necessarily be in the public domain unless sponsored by NIH or other governmental agencies. The Publications Committee has no review or control authority over the production of review articles, chapters, etc., that use only previously published PHCTC data.
Publication and presentation approval process
The Publications Committee will approve all publications and presentations before journal submission or public presentation. Review of submitted draft material by the Publications Committee will occur within four weeks of receipt (two weeks for abstracts). Majority vote by the Steering Committee will be used to resolve disputes in cases where the Publications Committee rejects publications or demands changes that conflict with the authors’ opinions.
ETHICS COMMITTEE
Ethics in clinical trials are governed in large part by Federal (each country's) regulations and by the IRB of each participating institution. The Ethics Committee's main job is to ensure compliance. The Ethics Committee is a subcommittee of the Steering Committee. The main duties are to maintain conflict of interest data for key personnel at all sites and to write the initial template of the consent form for each center. The ethics group reviews the approved consent forms from all PHCTC sites, copies of which are maintained by the CCC. Issues of ethical misconduct or simple issues of ethics interpretation can be heard and initiated by the Ethics Committee and referred to the Steering Committee.
DATA SHARING COMMITTEE
The purpose of the Data Sharing Committee (DSC) is to review and approve or disapprove the use of existing data by network investigators for purposes of ancillary or substudies or exploratory analyses. The DSC committee approves or denies data access for non-PHCTC investigators in the period after studies are completed, but before data become public domain (open period). Denials of use of data to PHCTC investigators can be appealed to the Steering Committee; however, the Steering Committee does not review approved studies. The DSC is designed to prevent scientifically invalid studies from being performed and to prevent more than one group of investigators from working on the same question at the same time. Investigators have 12 months of exclusivity to conduct approved studies before submitting a manuscript to the Publications Committee. If this time window is exceeded, the DSC may approve requests by other investigators to gain control of that data or sample set. Requirements to obtain data/samples through the CCC are as follows: Proposal approved by either DSC or Pathogenesis Committees (approval letter from the committee chair) and IRB approval (or exemption letter). Note: Significant changes to approved proposals will require approval from the corresponding committee. Data variables meeting health insurance protection and portability act (HIPAA) definition of Limited Access Data will require a data use agreement (DUA) signed by the PHCTC Sponsor of proposal (or center PI).
PATHOGENESIS COMMITTEE
The Pathogenesis Committee (PC) is a subcommittee of the Steering Committee. Similar to the DSC, the purpose of this committee is to review and approve or disapprove the use of banked biological samples by network investigators for purposes of substudies or exploratory analyses. The Pathogenesis Committee approves or denies data access for non-PHCTC investigators in the period after studies are completed, but before samples become public domain. Denials of use of samples to PHCTC investigators can be appealed to the Steering Committee; however, the SC does not review approved studies. The PC is designed to prevent scientifically invalid studies from being performed by PHCTC investigators, to prevent duplication of work and to act as good stewards of limited precious resources.
Requirements to obtain data/samples through the CCC include (1) proposal approved by either DS or Pathogenesis Committees (approval letter from the committee chair), and (2) IRB approval (or exemption letter). Note that significant changes to approved proposals will require approval from the corresponding committee. Data variables meeting HIPAA definition of Limited Access Data will require a DUA signed by the PHCTC Investigator Sponsor of proposal (or center PI).
INSTITUTIONAL REVIEW COMMITTEE
The Institutional Review Committee (IRC) is a subcommittee of the Steering Committee. The purpose of the IRC is to deal with suboptimal performance of sites, protocol noncompliance, or misconduct. For underperforming sites, efforts like mentoring, retraining, site visits and counseling are used to improve performance. The IRC can recommend to the sponsor the discontinuation of clinical sites. IRC decisions are not subject to Steering Committee approval.
AD HOC COMMITTEES
Protocol Committee
Each study will have a Protocol Committee consisting of a group of interested investigators, with an internally appointed or elected chair who will write and execute a Steering Committee approved protocol. The Protocol Committee must write the protocol, revise the protocol in accordance with Steering Committee member comments, develop a statistical plan with CCC biostatisticians, work with the Ethics Committee to draft the consent documents and with the protocol review committee and DSMB (see below) come to a final set of approved study documents. It would be the usual practice that the study protocol chair assume the role of the writing committee chair and select members of the committee to write the main study manuscript. Members of the Protocol Committee must be available during the conduct of the study to answer questions about patient's eligibility and protocol conduct and deviations (call coordinated by the CCC).
Protocol Review Committee
The Protocol Review Committee (PRC) is an ad hoc group independent of all PHCTC centers, assembled by and in cooperation with the funding body to review and comment on all penultimate protocols. The Steering Committee and Protocol Committee review PRC recommendations and negotiate an acceptable resolution of disagreements.
Data and safety monitoring board
The sponsor and Steering Committee appoint an independent DSMB comprising clinicians, relevant basic scientists and biostatisticians to provide additional oversight in protocol design and to conduct interim safety and efficacy analyses in accordance with protocol design specifications. The DSMB makes recommendations to the sponsor(s).
SUMMARY
This paper presents and develops what appears to be a highly prescriptive proposal for moving forward with a clinical trials consortium in PH. It is meant to be read as a starting point for discussion and negotiation, rather than as an endpoint with a fixed structure. The example that is fleshed out in this paper is the product of our experience in the current culture of PH clinical trials and an already functioning, highly successful consortium in another disease, ARDS. We hope that this paper will stimulate discussion and movement toward the next generation of clinical trials in PH.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al. ACCF/AHA 2008 Expert Consensus Document on Pulmonary Hypertension. J Am Coll Cardiol. 2009;53:1573–619. doi: 10.1016/j.jacc.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 2.Rich S. Future of Clinical Trials for Pulmonary Hypertension. Circulation. 2011;123:2919–21. doi: 10.1161/CIRCULATIONAHA.111.037762. [DOI] [PubMed] [Google Scholar]
- 3.Corris PA. Clinical Trials in Pulmonary Hypertension. [Last accessed on 28 Feb 2013]. Available from: http://www.pvri.info/content/oct-2012-clinical-trials-pulmonary-hypertension .
- 4.Munos BH, Chin WW. How to Revive Breakthrough Innovation in the Pharmaceutical Industry. Sci Transl Med. 2011;89:1–3. doi: 10.1126/scitranslmed.3002273. [DOI] [PubMed] [Google Scholar]
- 5.Pharma's Asian Syndicate. Nature Biotechnology. 2012;28:297. [Google Scholar]
- 6. Available from: http://www.ardsnet.org/ In order to hasten the development of effective therapy for Acute Respiratory Distress Syndrome (ARDS), the National Heart, Lung and Blood Institute, National Institutes of Health, initiated a clinical network to carry out multi-center clinical trials of ARDS treatments. The ARDS Network was established as a contract program in 1994 following a national competition .
- 7.The Cardiovascular Medical Research and Education Fund. [Last accessed date 28 Feb 2013]. Available from: http://www.ipahresearch.org .
- 8.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;3:261–72. doi: 10.1164/rccm.201201-0164OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.The National Heart, Lung and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Initial Trophic vs Full Enteral Feeding in Patients with Acute Lung Injury: The EDEN Randomized Trial. JAMA. 2012;307:137–46. doi: 10.1001/jama.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]