

Abstract
Pulmonary vascular remodeling and oxidative stress are common to many adult lung diseases. However, little is known about the relevance of lung mesenchymal stem cells (MSCs) in these processes. We tested the hypothesis that dysfunctional lung MSCs directly participate in remodeling of the microcirculation. We employed a genetic model to deplete extracellular superoxide dismutase (EC-SOD) in lung MSCs coupled with lineage tracing analysis. We crossed floxpsod3 and mT/mG reporter mice to a strain expressing Cre recombinase under the control of the ABCG2 promoter. We demonstrated In vivo that depletion of EC-SOD in lung MSCs resulted in their contribution to microvascular remodeling in the smooth muscle actin positive layer. We further characterized lung MSCs to be multipotent vascular precursors, capable of myofibroblast, endothelial and pericyte differentiation in vitro. EC-SOD deficiency in cultured lung MSCs accelerated proliferation and apoptosis, restricted colony-forming ability, multilineage differentiation potential and promoted the transition to a contractile phenotype. Further studies correlated cell dysfunction to alterations in canonical Wnt/β-catenin signaling, which were more evident under conditions of oxidative stress. Our data establish that lung MSCs are a multipotent vascular precursor population, a population which has the capacity to participate in vascular remodeling and their function is likely regulated in part by the Wnt/β-catenin signaling pathway. These studies highlight an important role for microenviromental regulation of multipotent MSC function as well as their potential to contribute to tissue remodeling.
Keywords: endothelial cell, extracellular superoxide dismutase, myofibroblast, niche, pericytes, pulmonary arterial hypertension, resident lung mesenchymal stem cells
During lung development, the mesenchyme influences the development of both the epithelium and distal vasculature.[1–8] The intimacy of this relationship persists into the adult tissue and is recapitulated during organ repair and regeneration.[7,9] However, the function of mesenchymal stem cells (MSCs) in the adult lung during adult pulmonary tissue homeostasis and disease remains to be determined. Understanding these processes is crucial to defining the role lung MSCs play during normal tissue function and the adverse effects of an abnormal microenvironment on the MSCs associated with pathological pulmonary remodeling.
We have previously defined lung MSCs using the side population (SP) phenotype and expression of the ATP-binding cassette subfamily G member 2 (ABCG2). These cells exhibit anti-inflammatory properties and are negatively impacted by bleomycin injury and subsequent fibrosis.[10] However, exogenously administered lung MSCs function to protect lung integrity following bleomycin injury and subsequent oxidative stress, illustrating the importance of this novel population during lung injury.[10] Therefore, similar to other tissue resident stem cells, lung MSCs may regulate their native tissue repair.[7,10–16]
In vitro, lung MSCs are multipotent and differentiate into the mesenchymal lineages including bone, fat and cartilage. Lung MSCs express cell surface determinants CD90, CD105, CD106, CD73, CD44 and ScaI, while lacking the hematopoietic markers CD45, CD14, CD11b, c-kit and CD34.[10,13,15,17,18] Taken together, these tissue resident MSCs are similar to bone marrow MSCs[10,19] as well as MSCs identified in other adult tissues including skeletal muscle, adipose tissue and pancreas.[20] To date, studies on the role of stem cells in lung repair and regeneration have focused almost entirely on exogenously introduced bone marrow mesenchymal stem cell (BM-MSC) or resident epithelial stem cell populations.[9] The role of other resident lung stem cell populations including perivascular MSCs in the vascular remodeling associated with pulmonary disease, including but not limited to pulmonary hypertension or fibrosis, has not been addressed.
One mechanism which may influence lung MSC function during lung disease is oxidative stress. Extracellular superoxide dismutase (EC-SOD) is a secreted enzyme prevalent in lung tissue, which is a major defense against oxidative damage caused by the superoxide anion and lung injuries resulting from oxidative stress including bleomycin-induced fibrosis, hypoxia and inflammation.[21–29] EC-SOD null mice exposed to these stimuli exhibit increased fibrosis and vascular remodeling relative to that observed in wild type (WT) rodents.[30] During pulmonary fibrosis and in response to hypoxia, expression levels of EC-SOD have been shown to decrease.[22,26,27,30,31] Conversely, overexpression of EC-SOD in rodents attenuates pulmonary hypertension and vascular remodeling as a result of bleomycin injury or hypoxic exposure.[26,27] Additional studies have suggested that the aforementioned models utilizing EC-SOD null mice promote compensatory mechanisms that allow mice born without EC-SOD to display only minor alterations in lung histology under basal conditions, including slight septal thickening.[22,30] In contrast, more severe effects are noted when EC-SOD is conditionally knocked out in lung tissue during adulthood In vivo.[22] Even in the absence of injury, conditional EC-SOD null mice exhibit histological properties of acute lung injury, demonstrating a loss of patent alveoli and increased inflammation and mortality.[22] It has therefore been predicted that EC-SOD is essential to protecting against oxidative damage.[27] Despite this knowledge, the causal relationship between EC-SOD, lung MSCs and progression of lung disease is unknown.
Furthermore, a downstream target of oxidative stress regulating the phenotype and function of multipotent MSCs is likely the Wnt signaling pathway. The Wnt family of proteins is a highly conserved group of signaling molecules, β-catenin protein being a central mediator of canonical signaling.[32–38] Activation of the pathway results in altered mesenchymal cell fate specification, as well as pathological angiogenesis, during development and in adulthood.[36,37,39] Chronic activation of β-catenin can result in hypercellularity of tissue and deregulated self-renewal.[34,38] In the lung, Wnt/β-catenin pathway modulates experimental emphysema,[40] and the Wnt inhibitor secreted frizzled related protein-1 (SFRP-1) upregulates the expression of proteases that are important in the development of human emphysema.[41] Conversely, during lung fibrosis, activation of the Wnt pathway is present in proliferative myofibroblast lesions and plays an important role in the fibrotic changes that occur.[42] Though study in this field is just emerging, here we present evidence indicating that the Wnt signaling pathway and its inhibitors will be key determinants of the effects of MSCs within the lung.
In this study, we set out to test two concurrent hypotheses. The first is that dysfunctional lung MSCs participate in remodeling of the distal lung microvasculature. The second one is that the maintenance of proper phenotype and function of lung MSCs is linked, at least in part, to their production of EC-SOD and subsequent regulation of the Wnt/β-catenin pathway. To test these hypotheses, we employed novel engineered murine strains and cell lines exploiting an EC-SOD conditional knockout targeted to lung MSCs using the ABCG2 gene promoter to drive expression of tamoxifen-inducible Cre recombinase.[43] In vivo we documented the participation of lung MSCs in microvascular remodeling and their differentiation to a contractile phenotype in response to oxidative stress. In vitro we show for the first time that lung MSCs are multipotent vascular precursors, capable of differentiation to myofibroblast, endothelial, smooth muscle and pericyte cells. The loss of the lung MSC phenotype and function in vivo as a result of EC-SOD knockout was translated in vitro, where we characterized a loss of stemness, accelerated proliferation and apoptosis, restricted multilineage differentiation potential and the transition to a contractile phenotype. Further studies in vitro and In vivo indicate that alteration from a lung MSC to a contractile phenotype was associated with differences in canonical Wnt/β-catenin signaling. Our data are the first to demonstrate a role for lung MSCs in microvascular remodeling in response to oxidative stress.
MATERIALS AND METHODS
Isolation of lung subpopulations
All procedures and protocols were approved by Institutional Animal Care and Use Committee at the University of Colorado and Vanderbilt University. Lung MSCs were isolated from ABCG2 Cre-ERT2 × mT/mG mice (termed ABCG2 mice, cells WT lung MSC) and ABCG2 Cre-ERT2 × mT/mG × floxpsod3 mice (termed A/SOD mice, cells KO lung MSC); lung fibroblasts (WT FB) were isolated from the same aliquots of this single cell suspension and presumptive pericytes, NG2 ds red lung cells, were isolated from Cspg4 ds red mice (JAX stock 008241; termed NG2) by flow cytometry. The cells were sorted, cultured and phenotyped as described previously.[10,30,17,24] Two to three separate analyses were performed on 30,000-50,000 cells per sample. Single cell suspensions of Hoechst-stained lung tissue, or tamoxifen-induced ABCG2 or A/SOD, were stained with antibodies to the cell surface markers indicated in the figure legend [Methods Table 1]. Gates were set using fluorescent minus one (FMO) controls. Phenotypic analyses were repeated twice independently. Gating strategies included FSC/SSC, dead cell exclusion with either propidium iodide or 4’,6-diamidino-2-phenylindole (DAPI), red blood cell exclusion with Ter119 and doublet discrimination. Controls for flow cytometry included Hoechst-stained BM, unstained cells and cell suspensions incubated with conjugated isotype-matched control antibodies. Hoechstlow CD45neg lung MSCs were sorted using a Legacy Moflo cell sorter with Summit 4.3 software (Beckman Coulter, Miami, Fla., USA). Sort mode was set to Purify 1. BAL and lung MSCs were analyzed on a CyAn ADP flow cytometer (Beckman Coulter). Multilineage differentiation and characterization of lung MSCs to adipocyte, chondrocyte and osteoblast lineages was performed as previously described.[10,17,44,45] To induce endothelial differentiation, the sorted cells were plated on gelatin-coated plastic and were cultured in endothelial growth medium (EGM) medium (Lonza Walkersville, MD,) until the cells were confluent. Cells were then incubated with Alexa 488-labeled AcDiLDL (Life Technologies Grand Island, NY) as previously described,[46] and positively stained cells were enriched by flow cytometry. Cells were expanded and phenotyped as presented. Rat lung microvascular endothelial cells (MVEC) were a gift from Dr. Alvarez. Smooth muscle differentiation was performed using smooth muscle growth medium-2 (Lonza, Walkersville, Inc.) for 24 days. NG2-positive pericyte differentiation was carried out using low oxygen (6%) and MEM-α + 20% fetal calf serum for 7-14 days.
Methods Table 1.
Reagents used in analysis
Multipotent differentiation analysis of lung subpopulations
Mesenchymal differentiation potential of cells at Passage 6 was performed to determine the multipotential ability as described.[10,17] For colony forming unit, fibroblast assay (CFU-F) cells were diluted in differentiation media (Stem Cell Technologies, Vancouver, BC, Canada) to a final concentration of 1 × 106 cells/mL. Serial dilutions were performed to obtain final concentrations of 6 × 104 cells/dish, 3 × 104 cells/dish, 1.5 × 104 cells/dish and 0.75 × 104 cells/dish, with 10 mL total media in each 100-mm dish. To increase the levels of β-catenin and activate Wnt signaling, lithium chloride was added to the media, which was changed on Day 5. Analyses were performed in duplicate and were repeated. Cells were cultured for 10 days at 37°C in an incubator with 5% CO2 and >95% humidity. On Day 10, cells were washed with phosphate-buffered saline (PBS) and then fixed using 100% methanol for five minutes at room temperature. CFU-F colonies were detected by staining with 0.4% w/v Giemsa staining solution (SIGMA Chemical Co, St. Louis, Mo., USA) diluted 1:20 with deionized water and an inverted microscope was used to quantify the number of colonies. T-cell proliferation assays were performed twice independently as described.[10]
Proliferation and apoptosis of lung mesenchymal stem cells
To detect differences in proliferation, cell cycle and apoptosis, four 6-well plates were seeded with 50,000 cells/well in Minimum Essential-alpha (MEM-α) media (Thermo Scientific, Logan, Utah, USA) supplemented with 20% fetal bovine serum (FBS) and were incubated overnight. Samples of WT and EC-SOD KO lung MSCs were analyzed in triplicate and assays repeated thrice. At 0 hours, 24 hours, 48 hours and 72 hours, the cells were trypsinized and washed with ice-cold PBS. Viable MSCs were counted using a hemocytometer and trypan blue exclusion.[47] Apoptosis was detected using the standard protocol for YO-PRO-1, Vybrant Apoptosis Assay (Invitrogen, Carlsbad, Calif., USA). The Krishan/propidium iodide method was used to perform cell cycle analysis. Data were collected using a FacsCalibur with Cellquest software (Becton Dickenson, San Jose, Calif., USA) and analyzed using Summit software (Cytomation, Ft. Collins, Colo., USA).[47]
In vivo model of hypobaric hypoxic pulmonary arterial hypertension
ABCG2 mice[43] and A/SOD[22] incorporated a fluorescent enhanced green fluorescent protein (eGFP) reporter (mT/mG) to facilitate lineage tracing analysis. Matched controls for the experiments were floxpsod3mice (floxp). Mice were injected intraperitoneally at six to eight weeks of age with 1 mg tamoxifen (T-5648; SIGMA, St. Louis, Mo., USA) in sesame oil or sesame oil alone (vehicle control). For lineage tracing analysis, mice were injected with 4 mg on five consecutive days (20 mg total) or 1 mg in a single dose. In all experiments, mice were exposed two weeks after injection to either ambient air (Pb = 630 mmHg; 5260 feet) or hypobaric hypoxia (Pb~400 mmHg; 16,000 feet) for 5 weeks. Right ventricular (RV) pressures were indirectly measured and analyzed as previously described.[10] Five independent experiments were pooled for the hemodynamic measurements. Independent mice were analyzed for each measurement (RA: A/SOD vehicle n = 15; floxp gene + TAM n = 17; A/SOD + TAM n = 18; HY: A/SOD vehicle n = 16; floxp gene + TAM n = 16; A/SOD + TAM n = 19). Peripheral blood was collected and analyzed to determine hematocrit (HCT).
Histological analysis, including ABCG2-positive lung vessel density, muscularization, vessel wall thickness and imaging was performed as described.[10,46,48,49] Paraffin-embedded mouse lung sections were treated using a standard processing method for immunostaining and incubated with primary antibody smooth muscle alpha actin (SMA), Factor VIII (1:500; DAKO, Ft. Collins, Colo., USA), NG2 (1:200; Millipore, Billerica, Mass., USA), or ABCG2 (1:100; BD Pharmingen, SanDiego, Calif., USA) overnight followed by Alexa 488 or 594 fluorescent secondary antibody (1:500, Invitrogen, Carlsbad, Calif., USA).[10,46,49] All antibodies were diluted in a blocking buffer (tris-buffered saline tween with 10% fetal calf serum) and controls consisted of primary isotype with secondary antibody or secondary antibody only.
Transcriptome analysis
Total RNA was prepared with PicoPure RNA Isolation Kit reagents (Arcturus Bioscience Inc., Mountain View, Calif., USA) from two independently isolated cultures of WT lung MSCs (n = 2), KO lung MSCs (n = 2), or WT FB (n = 2). Array analysis and quantitative reverse transcription polymerase chain reaction (qRT-PCR) validation was performed as described[44] (primer list is provided in Methods Table 1).
Data analysis/statistics
Data are expressed as mean ± standard error using the JMP 5.0 statistical package (SAS, Cary, N.C., USA). Comparisons between groups were made by one-way analysis of variance (ANOVA) followed by Tukey honestly significant difference (HSD) post-hoc analysis (two groups), or Fisher's protected least significant difference (PLSD) (more than two groups) was used to test if there were statistical differences between treatment groups at a significance level of α = 0.05. Differences between treatment groups were considered significant at P < 0.05, P < 0.05*, P < 0.01** and P < 0.001***.
RESULTS
Targeted deletion of EC-SOD in ABCG2pos lung mesenchymal stem cells promotes their direct contribution to vascular remodeling under ambient air and during hypobaric induced pulmonary arterial hypertension
The recent exploitation of the multidrug-resistant transporter ABCG2 expression by lung MSCs and other resident tissue stem cell populations has facilitated the study of these cells In vivo for the first time.[10,43,50,51] ABCG2 is a half-transporter, thus cytoplasmic protein expression does not constitute membrane function or active transcription, as necessary to achieve side population phenotype.[43,51] Generally, ABCG2 expression in lung has been characterized by immunostaining of paraffin tissues as present in the cytoplasm of epithelium, endothelium and mesenchyme.[52] However, functional ABCG2 transporter has been shown to identify progenitors, not differentiated cell types, for the tissues evaluated.[10,18,46,53–56] In addition, recent studies of muscle and hematopoietic cells exploiting ABCG2-driven Cre employed doses of tamoxifen ranging from 20 mg and greater[43] and labeled vascular endothelium and progenitors with lacZ. We therefore titrated our dose of tamoxifen and used a more efficient reporter, an inducible membrane eGFP, to identify single cell populations typically overlooked with conventional lacZ staining and low activity promoters. Here, using ABCG2 mouse lung, we show that high-dose tamoxifen labels both vascular EC and mesenchymal cells at the alveolar capillary interface of the distal lung (Fig. 1A and B) in contrast to low-dose tamoxifen which labels single mesenchymal cells also localized to the alveolar capillary interface (Fig. 1C and D). Colocalization of these cells with Factor VIII is likely a rare event in that dual labeled cells were not frequently detected by immunostaining (Fig. 1E and F) and coexpression of ABCG2 and VE-cadherin was not detectable by flow cytometry (Fig. 2C). These data suggest that the majority of cells labeled using the ABCG2 promoter in this model are lung MSCs. Therefore, the majority of cells detected are MSCs and for reference, the surfactant protein c (SPC) promoter also labels cells not considered alveolar epithelium. Our findings were further confirmed by the in vitro analyses presented in these studies.
Figure 1.
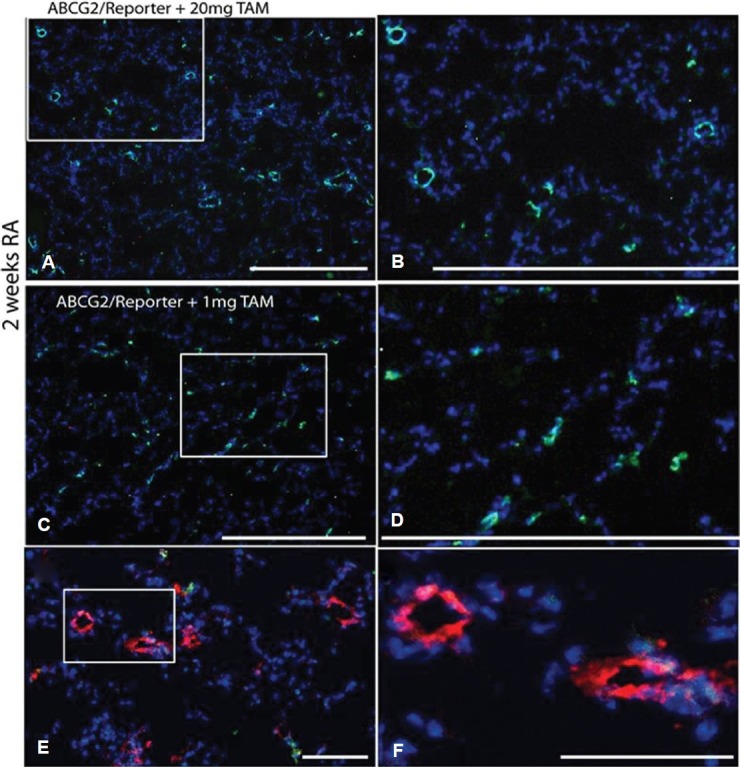
Optimization of lung MSC targeted recombination in vivo. ABCG2 mouse lung 2 weeks post induction using 20 mg or 1 mg of tamoxifen. Recombination results in the appearance of membrane eGFP expression. (A and B) 20 mg dose tamoxifen labels both vascular EC and mesenchymal cells. (C and D) 1 mg tamoxifen induction labels predominantly mesenchymal cells. Scale bars = 500 μM. (E and F) Factor VIII labeling of vascular endothelium (red) demonstrates lack of detectable ABCG2-positive endothelial cell with low-dose tamoxifen. Scale bars = 50 μM.
Figure 2.
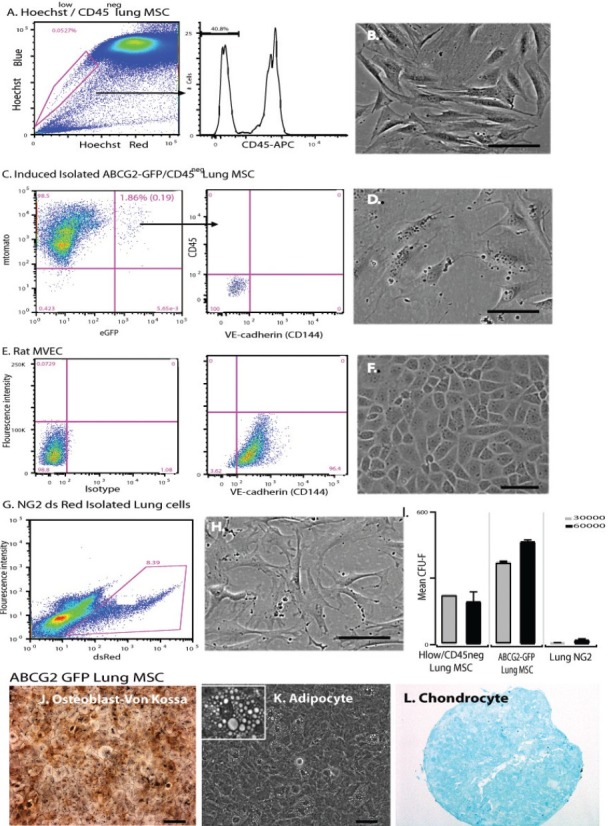
Lung MSCs are distinct from endothelial and NG-2 expressing pericyte populations. Isolation gating strategies and surface determinant analysis of (A) H33342lowCD45neg lung MSCs, (C) ABCG2-eGFPposCD45neg lung MSCs, (E) rat lung microvascular endothelium (MVEC) and (G) NG-2pos ds-red lung cells. Lung MSCs and rat MVEC were stained with anti-CD144. (B, D, F, H) Representative phase micrographs of the isolated cells. (I) Colony forming assay was performed to detect differences in MSC potential between Hoechst 33342lowCD45neg lung MSCs, ABCG2-eGFP lung MSCs and NG-2 ds red lung cells, n = 3, n = 4. Scale bar = 100 μM. (J–L) Multilineage differentiation of ABCG2 lung MSCs to osteocyte, adipocyte and chondrocyte lineages was performed. Osteoblasts were detected by Von Kossa stain (brown/black) and adipocytes by the appearance of fat droplets (100×, 200 × mag.). Chondrocytes were cultured in micromasses, fixed and stained to detect aggrecan (red). Scale bar = 200 μM.
Because turnover of vascular cells in the adult lung is slow, we genetically altered lung MSCs to promote a “dysfunctional” phenotype. We engineered an EC-SOD conditional knockout by crossing the ABCG2 mice with loxpsod3 mice.[22,43,50,51] This novel inducible system, along with titration of tamoxifen dosing, allowed the assay of lung MSC phenotype and function In vivo by knocking out EC-SOD in the cells of interest. The percentage of lung MSCs in a single cell suspension from whole lung tissue was analyzed by flow cytometry two weeks post deletion of EC-SOD. The percentage of lung MSCs was reduced twofold from 1.86% (0.19) in WT to 0.90% (0.19) in KO lung tissue suspensions, suggesting loss or transition of MSCs from this population.
To test the hypothesis that the decrease in lung MSCs was a result of their differentiation to participate in vascular remodeling, we employed the hypobaric hypoxia induced model of pulmonary arterial hypertension (PAH). Using the ABCG2 and A/SOD mice, the expression of eGFP was traced five weeks post exposure to room air or hypoxia (Fig. 3). Contribution of ABCG2-positive green cells to microvessels was evident in the A/SOD hypoxia exposed group (Fig. 3D–F). Conversely, no significant contribution of lung MSCs was detectable in either room air group or hypoxia-exposed ABCG2 mice (Fig. 3A–C). Interestingly, at this stage of analyses, the ABCG2-positive cell contribution to the vessels was only detectable in the A/SOD hypoxia-exposed group. This was due to the complete encircling of vessels by the eGFP-positive cells in this group. However, when colocalization was performed to detect SMA, significant levels of eGFP-positive cells were costained in the microvessels in the ABCG2 and A/SOD lung tissue from the hypoxia-exposed group (Fig. 3H–K), as well as the A/SOD group exposed to ambient air conditions (Fig. 3G and I). SMA labels the contractile cytoskeletal protein associated with pericytes, fibroblasts and smooth muscle. Following SMA staining, lower levels of engraftment were visible. These data support our hypothesis that perivascular lung MSCs contribute to remodeling of the distal lung microvasculature and that the degree of oxidative stress may influence their function and exacerbate their participation.
Figure 3.
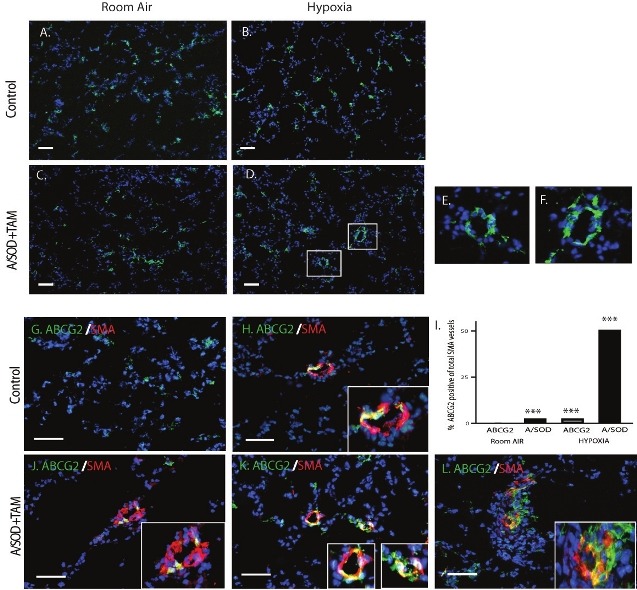
Dysfunctional lung MSCs directly contribute to microvascular remodeling in vivo. ABCG2 mice were exposed to either ambient air pressure (RA) or hypobaric hypoxia (HY) for 5 weeks, 2 weeks post induction. Representative fluorescent micrographs of frozen sections from tamoxifen-induced (A and B) ABCG2 mice and (C-F) A/SOD mice are presented. ABCG2-eGFP lung MSCs were lineage traced via their membrane green fluorescence. ABCG2-eGFPpositive cells were associated with the muscularized microvessels in the KO lung tissue from the hypoxia-exposed group. Boxes represent positive enlarged microvessels presented in panels E and F. (G-L) Dual localization of ABCG2-eGFP cells with the microvasculature was performed by immunostaining to localize SMA (red). Overlap is indicated by yellow color. Enlarged dual positive microvessels are presented in boxes. Scale bar = 30 μM (I). Contribution of ABCG2 cells to the microvasculature was quantitated by counting overlapping green/red signals per total SMA positive vessel 0-50 μM in 30 fov. n = 3-5 mice per group. ***P < 0.0001 relative to the ABCG2 RA group.
Because the deletion of EC-SOD in lung MSCs enhanced their contribution to the muscular layer of microvessels in response to hypobaric hypoxia, we sought to analyze subsequent alterations in physiology and muscularization in this model of PAH. Right ventricular systolic pressure (RVSP) was chosen as a functional measure of PAH. Histologic endpoints analyzed included muscularization and muscular thickening. RVSP was used as an indirect measure of pulmonary artery pressure with increased values indicative of PAH (Fig. 4A). Conditional knockdown of EC-SOD in lung MSCs resulted in increased RVSP associated with PAH following both ambient air exposure and more significantly following a five-week exposure to hypobaric hypoxia. The vehicle and floxed EC-SOD controls exhibited baseline RV pressures under room air conditions and responded appropriately to hypoxia with an increase in RVSP. KO of EC-SOD did not result in significantly altered HCT (not shown), while the expected increase in HCT was present in the RA versus hypoxia-exposed groups (52.5 (1.3) vs. 65.8 (1.6); P < 0.0001). Taken together, these results illustrate that the quantifiable increase in RVSP was not due to increased HCT.
Figure 4.
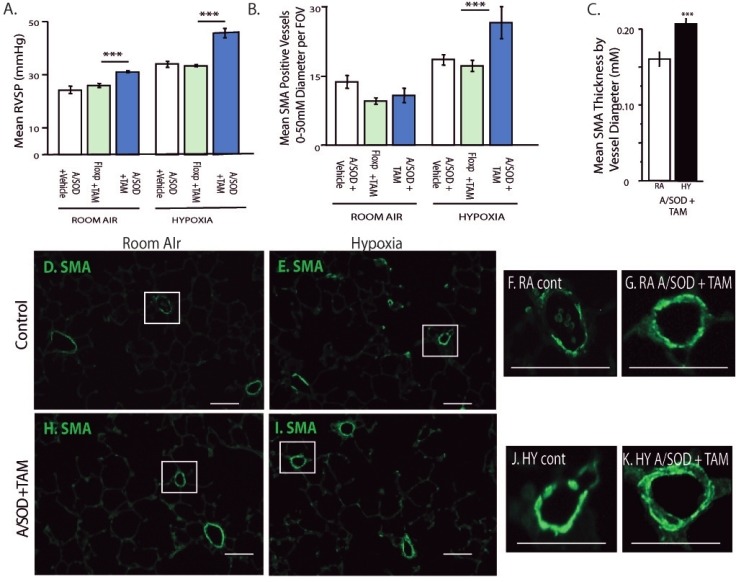
Functional consequences of EC-SOD knockout in lung MSCs include increased muscularization with exacerbated PAH. Physiologic parameters were analyzed to detect how remodeling by MSCs affected PAH. Comparisons are presented between vehicle controls, floxed sod3 + tamoxifen controls and tamoxifen-induced A/SOD mice. In all experiments, 2 weeks after induction, mice were exposed to either ambient air pressure or hypobaric hypoxia for 5 weeks. (A) RVSP was measured via a pressure transducer inserted into the right cardiac ventricle, n = 10, 8, 10, 8, 8, 11. Analysis of the lung tissue was performed to measure histological endpoints associated with MSC remodeling and PAH. (B) Quantitation of muscularization was performed by counting smooth muscle alpha actin (SMA) positive vessels per field of view in paraffin-stained lung sections. Differences were evident in the 0-50 μM diameter vessels; n = 5, 9, 8, 8, 8, 8. (C) The thickness of SMA-positive vessels under 30 μM was measured as a function of vessel diameter; n = 4, 4. (D-K) Representative fluorescent micrographs of SMA (green) stained paraffin lung tissue sections from control and induced (A/SOD + TAM) mice exposed to room air or hypoxia. Enlarged panels of boxed vessels left to right: SMA RA control, RA A/SOD + TAM, hypoxia control, hypoxia A/SOD + TAM; H and E hypoxia A/SOD + TAM. Scale bars = 25 μM.
Muscularization associated with PAH was quantitated by counting SMA-positive vessels ranging in diameter from 0 to 50 μM, from 50 to 100 μM and from 100 to 200 μM (Fig. 4B). As anticipated, with significantly increased RVSP following 5 weeks of hypobaric hypoxic exposure, lung MSC EC-SOD KO mice demonstrated significant increases in muscularization of 0-50 μM diameter microvessels over control groups following hypoxia exposure; no significant differences were observed in the other groups. Thickness of the smooth muscle layers was measured in 0-30 μM diameter microvessels, comparing room air and hypoxia-exposed samples by measuring thickness of the SMA layer relative to the vessel diameter. The thickness of muscularized vessels varied and was 1.3-fold greater in the hypoxia-exposed lung MSC EC-SOD KO mice (Fig. 4C). The contribution of MSCs to remodeling presented in Figure 3 was proportionate to the degree of muscularization associated with exacerbated RVSP and muscularization documented in Figure 4. Taken together, these results suggest that lung MSCs rendered sensitive to oxidative stress function abnormally, resulting in increased vascular remodeling and exacerbated PAH.
CD45neg ABCG2pos lung mesenchymal stem cells are multipotent vascular precursors
We have recently demonstrated that lung MSCs, first studied using H33342 dye efflux and the side population phenotype (Fig. 2A), could be localized in both murine and human tissues using the multidrug-resistant transporter expression, ABCG2.[10,18] However, recent studies have raised the possibility that resident lung MSCs, due to their multipotent nature, may be pericytes.[20,57] Here, we provide evidence that although similar, in the adult lung the multipotent ABCG2 population represents a perivascular endothelial and pericyte precursor population. Here, we confirm the MSC characteristics of ABCG2-positive lung cells, previously reported for the side population of H33342-stained cells. We also compare them to NG2-positive lung cells, presumptive pericytes.
Lung MSCs were isolated from single cell suspensions of ABCG2 lung and presumptive pericytes from NG2 lung cells by flow cytometry (Fig. 2A–H). Rat lung MVEC were used as a positive control for VE-cadherin (CD144) staining (Fig. 2E and F). The ABCG2-eGFP positive cells lack the endothelial marker CD144 which has been reported previously for the H33342-positive MSCs (Fig. 2C[17,46,54]), similar to what was demonstrated in lung tissue staining (Fig. 1). H33342low CD45neg and ABCG2-eGFP positive cells exhibited rigorous CFU-F activity which was absent in the NG-2 lung cells (Fig. 2I). Both ABCG2 and NG2 ds red positive cells differentiated to adipocytes and chondrocytes, but NG2-positive cells did not demonstrate differentiation to osteoblasts (Fig. 2J-L; not shown for NG2 cells).
Global gene expression analysis was then performed to compare the ABCG2 MSCs, WT MSCs (isolated via Hoechst 33342 staining) and NG2-positive population of lung cells (pericytes). A heat map analysis shows distinct differences occur between the lung MSC and NG2 populations of cells (Fig. 5A). Epithelial genetic signature including cytokeratin, keratins and surfactant proteins is absent. Macrophage lineage markers including CD14 and Lys M are also absent. The differentiated endothelial marker VE-cadherin is not detected. Relative expression analysis of genes over-represented in ontogeny and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis included adhesion and matrix production as well as development and are presented in Fig. 5B and C. These data demonstrate the similarities of WT MSCs and ABCG2 MSCs, as well as their distinct similarities and differences to NG2 pericytes. It is likely that MSCs have been inappropriately characterized as pericytes because they are both perivascular and coisolate with the pericyte population of cells. This theory is supported by the enrichment of CFU-F in the lung ABCG2 population over the NG2. Furthermore, multipotent lung MSCs may be a pericyte precursor population.
Figure 5.

Global gene expression profiling illustrates the divergence of lung MSCs from lung NG-2 expressing pericyte populations. Global gene profiling was performed to compare Hoechst 33342lowCD45neg lung MSCs, ABCG2-eGFP lung MSCs, WT FB and NG-2 ds red lung cells. (A) Heat map depiction of relative levels of genes expressed which were overrepresented in ontogeny and KEGG analysis. Ontogeny groups are indicated. Red indicates high levels and blue indicates low levels of gene expression. (B and C) Demonstrate the expression levels of representative genes selected from the analysis and include the Wnt pathway (wisp1, frzb, tbx 20, dlk1), angiogenesis (Flt1, id1, ephb1, ephb2, epha4, nrp2) and the matrix proteins (col18a1, col14a1).
Therefore, we then tested the ability of lung MSCs to differentiate to lineages which have the potential to contribute to vascular tissue homeostasis and disease, including myofibroblasts, endothelium (EC) and pericytes (Fig. 6). Prior to directed differentiation, the lung MSCs lacked markers of differentiated cells including macrophage F480, VE-cadherin, CD146 and NG-2 (Figs. 1, 4C, (Supplemental Figure 1] [Access [Supplemental Figure 1 at www.pulmonarycirculation.org] and Table 1[10,17]). Directed differentiation of MSCs was also performed to the EC lineage. EC were isolated from the mixed population of cells by sorting for DiLDL uptake, a key functional component of EC (Supplemental Fig. 1). The isolated cells demonstrated a cobblestone morphology and the presence of Weibel-Palade bodies by transmission electron microscopy (Fig. 6A and B). The endothelial cells expressed Flt-1, VE-cadherin and exhibited tube-forming ability in vitro (Fig 6C–E). In the presence of platelet derived growth factor B (PDGF)-BB lung, MSCs exhibit a proliferative phenotype with corresponding increased expression of smooth muscle actin, collagen and elastin, as well as decreased Flt-1 and Flk-1 Fig 6F–H and Table 2). Pericyte differentiation was confirmed by the upregulation of NG-2 protein expression (Fig. 6I) as well as increased SMA and NG2 message and expression of rgs5 (Fig. 7). Taken together, the multipotent nature of the ABCG2-positive population of lung MSCs may be considered a candidate population of cells that could participate in tissue repair or remodeling during disease. These results provide justification to the hypothesis that dysfunctional lung MSCs participate in remodeling of the distal lung microvasculature under stress conditions, specifically oxidative stress and also provide an in vitro model with which we may study the cellular mechanisms that regulate lung MSC phenotype and function.
Figure 6.
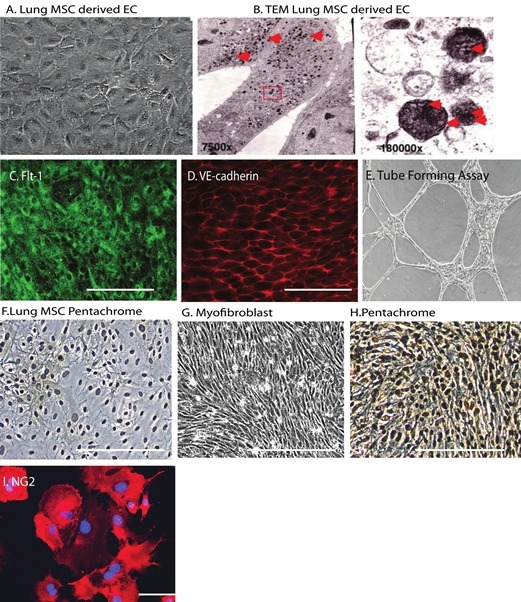
Lung MSCs represent a multipotent vascular precursor population. Endothelial differentiation was documented by (A) cobblestone morphology, the presence of (B) Weibel-Palade bodies by electron microscopy and immunostaining to detect (C) Flt-1 (green), (D) VE-cadherin (red) and an (E) in vitro tube forming assay. (F-H) Bright field image of control WT lung MSCs stained with pentachrome. Phase micrograph of PDGF-BB treated WT lung MSCs and (I) bright field image of pentachrome staining, brown = collagen, black = elastin. Differentiation to a pericyte lineage was documented by NG-2 expression (red). Scale bar = 100 μM.
Supplemental Figure 1.
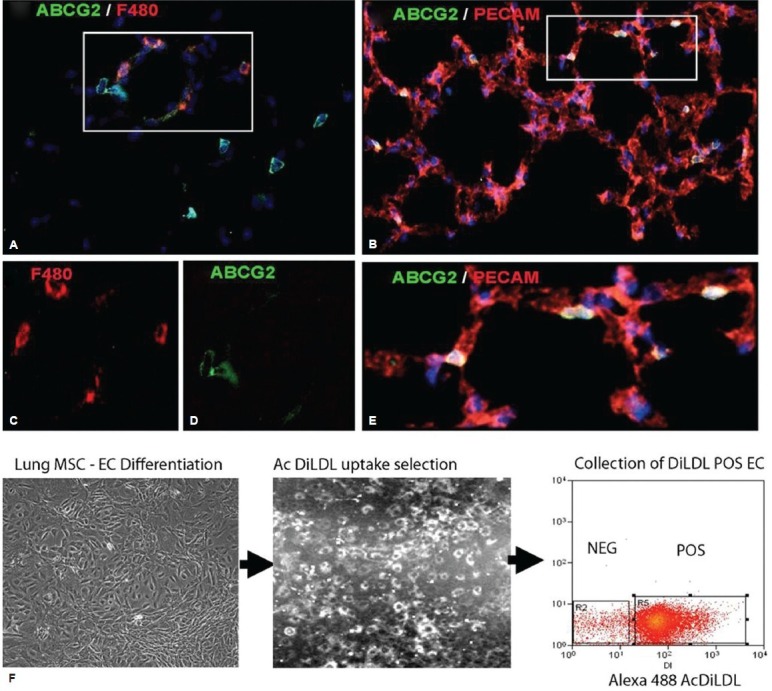
Lung MSC marker expression and selection. ABCG2 mouse lung 2 weeks post induction using 1 mg of tamoxifen. Recombination results in the appearance of membrane eGFP expression. (A, C and D) ABCG2 costaining to detect the macrophage marker F480 (red). (B and E) ABCG2 costaining to detect the PECAM (red). (F) AcDiLDL (conjugated to Alexa 488) was used to label putative MSC.derived EC and select the subpopulation by flow cytometry.
Table 1.
Flow cytometric characterization of lung mesenchymal stem cells
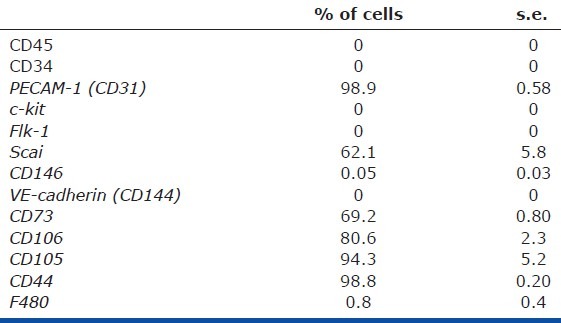
Table 2.
Quantitative polymerase chain reaction lung mesenchymal stem cells copies per 10,000 HPRT (s.e.)
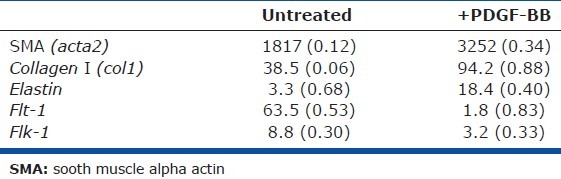
Figure 7.

The canonical WNT pathway regulates lung MSC phenotype and function in response to oxidative stress, driving myofibroblast transdifferentiation. WT and EC-SOD KO lung MSCs and lung FB were isolated from mouse lungs by flow cytometry and expanded in culture. cDNA from each sample was hybridized to Affymetrix mouse whole genome microarrays. (A) Principal component analysis of array data showing clustering of cell populations WT lung MSCs (SP1, 2) and EC SOD KO lung MSCs (SOD KO 1, 2) separate from lung FB (FB1, 2). Supervised hierarchical cluster analysis of genes involved in WNT or TGFb signaling and stemness, following normalization of all data sets. Two independently isolated pooled cultures of WT and KO lung MSCs (n = 2) or lung FB (n = 2) were used for these analyses. (B-D) Quantitative PCR was performed to validate gene expression patterns in isolated lung MSCs identified by microarray analysis. (E) Quantitative PCR was performed to analyze changes in WT versus EC-SOD KO lung MSC gene expression in response to standard conditions (21%; black) or relative hypoxia (6% oxygen; blue) exposure over a period of 72 h; n = 3, 3, 3. (F) Quantitative PCR was performed to detect differences in gene expression in murine lung tissue following room air (black) or hypobaric hypoxia (blue) exposure; n = 3, 3, 3. **Indicates a difference between room air and hypoxia exposed in the same group.
Extracellular superoxide dismutase expression is required to maintain lung mesenchymal stem cells phenotype and function
In order to determine whether loss of the antioxidant enzyme EC-SOD affected lung MSC phenotype, function and turnover in vitro, we isolated and cultured WT and EC-SOD KO lung MSCs. Upon comparison, the WT cells exhibited a characteristic MSC morphology (Fig. 8A) while the EC-SOD null cells appeared more elongated with spindle-like cell processes (Fig. 8B) more similar to a fibroblast (FB). The cells were then analyzed by flow cytometry relative to each other and primary lung FB to detect cell surface determinants characteristic of MSCs (Fig. 8C). The cell populations evaluated were negative for hematopoietic markers CD45, c-kit, CD34 and the macrophage markers CD14, CD11b and F480. All of the cells expressed the mesenchymal markers ScaI at varying levels CD90 and CD44. WT and EC-SOD KO lung MSCs were positive for CD105 and 106, while lung FB did not express significant levels of these characteristic mesenchymal markers. Divergence in the cell surface phenotype between WT and EC-SOD KO lung MSCs was in the expression of CD80, a T-cell regulatory molecule, which was undetectable on EC-SOD KO cells. Interestingly, the KO lung MSCs did not inhibit T-cell proliferation relative to their WT counterparts (Fig. 8D). The KO lung MSC group exhibited the same T-cell proliferation as the group lacking MSCs. Therefore, these results suggest that the previously described[10] anti-inflammatory properties of lung MSCs lacking EC-SOD are altered in vivo.
Figure 8.

EC-SOD regulates the function of lung MSCs. Bright field images of (A and B) representative bright field images of WT and EC-SOD KO lung MSCs; scale bar = 100 μM. (c) WT, KO and lung FB were stained with antibodies to detect cell surface determinants characteristic of MSCs and the resulting histograms overlayed for comparison (blue, red and green, respectively). The gates were set based on FMO controls. (D) In vitro analysis of the effects of WT or KO lung MSCs on T-cell proliferation. (E) Changes in WT (blue) or KO (red) cell number over a period of 0-72 h were quantitated via trypan blue exclusion and automated cell counting of a hemocytometer. Results are presented as total numbers of cells per time point. (F) Apoptosis was measured in the same cell preparations by staining with YoPro/PI and analyzing fluorescence intensity by flow cytometry. WT (blue) or KO (red) is presented as a percentage YoPro-positive cells in early apoptosis of total cells over time. Each experiment was performed in triplicate thrice. (G and H) Colony forming assay was performed to detect differences in MSC potential between WT and KO lung MSCs. Representative bright field photos of assay plates following Giemsa staining for WT and KO are depicted; n = 3, n = 4. Colony morphology varied between the two groups. Scale bar = 0.5 cm. (J-M) Multilineage differentiation of EC-SOD KO lung MSCs to osteocyte, adipocyte and chondrocyte lineages was performed. Osteoblasts were detected by Von Kossa stain (brown/black) and adipocytes by the appearance of fat droplets (100×, 200 × mag.). Chondrocytes were cultured in micromasses, fixed and stained to detect aggrecan (red). Scale bar = 200 μM.
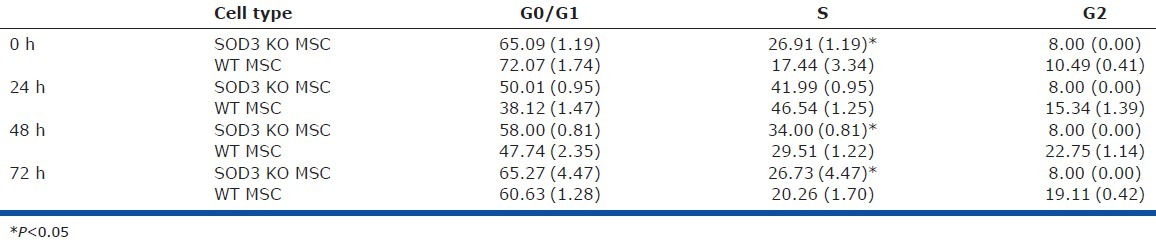
We then analyzed the relative rate of cell turnover by comparing the growth curves of WT and EC-SOD KO lung MSCs in parallel to apoptosis and cell cycle analysis using cell counts and either a YoPro dye-based or propidium iodide-based fluorescent flow cytometric assay (Fig. 8E and F and Table 3). An equal number of cells were plated 12 h prior to the start of the cell counting at time zero and the cells were analyzed to 72 h in culture. We found that EC-SOD KO MSCs had increased cell numbers on 48- and 72-h time points and had significantly higher numbers of cells undergoing apoptosis at 24, 48 and 72 h. These results correlated with cell cycle analysis that demonstrated EC-SOD KO MSCs had a significantly increased number of cells in the S phase of cell cycle at time 0, 48 and 72 h (Table 3).
Table 3.
SOD3 KO lung mesenchymal stem cells have Increased numbers of cells in S phase of the cell cycle
CFU-F assay was performed to enumerate the presence of MSCs in the cultures, which correlates to their potential to propagate and differentiate. The analysis demonstrated a robust decrease in CFU-F potential in the EC-SOD KO MSCs when compared to WT lung MSCs (Fig. 8G–I). In addition, the cell types demonstrated visibly different colony and cell morphology. While the WT MSCs were confluent and compact in a colony, the EC-SOD KO MSCs appeared spindle shaped or fusiform and not uniformly organized within the colony. This significant difference in CFU-F was supported by further multilineage differentiation analyses of the cell populations. WT lung MSCs were able to differentiate into the osteocyte, adipocyte and chondrocyte lineages (Fig. 2J–L)[10,18]), whereas the EC-SOD KO cells were limited to robust differentiation toward the chondrocyte lineage evidenced by the increased size of the micromass (Fig. 8J-M).
Extracellular superoxide dismutase regulates the lung mesenchymal stem cells-contractile cell transition via Wnt signaling
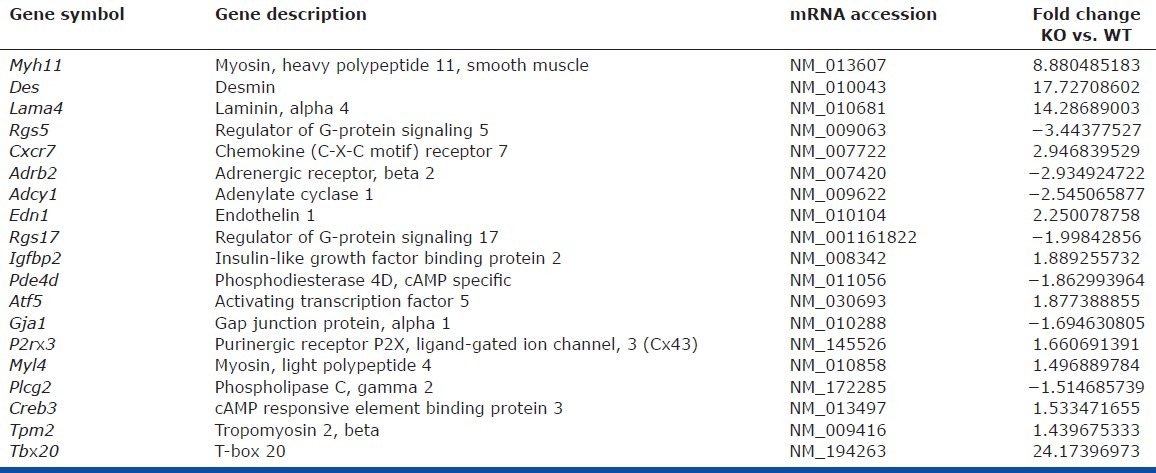
We previously demonstrated that lung MSCs were indeed a distinct population of cells present in the pulmonary stroma relative to lung FB, using global gene expression analysis.[10] In these studies, we further compared the genetic signature of lung MSCs following deletion of EC-SOD. Distinct cell populations were isolated and total RNA was prepared from two independently isolated cultures of WT MSCs, KO lung MSCs, or WT FB. Complimentary DNA generated from amplified RNA was hybridized to duplicate Affymetrix Mouse gene 1.0st chips. Principal component analysis showed distinct differences between each of the three cell populations (Fig. 7A). Each set of pooled duplicate samples segregated to a different region of the diagram, with the KO MSCs falling between the WT MSCs and FB. We also confirmed that EC-SOD KO resulted in the appearance of more contractile-like cells by their increased expression of SMA (acta2) as well as the pericyte marker NG2, relative to WT MSCs (Fig. 7B and C and Table 4). The intermediate phenotype and transcriptional differences in KO MSCs identified by the principle component analysis were also reflected in supervised hierarchical analysis and validation by quantitative polymerase chain reaction (qPCR) of genes associated with Wnt/Tgfβ/stemness (Fig. 7D), immune response and angiogenesis/vasculogenesis. This revealed the KO lung MSC expression of inflammatory mediators (including CxCl14, CxCl12), higher levels of profibrotic genes and decreased levels of angiogenic genes (including col3a1, postn and Flk-1, respectively) relative to WT MSCs.
Table 4.
Differences in contractile gene expression
To further elucidate Wnt signaling as mediating differences between WT and KO lung MSC phenotype and function, we analyzed the cells for expression of key pathway components. All cells expressed the Wnt pathway genes sfrp1, wnt5a and rspo2, although differences in the trends of expression appeared significantly different (Fig. 7D). To better understand how the additional burden of oxidative stress in the absence of EC-SOD expression affects the Wnt signaling pathway, expression levels of cntb (β-catenin), rspo2, sfrp1, wnt5a and sod3, pericyte markers (PDGFRβ, rgs5) as well as foxO1 were analyzed in response to oxidative stress in vitro. We exposed both WT and KO cultured cells to 6% oxygen or relative hypoxia and from time zero to 72 h. RNA was extracted for qPCR analysis used to measure gene expression levels (Fig. 7E). Isolated WT lung MSCs (black) exhibited steadily increased levels of the genes assayed, while the KO lung MSCs (blue) either remained the same as baseline or decreased, illustrating their inability to deal with exogenous oxidative stress. The most significant decreases in the EC-SOD KO lung MSCs were in β-catenin, wnt5a, PDGFRβ and foxO1 levels, while rgs5 was absent. Lung tissue isolated from the mice was analyzed by qPCR following room air or hypobaric hypoxia exposure to compare Wnt signaling effectors cntb (β-catenin), sfrp1 and foxO1 (Fig. 8F). Trends in gene expression following 5 weeks of room air exposure demonstrated an increase in Wnt molecules downstream of EC-SOD deletion in lung MSCs. The converse was found after exposure to hypoxia. Trends in sfrp1 and foxO1 expression were similar to hypoxia exposed EC-SOD KO lung tissue and isolated EC-SOD KO lung MSCs. Both sfrp1 and foxO1 are known to play a role in the Wnt response to oxidative stress in stem cell populations.[58–60]
Because Wnt signaling was implicated in the regulation of lung MSC phenotype and function, we localized β-catenin in conjunction with increased in SMA and NG2 protein. The presence of β-catenin in the nucleus of EC-SOD KO cells and fibroblasts was demonstrated in contrast to the cytoplasmic distribution in WT cells (Fig. 9A and B). The presence of nuclear β-catenin was correlated with increased actin filaments (Fig. 9C) or expression of NG2 in WT MSCs (Fig. 9D). To establish a direct link between increased canonical Wnt signaling and functional alterations in lung MSCs, including loss of stemness, we exposed MSCs to titrated lithium chloride (LiCl) in a luciferase activity assay and CFU-F assay (Fig. 9D–F). LiCl increases the levels of β-catenin by preventing its degradation, subsequently activating Wnt signaling. This was confirmed in the lung MSCs by measuring Wnt signaling activity using a luciferase reporter assay (Fig. 9D). LiCl treatment resulted in decreased CFU activity relative to control. Interestingly, 10 mM LiCl treatment resulted in significantly larger colonies (Fig. 9E and F).
Figure 9.
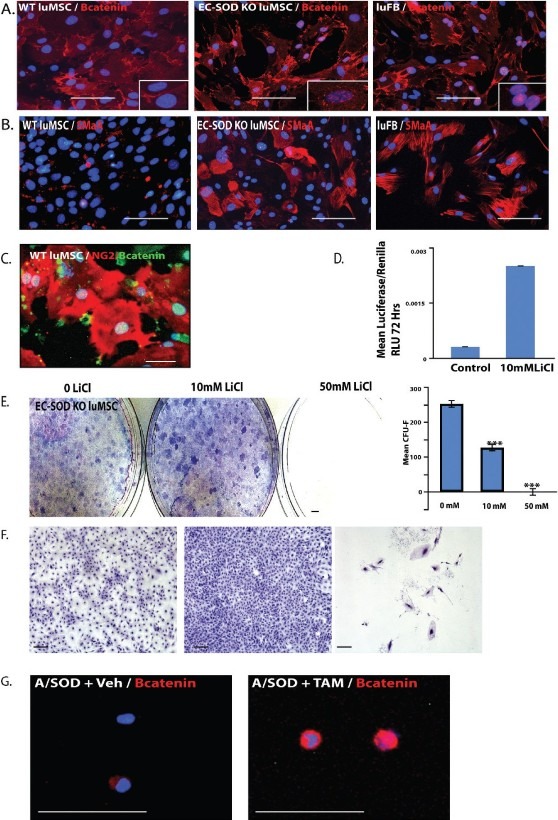
The canonical WNT pathway regulates lung MSC phenotype and function in response to oxidative stress, driving transdifferentiation to a contractile phenotype in vitro and in vivo. (A) WT and KO MSCs and WT FB were immunostained to detect β-catenin protein (red), (B) to localize SMA protein (red) and (C) colocalize nuclear β-catenin with NG-2 expression in WT MSCs. (D and F) Lung MSCs were exposed to LiCl to activate canonical Wnt signaling in a luciferase reporter and CFU-F assay. Representative images and quantitation are presented; n = 3. Scale bars = 0.5 cm or 100 μM. (F) A/SOD mice were induced with vehicle (A/SOD + veh) or tamoxifen (A/SOD + TAM) and lung MSCs isolated from total lung cells by flow cytometry after 96 h. The isolated cells were spun onto slides and stained with antibody to detect β-catenin protein (red); n = 3, 3. Nuclei were costained with DAPI (blue). Scale bar = 30 μM.
To translate these findings In vivo, we employed the novel model of lung MSC conditional EC-SOD KO. We induced A/SOD mice with vehicle or tamoxifen and isolated lung MSCs 96 h later by flow cytometry. Isolated cells were spun onto slides and β-catenin protein localization, analyzed by immunostaining (Fig. 9G). Elevated β-catenin protein was detected in the tamoxifen-induced A/SOD lung MSCs. Taken together, our data indicate that a link exists between the myofibroblast transition of lung MSCs and Wnt signaling.
DISCUSSION
Underlying defects in pulmonary microvasculature contribute to a number of lung diseases including PAH. Although interactions between mesenchyme, endothelium and epithelium are critical in the homeostatic maintenance of the surfaces for gas exchange in the adult organ, little is known about the relevance of lung MSCs in these processes. Therefore, the current study documents several important new findings. We demonstrate that lung MSCs are a multipotent population of vascular precursors which contribute to vascular remodeling in the presence of oxidative stress. Our data show that the expression of EC-SOD by lung MSCs is required for the maintenance of their phenotype and function both In vivo and in vitro. In these studies, we show both in vitro and In vivo that targeted deletion of EC-SOD in lung MSCs promotes their transdifferentiation to a contractile phenotype. We also establish the link between Wnt/β-catenin signaling and the transition of lung MSCs to a contractile cell phenotype. In doing so, we show for the first time that resident lung MSCs play a critical role in the maintenance of normal lung structure and function. Moreover, our data mechanistically link oxidative stress to changes in lung MSC phenotype, via Wnt signaling, that compromise their ability to participate in reparative activities and contribute to the onset of PAH.
In both murine and human lung tissue, MSCs colocalize with the alveolar capillary network in the distal lung, making lung MSCs anatomically similar to adult angioblasts, pericyte and endothelial precursors.[1,20,57,61] This hypothesis was further confirmed by lung MSC perivascular localization, proangiogenic properties, multilineage potential and expression of PDGFRβ smooth muscle alpha actin and acquired expression of VE-cadherin and NG2.[20,57,61–64] Taken together, our phenotyping and functional data both in vitro and In vivo suggest that these lung MSCs participate in vascular homeostasis as well as contribute to vascular remodeling during disease. Targeted knockout of EC-SOD in lung MSCs exacerbated remodeling during PAH and MSC contribution to microvessel. This was demonstrated by enhanced ABCG2-positive cell contribution to thickened actin-positive microvessels and increased RVSP. These results are highly significant because while MSCs from various tissues have been shown to express pericyte markers, they have not been linked functionally to disease pathogenesis.[20,57,63] Therefore, lung MSC expression of EC-SOD plays a pivotal role in these processes.
An important aspect of these studies is the demonstration that lung MSCs respond to oxidant stress and knockdown of the antioxidant enzyme, EC-SOD, via changes in β-catenin/Wnt signaling. Wnt/β-catenin signaling is critical for the maintenance, functionality and lineage specification of bone marrow MSCs[36] and the development and specification of early lung mesenchyme;[1,8] however, its importance in the regulation of lung MSCs is unknown. Our studies show that with genetic deletion of EC-SOD in isolated lung MSCs, there is a concomitant change in cell morphology, increased proliferation, entry into cell cycle and apoptosis, indicative of loss of self-renewal. Additively, the in vitro and In vivo changes in cell properties indicate a transition to actively cycling, more contractile, less stem cell-like cells. Further alterations in lung MSC function as a result of EC-SOD KO and activation of canonical Wnt signaling were demonstrated in vitro. First, EC-SOD KO lung MSCs or MSCs in the presence of LiCl exhibited a loss of CFUs, a hallmark of MSC potential.[64] Secondly, KO cells demonstrated chondrocyte lineage restricted differentiation, in contrast to WT, which differentiated to chondrocytes, osteocyte and adipocyte lineages. These changes, in part, can be attributed to activation of the canonical Wnt signaling pathway, which has been shown to both stimulate chondrogenic and inhibit adipogenic and osteogenic differentiation of bone marrow MSCs and pericytes.[35,36] The studies of isolated MSCs show the direct effect that loss of EC-SOD has on Wnt signaling, regulating their phenotype and function. Further studies will be needed to determine if the loss of EC-SOD leads to altered Wnt signaling as a result of increased extracellular superoxide, secondary to its effects on nitric oxide metabolism or hydrogen peroxide formation, or if loss of EC-SOD activity is distinct from its known enzymatic function, as has been recently proposed.[65,66]
Finally, gene expression analysis was used to explore the underlying mechanisms by which loss of EC-SOD stimulates Wnt signaling and negatively impacts MSC function. In the absence of EC-SOD expression, there was a significant decrease in PDGFRβ and rgs5.[67] In parallel to loss of pericyte markers, we detected a corresponding increase in inflammatory and myofibroblast markers including SMA (acta2), col3a1 and postn. The increases in SMA gene and protein levels by EC-SOD KO cells were coupled to increased nuclear localization of β-catenin, more similar to lung FB than to WT lung MSCs. Interestingly, both of these cells expressed similar levels of wnt5a ligand which inhibits canonical Wnt signaling.[68] Exposure of isolated WT and KO lung MSCs to the additional oxidative stress, chronic hypoxia, demonstrated WT increases in gene expression of β-catenin/Wnt signaling targets including ctnb1, rspo2, sfrp1 and wnt5a. KO cells did not respond appropriately. Interestingly, WT lung MSC expression of sod3 and foxO1 were increased as well. The foxO1 is an important coregulator of β-catenin activity in response to oxidative stress in stem cells.[58,60] Reactive oxygen species increased in the absence of foxO1 expression, whereas, conversely, increased foxO1 transcription as a result of β-catenin/foxO1 binding increases the expression of antioxidant genes including sod3.[58] These interactions between β-catenin/foxO1 also decrease Wnt signaling by diverting the pool of β-catenin from T-cell factor/lymphoid enhancer factor hypoxanthine phosphoribosyltransferase (TCF/LEF) to foxO1 transcription. Increased levels of sfrp1 and wnt5a (as observed in WT MSC) may also antagonize Wnt/β-catenin signaling. These data also suggest that rspo2 may be a modulatory ligand binding frzd8 to activate Wnt signaling in response to oxidative stress and that increased β-catenin/Wnt signaling in the absence of foxO1 influences the lung MSC transition to a prosynthetic and inflammatory myofibroblast. Therefore, both the canonical Wnt/β-catenin pathway and foxO1 likely regulate lung MSC function in response to oxidative stress. While Wnt signaling is necessary to maintain a reparative MSC/pericyte phenotype, a balance in signaling is clearly required to inhibit the pathological transition to a myofibroblast. Further lineage tracing and in vitro molecular analyses will address this possibility.
We have confirmed that lung MSC function as vascular precursors and that targeted deletion of EC-SOD expression increases canonical Wnt signaling which modulates their function during response to oxidative stress both in vitro and in vivo. Alteration of this mechanism results in loss of stemness and the transition to a contractile pericyte/myofibroblast phenotype by lung MSCs and subsequent pathological alterations in pulmonary tissue structure and function. Therefore, multiple functions of lung MSCs may be necessary to maintain tissue integrity. These studies indicate that a broader understanding of the interplay of lung MSCs with the vasculature and epithelium will be required to fully appreciate how paracrine expression of Wnt ligands may alter their function and differentiation during the development of pulmonary diseases.
ACKNOWLEDGMENTS
The authors would like to thank Drs. Patricia D’Amore and Rubin Tuder for critical feedback and discussion, Drs Brian Sorrentino and Soghra Fatima for the gift of ABCG2 CreERT2 mice, as well as Du Hyun Jun and John Psilas for expert technical assistance.
Footnotes
Source of Support: This work was funded by grants to Susan M. Majka: American Heart Association Grant in Aid 0855953G, NIH 1R01 HL091105.01. Additional support was provided by grants to Joshua P. Fessel: NIH T32 HL 094296.02; EPS: NIH 1P30HL101295; EN.G: HL086680; the UCCC Flow Cytometry Core (NIH 5 P30 CA 46934.15), the UCCC Microarray core (NCI P30 CA 46934.14)
Conflict of Interest: None declared.
REFERENCES
- 1.De Langhe SP, Carraro G, Tefft D, Li C, Xu X, Chai Y, et al. Formation and Differentiation of Multiple Mesenchymal Lineages during Lung Development Is Regulated by β-catenin Signaling. PLoS ONE. 2008;3:e1516. doi: 10.1371/journal.pone.0001516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Langhe SP, Carraro G, Warburton D, Hajihosseini MK, Bellusci S. Levels of mesenchymal FGFR2 signaling modulate smooth muscle progenitor cell commitment in the lung. Devel Biol. 2006;299:52–62. doi: 10.1016/j.ydbio.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 3.El-Hashash AH, Al Alam D, Turcatel G, Rogers O, Li X, Bellusci S, et al. Six1 transcription factor is critical for coordination of epithelial, mesenchymal and vascular morphogenesis in the mammalian lung. Dev Biol. 2011;353:242–58. doi: 10.1016/j.ydbio.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mailleux AA, Kelly R, Veltmaat JM, De Langhe SP, Zaffran S, Thiery JP, et al. Fgf10 expression identifies parabronchial smooth muscle cell progenitors and is required for their entry into the smooth muscle cell lineage. Development. 2005;132:2157–66. doi: 10.1242/dev.01795. [DOI] [PubMed] [Google Scholar]
- 5.Ramasamy SK, Mailleux AA, Gupte VV, Mata F, Sala FG, Veltmaat JM, et al. Fgf10 dosage is critical for the amplification of epithelial cell progenitors and for the formation of multiple mesenchymal lineages during lung development. Devel Biol. 2007;307:237–47. doi: 10.1016/j.ydbio.2007.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shannon JM, Nielsen LD, Gebb SA, Randell SH. Mesenchyme specifies epithelial differentiation in reciprocal recombinants of embryonic lung and trachea. Dev Dyn. 1998;212:482–94. doi: 10.1002/(SICI)1097-0177(199808)212:4<482::AID-AJA2>3.0.CO;2-D. 1998. [DOI] [PubMed] [Google Scholar]
- 7.Volckaert T, Dill E, Campbell A, Tiozzo C, Majka S, Bellusci S, et al. Parabronchial smooth muscle constitutes an airway epithelial stem cell niche in the mouse lung after injury. J Clin Invest. 2011;121:4409–19. doi: 10.1172/JCI58097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yin Y, White AC, Huh SH, Hilton MJ, Kanazawa H, Long F, et al. An FGF-WNT gene regulatory network controls lung mesenchyme development. Dev Biol. 2008;319:426–36. doi: 10.1016/j.ydbio.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beers MF. The three R's of lung health and disease: Repair, remodeling and regeneration. J Clin Invest. 2011;121:2065–73. doi: 10.1172/JCI45961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jun D, Garat C, West J, Thorn N, Chow K, Cleaver T, et al. The Pathology of Bleomycin-Induced Fibrosis Is Associated with Loss of Resident Lung Mesenchymal Stem Cells That Regulate Effector T-cell Proliferation. Stem Cells. 2011;29:725–35. doi: 10.1002/stem.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aslam M, Baveja R, Liang OD, Fernandez-Gonzalez A, Lee C, Mitsialis SA, et al. Bone Marrow Stromal Cells Attenuate Lung Injury in a Murine Model of Neonatal Chronic Lung Disease. Am J Respir Crit Care Med. 2009;180:1122–30. doi: 10.1164/rccm.200902-0242OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benvenuto F, Ferrari S, Gerdoni E, Gualandi F, Frassoni F, Pistoia V, et al. Human Mesenchymal Stem Cells Promote Survival of T Cells in a Quiescent State. Stem Cells. 2007;25:1753–60. doi: 10.1634/stemcells.2007-0068. [DOI] [PubMed] [Google Scholar]
- 13.Chateauvieux S, Ichanté JL, Delorme B, Frouin V, Piétu G, Langonné A, et al. Molecular profile of mouse stromal mesenchymal stem cells. Physiol Genomics. 2007;29:128–38. doi: 10.1152/physiolgenomics.00197.2006. [DOI] [PubMed] [Google Scholar]
- 14.Lee RH, Pulin AA, Seo MJ, Kota DJ, Ylostalo J, Larson BL, et al. Intravenous hMSCs Improve Myocardial Infarction in Mice because Cells Embolized in Lung Are Activated to Secrete the Anti-inflammatory Protein TSG-6. Cell Stem Cell. 2009;5:54–63. doi: 10.1016/j.stem.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McQualter JL, Brouard N, Williams B, Baird BN, Sims-Lucas S, Yuen K, et al. Endogenous Fibroblastic Progenitor Cells in the Adult Mouse Lung Are Highly Enriched in the Sca-1 Positive Cell Fraction. Stem Cells. 2009;27:623–33. doi: 10.1634/stemcells.2008-0866. [DOI] [PubMed] [Google Scholar]
- 16.Tsai MS, Hwang SM, Chen KD, Lee YS, Hsu LW, Chang YJ, et al. Functional Network Analysis of the Transcriptomes of Mesenchymal Stem Cells Derived from Amniotic Fluid, Amniotic Membrane, Cord Blood and Bone Marrow. Stem Cells. 2007;25:2511–23. doi: 10.1634/stemcells.2007-0023. [DOI] [PubMed] [Google Scholar]
- 17.Martin J, Helm K, Ruegg P, Varella-Garcia M, Burnham E, Majka S. Adult lung side population cells have mesenchymal stem cell potential. Cytotherapy. 2008;10:140–51. doi: 10.1080/14653240801895296. [DOI] [PubMed] [Google Scholar]
- 18.Summer R, Fitzsimmons K, Dwyer D, Murphy J, Fine A. Isolation of an Adult Mouse Lung Mesenchymal Progenitor Cell Population. Am J Respir Cell Mol Biol. 2007;37:152–9. doi: 10.1165/rcmb.2006-0386OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Méndez-Ferrer SM, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–34. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, et al. A Perivascular Origin for Mesenchymal Stem Cells in Multiple Human Organs. Cell Stem Cell. 2008;3:301–13. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 21.Baber SR, Deng W, Master RG, Bunnell BA, Taylor BK, Murthy SN, et al. Intratracheal mesenchymal stem cell administration attenuates monocrotaline-induced pulmonary hypertension and endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2007;292:H1120–8. doi: 10.1152/ajpheart.00173.2006. [DOI] [PubMed] [Google Scholar]
- 22.Gongora MC, Lob HE, Landmesser U, Guzik TJ, Martin WD, Ozumi K, et al. Loss of Extracellular Superoxide Dismutase Leads to Acute Lung Damage in the Presence of Ambient Air: A Potential Mechanism Underlying Adult Respiratory Distress Syndrome. Am J Pathol. 2008;173:915–26. doi: 10.2353/ajpath.2008.080119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JW, Fang X, Gupta N, Serikov V, Matthay MA. Allogeneic human mesenchymal stem cells for treatment of E.coli endotoxin-induced acute lung injury in the ex vivo perfused human lung. Proc Natl Acad Sci. 2009;106:16357–62. doi: 10.1073/pnas.0907996106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu Z, Xu X, Hu X, Zhu G, Zhang P, van Deel ED, et al. Extracellular Superoxide Dismutase Deficiency Exacerbates Pressure Overload-Induced Left Ventricular Hypertrophy and Dysfunction. Hypertension. 2008;51:19–25. doi: 10.1161/HYPERTENSIONAHA.107.098186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nozik-Grayck E, Suliman HB, Majka S, Albietz J, Van Rheen Z, Roush K, et al. Lung EC-SOD overexpression attenuates hypoxic induction of Egr-1 and chronic hypoxic pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol. 2008;295:L422–30. doi: 10.1152/ajplung.90293.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oury TD, Schaefer LM, Fattman CL, Choi A, Weck KE, Watkins SC. Depletion of pulmonary EC-SOD after exposure to hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2002;283:L777–84. doi: 10.1152/ajplung.00011.2002. [DOI] [PubMed] [Google Scholar]
- 27.Van Rheen Z, Fattman C, Domarski S, Majka S, Klemm D, Stenmark KR, et al. Lung EC-SOD Overexpression Lessens Bleomycin-Induced Pulmonary Hypertension and Vascular Remodeling. Am J Respir Cell Mol Biol. 2010 doi: 10.1165/rcmb.2010-0065OC. 2010-0065OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu D, Guo H, Xu X, Lu Z, Fassett J, Hu X, et al. Exacerbated Pulmonary Arterial Hypertension and Right Ventricular Hypertrophy in Animals With Loss of Function of Extracellular Superoxide Dismutase. Hypertension. 2011;58:303–09. doi: 10.1161/HYPERTENSIONAHA.110.166819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yao H, Arunachalam G, Hwang JW, Chung S, Sundar IK, Kinnula VL, et al. Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc Natl Acad Sci. 2010;107:15571–6. doi: 10.1073/pnas.1007625107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fattman CL, Chang LY, Termin TA, Petersen L, Enghild JJ, Oury TD. Enhanced bleomycin-induced pulmonary damage in mice lacking extracellular superoxide dismutase. Free Radic Biol Med. 2003;35:763–71. doi: 10.1016/s0891-5849(03)00402-7. [DOI] [PubMed] [Google Scholar]
- 31.Gao F, Kinnula VL, Myll√§rniemi M, Oury TD. Extracellular Superoxide Dismutase in Pulmonary Fibrosis. Antioxid Redox Signal. 2008;10:343–54. doi: 10.1089/ars.2007.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baksh D, Boland GM, Tuan RS. Cross-talk between Wnt signaling pathways in human mesenchymal stem cells leads to functional antagonism during osteogenic differentiation. J Cell Biochem. 2007;101:1109–24. doi: 10.1002/jcb.21097. [DOI] [PubMed] [Google Scholar]
- 33.Etheridge SL, Spencer GJ, Heath DJ, Genever PG. Expression Profiling and Functional Analysis of Wnt Signaling Mechanisms in Mesenchymal Stem Cells. Stem Cells. 2004;22:849–60. doi: 10.1634/stemcells.22-5-849. [DOI] [PubMed] [Google Scholar]
- 34.Kalani MY, Cheshier SH, Cord BJ, Bababeygy SR, Vogel H, Weissman IL, et al. Wnt-mediated self-renewal of neural stem/progenitor cells. Proc Natl Acad Sci. 2008;105:16970–5. doi: 10.1073/pnas.0808616105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kirton JP, Crofts NJ, George SJ, Brennan K, Canfield AE. Wnt/β-Catenin Signaling Stimulates Chondrogenic and Inhibits Adipogenic Differentiation of Pericytes. Circ Res. 2007;101:581–9. doi: 10.1161/CIRCRESAHA.107.156372. [DOI] [PubMed] [Google Scholar]
- 36.Ling L, Nurcombe V, Cool SM. Wnt signaling controls the fate of mesenchymal stem cells. Gene. 2009;433:1–7. doi: 10.1016/j.gene.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 37.Reya TC. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–50. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 38.Reya TD, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–14. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- 39.Dejana E. The Role of Wnt Signaling in Physiological and Pathological Angiogenesis. Circ Res. 2010;107:943–52. doi: 10.1161/CIRCRESAHA.110.223750. [DOI] [PubMed] [Google Scholar]
- 40.Kneidinger N, Yildirim AO, Callegari L, Takenaka S, Stein MM, Dumitrascu R, et al. Activation of the WNT/beta-Catenin Pathway Attenuates Experimental Emphysema. Am J Respir Crit Care Med. 2011;183:723–33. doi: 10.1164/rccm.200910-1560OC. [DOI] [PubMed] [Google Scholar]
- 41.Foronjy R, Imai K, Shiomi T, Mercer B, Sklepkiewicz P, Thankachen J, et al. The divergent roles of secreted frizzled related protein-1 (SFRP1) in lung morphogenesis and emphysema. Am J Pathol. 2010;177:598–607. doi: 10.2353/ajpath.2010.090803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Konigshoff M, Kramer M, Balsara N, Wilhelm J, Amarie OV, Jahn A, et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J Clin Invest. 2009;119:772–87. doi: 10.1172/JCI33950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fatima S, Zhou S, Sorrentino BP. Abcg2 Expression Marks Tissue-Specific Stem Cells in Multiple Organs in a Mouse Progeny Tracking Model. Stem Cells. 2012;30:210–21. doi: 10.1002/stem.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Majka SM, Fox KE, Psilas JC, Helm KM, Childs CR, Acosta AS, et al. De novo generation of white adipocytes from the myeloid lineage via mesenchymal intermediates is age, adipose depot and gender specific. Proc Natl Acad Sci. 2010;107:14781–6. doi: 10.1073/pnas.1003512107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Haaften T, Byrne R, Bonnet S, Rochefort GY, Akabutu J, Bouchentouf M, et al. Airway Delivery of Mesenchymal Stem Cells Prevents Arrested Alveolar Growth in Neonatal Lung Injury in Rats. Am J Respir Crit Care Med. 2009;180:1131–42. doi: 10.1164/rccm.200902-0179OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Irwin D, Helm K, Campbell N, Imamura M, Fagan K, Harral J, et al. Neonatal lung side population cells demonstrate endothelial potential and are altered in response to hyperoxia-induced lung simplification. Am J Physiol Lung Cell Mol Physiol. 2007;293:L941–51. doi: 10.1152/ajplung.00054.2007. [DOI] [PubMed] [Google Scholar]
- 47.Majka S, Fox K, McGuire B, Crossno J, McGuire P, Izzo A. Pleiotropic role of VEGF-A in regulating fetal pulmonary mesenchymal cell turnover. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1183–92. doi: 10.1152/ajplung.00175.2005. [DOI] [PubMed] [Google Scholar]
- 48.Jackson KA, Majka SM, Wang H, Pocius J, Hartley CJ, Majesky MW, et al. Regeneration of ischemic cardiac muscle and vascular endothelium by adult stem cells. J Clin Investig. 2001;107:1395–402. doi: 10.1172/JCI12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klemm DJ, Crossno JT Jr, Psilas JC, Reusch JE, Garat CV. Reduction of reactive oxygen species prevents hypoxia-induced CREB depletion in pulmonary artery smooth muscle cells. J Cardiovasc Pharmacol. 2011;58:181–91. doi: 10.1097/FJC.0b013e31821f2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tadjali M, Zhou S, Rehg J, Sorrentino BP. Prospective Isolation of Murine Hematopoietic Stem Cells by Expression of an Abcg2/GFP Allele. Stem Cells. 2006;24:1556–63. doi: 10.1634/stemcells.2005-0562. [DOI] [PubMed] [Google Scholar]
- 51.Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7:1028–34. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
- 52.Summer R, Kotton DN, Sun X, Ma B, Fitzsimmons K, Fine A. Side population cells and Bcrp1 expression in lung. Am J Physiol Lung Cell Mol Physiol. 2003;285:L97–104. doi: 10.1152/ajplung.00009.2003. [DOI] [PubMed] [Google Scholar]
- 53.Giangreco A, Shen H, Reynolds SD, Stripp BR. Molecular phenotype of airway side population cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L624–30. doi: 10.1152/ajplung.00149.2003. [DOI] [PubMed] [Google Scholar]
- 54.Majka SM, Beutz MA, Hagen M, Izzo AA, Voelkel N, Helm KM. Identification of Novel Resident Pulmonary Stem Cells: Form and Function of the Lung Side Population. Stem Cells. 2005;23:1073–81. doi: 10.1634/stemcells.2005-0039. [DOI] [PubMed] [Google Scholar]
- 55.Reynolds SD, Shen H, Reynolds PR, Betsuyaku T, Pilewski JM, Gambelli F, et al. Molecular and functional properties of lung SP cells. Am J Physiol Lung Cell Mol Physiol. 2007;292:L972–83. doi: 10.1152/ajplung.00090.2006. [DOI] [PubMed] [Google Scholar]
- 56.Summer R, Kotton DN, Liang S, Fitzsimmons K, Sun X, Fine A. Embryonic Lung Side Population Cells Are Hematopoietic and Vascular Precursors. Am J Respir Cell Mol Biol. 2005;33:32–40. doi: 10.1165/rcmb.2005-0024OC. [DOI] [PubMed] [Google Scholar]
- 57.Armulik A, Genové G, Betsholtz C. Pericytes: Developmental, Physiological and Pathological Perspectives, Problems and Promises. Dev Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 58.Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. Functional Interaction Between ß-Catenin and FOXO in Oxidative Stress Signaling. Science. 2005;308:1181–4. doi: 10.1126/science.1109083. [DOI] [PubMed] [Google Scholar]
- 59.Goertz MJ, Wu Z, Gallardo TD, Hamra FK, Castrillon DH. Foxo1 is required in mouse spermatogonial stem cells for their maintenance and the initiation of spermatogenesis. J Clin Investig. 2011;121:3456–66. doi: 10.1172/JCI57984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, et al. FoxOs Are Critical Mediators of Hematopoietic Stem Cell Resistance to Physiologic Oxidative Stress. Cell. 2007;128:325–39. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 61.Fuxe J, Tabruyn S, Colton K, Zaid H, Adams A, Baluk P, et al. Pericyte Requirement for Anti-Leak Action of Angiopoietin-1 and Vascular Remodeling in Sustained Inflammation. Am J Pathol. 2011;178:2897–909. doi: 10.1016/j.ajpath.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Feng JM, Sharpe PT. Perivascular cells as mesenchymal stem cells. Expert Opin Biol Ther. 2010;10:1441–51. doi: 10.1517/14712598.2010.517191. [DOI] [PubMed] [Google Scholar]
- 63.Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM. Abnormalities in Pericytes on Blood Vessels and Endothelial Sprouts in Tumors. Am J Pathol. 2002;160:985–1000. doi: 10.1016/S0002-9440(10)64920-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nombela-Arrieta CR, Silberstein LE. The elusive nature and function of mesenchymal stem cells. Nat Rev Mol Cell Biol. 2011;12:126–31. doi: 10.1038/nrm3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee JS, Lee Ys, Jeon B, Jeon Yj, Yoo H, Kim TY. EC-SOD induces apoptosis through COX-2 and galectin-7 in the epidermis. J Dermatol Sci. 2012 doi: 10.1016/j.jdermsci.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 66.Lob HE, Vinh A, Li L, Blinder Y, Offermanns S, Harrison DG. Role of Vascular Extracellular Superoxide Dismutase in Hypertension. Hypertension. 2011;58:232–9. doi: 10.1161/HYPERTENSIONAHA.111.172718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Manzur M, Ganss R. Regulator of G Protein Signaling 5: A New Player in Vascular Remodeling. Trends Cardiovasc Med. 2009;19:26–30. doi: 10.1016/j.tcm.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 68.Nemeth MJ, Topol L, Anderson SM, Yang Y, Bodine DM. Wnt5a inhibits canonical Wnt signaling in hematopoietic stem cells and enhances repopulation. Proc Natl Acad Sci. 2007;104:15436–41. doi: 10.1073/pnas.0704747104. [DOI] [PMC free article] [PubMed] [Google Scholar]