

Abstract
Pulmonary heart disease (PHD) refers to altered structure or function of the right ventricle occurring in association with abnormal respiratory function. Although nearly always associated with some degree of PH, the degree, nature, severity, and causality of PH in relation to the PHD is not necessarily linear and direct. Abnormal gas exchange is a fundamental underpinning of PHD, affecting pulmonary vascular, cardiac, renal, and neurohormonal systems. Direct and indirect effects of chronic respiratory disease can disrupt the right ventricular-pulmonary arterial (RV-PA) interaction and, likewise, factors such as sympathetic nervous system activation, altered blood viscosity, and salt and water retention can function in a feedback loop to further influence RV-PA function. Left heart function may also be affected, especially in those with pre-existing left heart disease. Thus, the physiologic interactions between abnormal respiratory and cardiovascular function are complex, with PHD representing a heterogeneous end organ effect of an integrated multisystem process. In this review, we propose to separate PHD into two distinct entities, “Type I” and “Type II” PHD. Type I PHD is most common, and refers to subjects with chronic respiratory disease (CRD) where the perturbations in respiratory function dominate over more mild cardiac and circulatory disruptions. In contrast, Type II PHD refers to the smaller subset of patients with more severe pulmonary vascular and right heart dysfunction, whom often present in a fashion similar to patients with PAH. Phenotypic differences are not made by PA pressure alone, but instead by differences in the overall physiology and clinical syndrome. Thus, key differences can be seen in symptomatology, physical signs, cardiac imaging, hemodynamics, and the cardiovascular and gas exchange responses to exercise. Such key baseline differences in the overall physiologic phenotype are likely critical to predicting response to PH specific therapy. Recognizing PHD as distinct phenotypes assists in the necessary distinction of these patients, and may also provide a key clinical and pathophysiologic framework for improved patient selection for future studies investigating the role of pulmonary hypertension-specific therapies in PHD.
Keywords: pulmonary heart disease, cor pulmonale, pulmonary hypertension, group 3 pulmonary hypertension, pulmonary vascular disease, chronic respiratory disease, right ventricular function, exercise physiology, ventilatory inefficiency
The term “cor pulmonale” classically refers to the associated hypertrophic remodeling of the right ventricle (RV) that may accompany a variety of chronic respiratory diseases.[1] Pulmonary heart disease (PHD) may be considered a more modern definition of this entity, generally referring to altered structure and/or function of the right ventricle occurring in association with abnormal respiratory function. Pulmonary heart disease is nearly always associated with pulmonary hypertension (PH), which falls under the World Health Organization's Group III category-pulmonary hypertension “Associated with Lung Diseases and/or Hypoxemia.” Many prefer the term chronic respiratory disorder (CRD) as this description accounts for chronic obstructive pulmonary diseases (COPD), interstitial lung diseases (ILD), and the hypoventilatory disorders.[2,3] Pulmonary arterial hypertension (PAH; WHO Group I) and chronic thromboembolic disease (WHO Group IV) are diseases largely isolated to the pulmonary vasculature, and are thus excluded from discussions on PHD. However, PAH and thromboembolic-related PH are “prototype” conditions, where pulmonary vascular disease and its imposed limitations on the RV in part provide a framework for our understanding of the ways in which altered lung structure and function can affect the heart and, moreover, how treatment of these conditions leads to predictable improvements in right heart function, thus translating into improvements in functional status and outcome in these patients.
The physiological interactions between abnormal respiratory and cardiac function are complex, with pulmonary heart disease often representing an end-organ effect of an integrated multisystem process. The phenotypes of PHD vary widely, owed to the heterogeneity of CRD, the degree and nature of associated pulmonary hypertension, and the varied manner in which these processes interact with and alter cardiac function. An appreciation for the direct and indirect physiological consequences of abnormal respiratory function on cardiac function is required to best appreciate PHD (Fig. 1). Although PHD is primarily a “right heart” issue leading to perturbations in RV size, shape, and function, we will also discuss ways in which abnormal respiratory function can exacerbate left heart disease.
Figure 1.
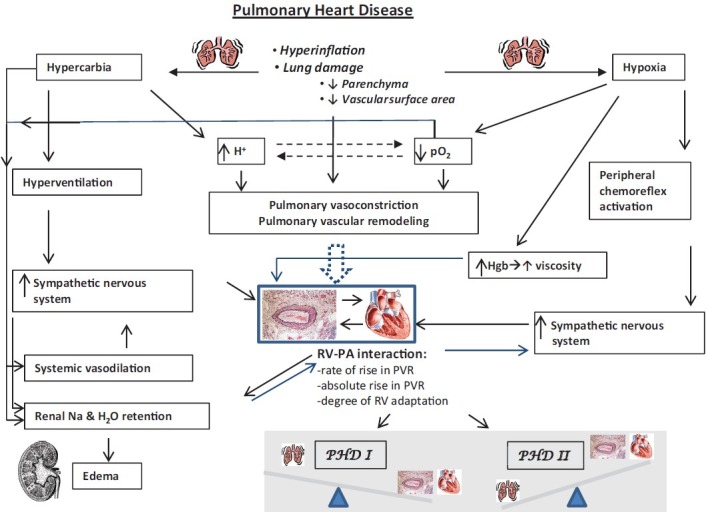
Flow diagram depicts the varying effects that abnormal lung structure and function may have an integrated physiology. Note the numerous ways in which abnormal respiratory structure and function can affect the pulmonary vascular structure and function. Similarly, how these perturbations can either directly, indirectly, or via feedback mechanisms, effect cardiac (especially right ventricular) function. A key concept in this diagram is how these varying pathophysiologic stimuli affect the cardiopulmonary unit, and namely, the right ventricular-pulmonary arterial interaction (RV-PA interaction). The nature of the RV-PA interaction dictates the relative impact of any given degree of pulmonary hypertension and pulmonary vascular disease on overall circulatory homeostasis. If the RV-PA interaction permits for relative adaptation of the RV to RV afterload, typically, the degree of lung pathology will predominate over the degree of RV-PA pathology. In this case, the type I PHD phenotype is present, where the respiratory pathology dominates the clinical picture. If there is a maladaptive RV-PA interaction, the degree of RV dysfunction and pulmonary vascular disease will often override the lung pathology, and the type II PHD phenotype is present.
NORMAL CARDIOPULMONARY ANATOMY AND PHYSIOLOGY
The RV is a thin-walled, distensible structure approximately one-sixth the mass of the left ventricle. It is anatomically linked to the left ventricle via the shared interventricular septum and interlacing epicardial muscle fibers. The pulmonary vasculature is comprised of thin-walled proximal pulmonary arteries and less prominent resistance arterioles. Pulmonary artery compliance is one-half and vascular resistance one-tenth systemic values. Normal respiratory function requires normal parenchymal lung structure, thus normal airways and terminal alveoli, a normal alveolar-capillary interface, and normal lung and chest wall compliance. Descent of the diaphragm and chest wall expansion with inspiration typically generate a fall in pleural pressure of 3-5 cm H2O, which is sufficient to generate a normal tidal volume of 5-7 cc per kilogram. The normal fall in pleural pressure is partially transmitted across the thin-walled pulmonary vessels, causing modest differences between right atrial, pulmonary arterial, and left atrial (also, pulmonary artery wedge pressure, PAWP) pressures between inspiration and end-expiration. Correct measurement of these pressures should be done at end-expiration, when gas flow and pressure in the lung is near zero, allowing for pressure estimation in the absence of respiratory influence. The appropriate matching of ventilation and perfusion as well as normal alveolar-capillary diffusion distance typically ensure normal oxygenation, while normal ventilation requires appropriate central control and peripheral (airway, chest wall) function to regulate arterial carbon dioxide tension.
It is useful to think of the heart and lung as functioning as a coupled unit. We will consider coupling at two anato-physiological interfaces. First, there is the right heart-pulmonary vascular interface. The compliant and relatively nonmuscular RV is normally coupled to a low impedence pulmonary circulation, permitting the RV to eject large volumes of blood at low filling pressures and at a low energetic cost. As such, the RV can be thought of as a flow generator, in contrast to the LV which generates a great deal more pressure at the same level of flow. The second interface occurs at the alveolar-pulmonary vascular level. With elastic recoil of the chest-lung unit, alveolar pressure is ≈0 mmHg at end-expiration. Thus, at the base of the lung, or West Lung Zone 3 (where the bulk of ventilation and perfusion occur in upright posture), alveolar pressure does not exert any resistance effect on the pulmonary vasculature.[4] Changes in pleural pressure generation and transmission can be thought of as another interface between the heart and lung and will be incorporated into the discussion.
Considering the conceptual framework of normal heart-lung coupling, it becomes more apparent how various perturbations, or uncoupling, of the heart-lung interaction can lead to various expressions and degrees of PHD.
EFFECTS OF CRD ON LUNG FUNCTION
Please refer to the flow diagram (Fig. 1) which illustrates the varying physiological perturbations and integrated physiological changes that occur in response to an abnormal respiratory structure and function. Chronic obstructive pulmonary disease, ILD, and hypoventilatory disorders may lead to PHD. In each case, there is an invariable association between abnormal gas exchange, pulmonary hypertension, and altered right heart structure and/or function. Thus, it is distinctly uncommon for CRD to lead to PHD without concomitant hypoxemia and/or hypercapnia. Thus, an abnormal arterial blood gas may be as much a sine qua non for PHD as pulmonary hypertension.
Hypoxia leads to vasodilation in systemic arteries and vasoconstriction in the pulmonary vascular bed. Hypoxic pulmonary vasoconstriction (HPV) is a physiological response designed to maintain ventilation-perfusion matching in response to alveolar hypoxia. This unique vascular response was first noted in the feline circulation, but is present in varying intensity across mammalian species once the alveolar pO2 drops below 50-60 mmHg.[5,6] Mechanisms involved in HPV include direct oxygen sensing within the vascular smooth muscle, changes in redox state, and altered paracrine signaling from the vascular endothelium.[6] Generally, HPV leads to a rise in the PA pressure; however, the pulmonary hemodynamic responses to hypoxia are quite variable. Studies in healthy human volunteers and patients with COPD have shown that pulmonary hypertension in response to acute hypoxia represents an aggregate effect of both a modest increase in the cardiac output (CO) and increased pulmonary vascular resistance (PVR).[7–9] The variability in the HPV response is likely multifactorial, but seems to relate in part to the degree of muscularization of the pulmonary arteries at baseline, modulating effects of blood pH, and possibly genetic predisposition.[10] Acute correction of hypoxia with supplemental oxygen exerts the opposite hemodynamic effects, with the fall in pulmonary artery pressure typically occurring due to reductions in heart rate, stroke volume, and PVR; as with HPV, changes in mean pulmonary artery pressure (mPAP) to oxygen are variable. In response to hypoxia or hyperoxia, the respective rise and fall in mPAP varies in proportion to the magnitude change in pO2.[9]
Chronic hypoxia effects the pulmonary vasculature through both tonic vasoconstriction and likely to a greater degree, vascular remodeling with muscularization and proliferation of the vascular media and intima, respectively.[11] Patients with COPD and healthy natives at altitudes > 3,500 meters demonstrate similar pulmonary vascular morphology.[12] Vascular remodeling likely explains why there is only partial reversal of the increased PVR in response to oxygen administration or inhaled nitric oxide in patients with PH and CRD.[2,13] Interestingly, long-term oxygen therapy (LTOT) in patients with COPD did not lead to significant reductions in mPAP or PVR over a six-year period; however, despite progressive worsening of FEV1 and hypoxemia, the hemodynamics remained stable, suggesting that part of the survival benefit for LTOT relates to interrupted hemodynamic decline in these patients as lung function progressively deteriorates.[13]
Hypercapnia seems to have a comparatively lesser effect on pulmonary vascular tone as hypoxia, with the increase in hydrogen ion concentration, and not the pCO2 itself mediating the vasoconstriction. However, the ventilatory response to hypoxia seems to exert an important modulating effect on the degree of HPV, as changes in minute ventilation and arterial pCO2 correlate inversely with the vascular response to hypoxia; more robust ventilatory responses to hypoxia both abrogate the fall in pO2 and maintain a normal or low pCO2 during hypoxia. This serves to lessen the amplifying effect of a falling pH on HPV.[7,14] Thus, patients with both hypoxia and hypercapnia (i.e., obesity hypoventilation) are at greater risk of developing PH in response to hypoxia; this may explain why patients with sleep-disordered breathing generally require both daytime hypoxia and hypercapnia to develop significant daytime pulmonary hypertension.[15] We will discuss important extracardiac physiological effects of hypercapnia below.
More directly, PVR varies in a U-shaped distribution with lung volumes. As such, at very low lung volumes, or in the setting of widespread atelectasis, a derecruitment of the passive pulmonary vasculature leads to relative loss vascular surface area and can increase the PVR. This may occur in patients with sleep-disordered breathing during an apneic event. The more common scenario in clinical medicine is hyperinflation; at extremes of hyperinflation, such as severe air trapping from reactive airway disease or during the process known as “auto-peep” or “breath stacking” in mechanically ventilated patients, alveolar pressure may increase quite substantially, greatly exceeding pulmonary capillary and arteriolar pressure, thus compressing the pulmonary vasculature and increasing vascular resistance.[16–18]
Longstanding hypoxemia leads to secondary erythrocytosis and a rise in blood viscosity. An increase in the hemoglobin concentration from 12-17 g/dl can increase the blood viscosity by 50-75%. At hemoglobin concentrations above 17 g/dl, blood viscosity increases exponentially to values three-fold normal.[19] Owed to the direct relationship between blood viscosity (η) and vascular resistance (R = η L/r4), pulmonary vascular resistance will rise in direct proportion.
Lastly, loss of pulmonary vascular surface area due to lung destruction serves to increase the pulmonary vascular resistance. In the relative absence of pulmonary vascular disease in viable lung tissue, extensive lung destruction is required to cause a substantive increase in the PVR. However, if pulmonary vascular disease is present, vascular loss and vasoconstriction will have additive effects, typically leading to marked increases in the PVR (Fig. 1).
Thus, in the setting of CRD, varying degrees of hypoxia, hypercapnia, lung hyperinflation, secondary erythrocytosis, and loss of pulmonary vascular surface area lead to the inevitable association of CRD with varying degrees of pulmonary hypertension. Despite the invariable association between PH and PHD, the degree to which the PH itself is directly causal to the PHD varies quite substantially. Although most patients with CRD, PH, and PHD have a normal left atrial pressure, this should not be assumed, especially in older subjects and those with obesity and sleep-disordered breathing, where left atrial hypertension is quite common and may be the predominant cause of PH.[20,21] Pulmonary venous congestion is often very poorly tolerated in these patients, likely in part to the added deleterious effect on already compromised lung mechanics and gas exchange. In this setting, the diagnosis and treatment of underlying left heart congestion can lead to marked clinical improvements. The varying hemodynamics within PHD will be discussed in greater detail in the sections on cardiopulmonary interaction, as well as when we discuss the phenotypes of Type I and II PHD.
EFFECTS OF CRD ON CARDIOVASCULAR FUNCTION
Basic physiology dictates that changes in ventricular preload, afterload, and contractility modulate biventricular performance. Cardiac and renal functions are linked by the effects of cardiovascular reflexes on renal blood flow distribution and neurohormonal control of salt and water handling, which thus alters cardiac preload. Chronic respiratory diseases may affect any or all aspects of cardiorenal function with cardiac performance in the individual patient being determined by a complex interplay between these factors.
Arterial hypoxemia (pO2 < 60 mmHg) leads to stimulation of peripheral chemoreceptors in the carotid body and aorta. The primary physiological effects of peripheral chemoreceptor activation include intense systemic vasoconstriction (this overrides the direct systemic vasodilatory effect of hypoxia) to the renal, splanchnic, skeletal muscle, and cutaneous circulations, with cerebral and coronary artery vasodilation (“head and heart” circulation) as well as bradycardia. This redistributes relatively desaturated blood to the most vital organs and reduces myocardial oxygen demand. However, in the intact circulation, arterial hypoxia also stimulates increased ventilation (to correct hypercapnia and hypoxemia), which stimulates pulmonary stretch receptors that override the chemoreceptor vasoconstrictor response, leading to net systemic vasodilation and increased sympathetic tone to the heart. As a result, hypoxemia typically results in a fall in systemic vascular resistance (SVR) and increased sympathetic input, both of which lead to increases in stroke volume, heart rate, and cardiac output. Chronic stimulation of the peripheral chemoreflex is likely an important reason for why patients with advanced lung disease commonly are in a relatively high cardiac output state. The increased cardiac output is also likely to be driven in part by reflex-mediated and direct sympathomimetic effects of dyspnea. In fact, patients with advanced COPD are in a constant state of increased sympathetic tone.[22yn24] In turn, acute oxygen administration in the setting of hypoxemia typically leads to rapid reductions in heart rate and cardiac output, likely from chemoreceptor deactivation and thus, sympatholysis.[9]
An often overlooked physiological effect of hypoxemia, and in particular, hypercapnia is on renal blood flow and ultimately, salt and water reabsorption. Patients with PHD are often edematous, and edema is often used synonymously with right heart failure. However, this deserves clarification. In the context of heart failure, edema results from net expansion of the extracellular volume compartment as a result of a fall in effective circulating volume, leading to arterial and renal baroreceptor stimulation and activation of the renin-angiotensin-aldosterone-sympathetic (RAAS) cascade. Classically, this is associated with a reduced cardiac stroke volume and output as well as cardiac congestion. However, most patients with CRD and edema have normal or high cardiac outputs in the context of edema, and many have normal right atrial pressure. Moreover, following resolution of the edema, patients with CRD typically have a fall in cardiac output; taken together, these findings make a pure “cardiocentric” explanation for edema in PHD far less likely.
Early observations of patients with COPD and edema noted the poor correlation between edema, cardiac output, and right atrial pressure, as well as pulmonary artery pressures. A far stronger correlation was seen between edema and hypercapnia. Campbell et al., state “oedema is rarely present if the pCO2 is normal. If oedema is present, the pCO2 is almost always raised.” Typically, hypoxemia is more severe in the edematous patient with CRD; however, without concomitant hypercapnia, edema is usually not present.[25,26] Indeed, there is evidence to suggest that hypercapnia, and likely hypoxemia, through direct effects and peripheral chemoreceptor reflex stimulation, leads to marked reductions in renal blood flow, even in the presence of a normal or increased cardiac output. In the context of reduced renal blood flow, the glomerular filtration rate typically remains normal, indicating that renal filtration fraction increases which promotes increased proximal tubular salt/water retention. This process is aided by RAAS activation, which is known to occur in CRD, likely primarily from the effects of hypoxemia and hypercapnia on chemoreceptor activation and disruptions in renal blood flow. Some have theorized that the associated systemic vasodilation of hypercapnia leads to a reduction in effective circulating volume (similar to cirrhosis or nephrotic syndrome), RAAS activation, altered renal blood flow, and net renal sodium/water reabsorption.[27] Presumably, the added stimulus of an impaired cardiac output, seen in some patients with PHD, would only serve to amplify this feedback loop. It generally holds true that patients with chronic respiratory diseases typically associated with hypercapnia (i.e., COPD, thoracic cage abnormalities, obesity hypoventilation syndrome) are more likely to present with edema, as opposed to the diseases associated with hypoxia alone (i.e., idiopathic pulmonary fibrosis).
Correction of hypercapnia and hypoxemia often leads to decreased renal vascular resistance, thus restoring renal blood flow to normal.[28,29] This typically results in diuresis and natriuresis as has been reported by some, and consistently observed by clinicians in edematous patients with decompensated CRD shortly after correction of hypoxemia and hypercapnia even in the absence of diuretics.
Right heart congestion and right heart failure
Is there a difference between right heart congestion and right heart failure? Many conditions can lead to increased right atrial pressure or congestion. However, this may not necessarily indicate that the right heart is indeed failing. This seems to be an important distinction, as the term right heart failure often implicates the RV as the root cause, when in fact, the RV may be an innocent bystander in many subjects with CRD and mildly increased jugular venous pressure. Thus, we prefer to use the term right heart failure when there is evidence of both right heart congestion and impaired RV systolic function. Ideally, right heart failure should be supported by both echocardiographic evidence of impaired RV systolic function and hemodynamics showing an increased right atrial pressure and depressed stroke volume index. Moreover, there should be a relative absence of echocardiographic and hemodynamic features of left heart dysfunction. In the context of mildly increased right heart filling pressures and normal RV systolic function, the right heart is congested, but not failing.
Edema is variably present with RHF, often dictated by the tempo of onset of the RV failure. Thus, the presence or absence of edema should not be used as the primary judge of whether RHF is present. Moreover, edema should be considered as an integrated cardiorenal response, which, in the setting of CRD, may primarily reflect acute or chronic hypoxemia and hypercapnia even if cardiac function is normal. In most patients with PHD, when edematous, there is either no evidence, or mild evidence, of right heart congestion in the context of a normal cardiac output. A smaller subset of patients with PHD have a more traditional presentation of RHF, with overt right heart congestion, objective evidence of RV dysfunction, and a depressed cardiac output, typically with a markedly increased PVR.
EFFECTS OF CRD ON THE CARDIOPULMONARY INTERACTION
Diastolic interactions
Right ventricular hypertrophy is inherent to PHD, likely owed to an overall increase (in varying proportion) in pressure-volume work imposed by CRD and associated PH, and possibly other factors such as increased RV afterload related to extremes of pleural pressure, and possibly the hypertrophic effects of chronic neurohormonal activation on the RV. Volume expansion is common in PHD for the reasons highlighted above. Even in the presence of normal RV systolic function, the distensibility of the right heart in PHD is abnormal, thus predisposing the patient to mild-to-moderate RV congestion in parallel with increased ECV status. This is analogous to the effects of the interaction between impaired renal function and LV “diastolic dysfunction” in patients with hypertensive heart disease. The imposition of overt RV systolic dysfunction typically lends to more severe RV congestion, both by providing an increased stimulus for RAAS activation through a reduced cardiac output as well as a lesser ability to compensate for the increased preload due to a flattened cardiac function curve.
Owed to shared risk factors, such as tobacco abuse and older age, many patients with COPD have concomitant left heart disease, including coronary artery disease, systemic arteriosclerosis, and hypertensive heart disease. As such, volume retention related to their respiratory disease will often expose or exacerbate concomitant left heart disease, and thus symptomatic left heart congestion must be considered in these patients, especially during acute respiratory exacerbations. This same paradigm holds true in patients with obesity and sleep-disordered breathing, where hypertensive heart disease and nonsystolic heart failure are quite common, and in fact, may dominate the clinical picture.[21,30]
Systolic interactions
As detailed above, the right heart and pulmonary circulation function as a coupled unit such that the ventricular-vascular interaction, or relationship between intrinsic contractility of the RV and its afterload, ultimately dictates RV systolic performance (Fig. 1). In most patients with PHD, RV contractility is normal or increased due to the lack of primary insults to the RV such as ischemia, infiltrative disease, or other forms of myopathy. Thus, RV afterload is the critical determinant of RV performance in the setting of CRD.[31,32] Naturally, if the RV is intrinsically dysfunctional, then any added vascular load is especially devastating.
Varying degrees of pulmonary hypertension often result from an abnormal RV-pulmonary vascular interaction. However, the increased pulmonary artery pressure is the result, not the cause, of the RV-PA mismatch. Therefore, pulmonary artery pressure should not be used interchangeably with RV afterload. Afterload represents the opposition to blood flow, and is accounted for by proximal artery stiffness (which dictates the degree and timing of arterial wave reflection) and distal pulmonary vascular resistance. Pulmonary vascular resistance accounts for approximately 80% of afterload in most patients and represents the mostly clearly quantifiable aspect of afterload in clinical medicine. Thus, PVR is a far better measure of afterload than pressure. Practically speaking, this is why PH is far more common than the combination of PH and RV systolic failure in the context of PHD.
It is best not to consider the pressure in isolation, but rather, to consider the components of the pressure (flow, resistance, left atrial pressure) in order to understand its “upstream” effect on the RV. Naturally, invasive quantitation of hemodynamics is not plausible in every patient with pulmonary heart disease. However, the noninvasive assessment provides important insights into the pathophysiology of PH in an individual patient, and allows direct visualization of size, shape, and function changes that are fundamental to relative degrees of uncoupling of the right heart from the pulmonary circulation. Most patients with PHD have relatively mild PH related to mildly increased pulmonary vascular resistance with preserved RV systolic function, and thus, preserved RV stroke volume and cardiac output. In contrast, a small but important subset of patients with PHD has PH related to marked increases in the pulmonary vascular resistance, such that the RV can no longer adapt, and over RV systolic dysfunction results. In essence, the fundamental distinction between these two phenotypes is the degree of pulmonary vascular disease, which provides an important pathophysiologic foundation for the two different phenotypes of PHD, which will be discussed below.
Respiratory interactions
As mentioned above, pulmonary mechanics can affect cardiac function. In the context of airway obstruction, alveolar pressure increases and can exert a compressive effect on the neighboring pulmonary vasculature. As such, with marked hyperinflation, the pulmonary vascular resistance will increase. This scenario is especially relevant during extremes of hyperinflation, as seen in patients with acute exacerbations of asthma or COPD. Following the principles of ventricular-vascular coupling, the greater the degree of RV dysfunction at baseline, the greater the hemodynamic significance of any added vascular load imposed by the Starling resistor effect of airway pressure on pulmonary resistance. Some have theorized that the diseased lungs themselves may exert an external constraint on the heart, particularly when hyperinflated, thus modifying cardiac performance.[33] Moreover, marked changes in pleural pressure can also be transmitted to the heart and circulation, and may lead to significant increases in ventricular wall tension. Echocardiographic studies in patients with acute bronchoconstriction have shown acute RV dilatation during inspiration, with return of normal RV dimension during expiration, likely owed to the combined effects of hyperinflation on PVR and negative pleural pressure on RV wall tension. These same studies have shown that LV cavity dimensions reciprocally diminish during RV dilatation, which may explain the frequent finding of pulsus paradoxus in patients with acute bronchoconstriction.[17,34]
Similarly, patients with obstructive sleep apnea often generate markedly negative pleural pressures during periods of upper airway obstruction, analogous to the physiology of a Mueller maneuver. Cyclic, repeated increases in ventricular wall tension are thought to be one of the mechanisms responsible for sympathetic nervous system activation in these patients, perhaps contributing to their propensity to salt and water retention, as well as the development of systemic hypertension and left ventricular hypertrophy.[30]
More practically, patients with obesity, as well as obstructive and restrictive lung disease, must generate more negative pleural pressures to generate a normal tidal volume, given increased stiffness in the lung parenchyma itself or chest/abdominal constraint to normal ventilation. These larger swings in pleural pressure are transmitted to the heart and pulmonary vasculature, leading to relatively marked variation in pressure recordings obtained at end expiration versus inspiration. This is exaggerated further by lying supine, which adds further mechanical disadvantage to respiration in these patients. Thus, it is imperative that cardiac filling pressures and pulmonary artery pressures are obtained at end expiration, when respiratory influence on hemodynamics is near zero, and not taken as the electronic average of pressures across the respiratory cycle. The latter method is a common default in the modern era, where pressure recordings are often measured by the catheterization laboratory's hemodynamic acquisition software and not over-read manually by the performing physician. This can lead to marked errors in the accuracy of the hemodynamic recordings. A common scenario would be the case of an obese patient with COPD, with a pulmonary artery occlusion (wedge) pressure of 28 mmHg at end expiration and 5 mmHg with inspiration, leading to an “average” wedge pressure recorded of 15 mmHg. The result is marked underestimation of the left atrial pressure, and thus, overestimation of the PVR, leading to erroneous diagnosis of pulmonary arterial hypertension in a patient with decompensated left heart failure, with the potential for misguided pharmacotherapy.
PATIENT PHENOTYPES
As discussed above, PHD is a heterogeneous condition. In many patients, PHD is a marker of a severe underlying respiratory condition, with the direct and indirect effects of abnormal gas exchange being the critical determinants of altered cardiac performance. In others with PHD, cardiac size and function are more severely affected, and the (right) heart disease itself can dominate the clinical picture. Recognition of these different patient phenotypes is critically important to fully appreciate the clinical and potential therapeutic approaches to these patients.
Although not a customary schema, we will discuss PHD as occurring as two distinct entities, “Type I” and “Type II” PHD, the goal being to highlight the typical clinical, echocardiographic, and hemodynamic findings of two different phenotypes of PHD. Type I PHD will refer to subjects with chronic respiratory disease (CRD) and less than moderate degrees of RV systolic dysfunction, relatively mild increases in the pulmonary vascular resistance, with a cardiac index maintained within the normal range. This phenotype comprises the majority of PHD patients. In contrast, Type II PHD will refer to the smaller subset of patients with more marked increases in the pulmonary vascular resistance leading to a more advanced degree of right heart dysfunction, often characterized by marked RV enlargement, right heart congestion, and overt RV systolic dysfunction. Patients with this latter phenotype of PHD can often present in a fashion similar to patients with PAH. We will also discuss the treatment of PHD; however, we will do so largely in a theoretical context as the role for direct treatment of PHD and PH in CRD remains vastly understudied, and thus, poorly understood.
The fundamental distinction between the Type I and Type II PHD patient is the degree of pulmonary vascular disease present. Pulmonary vascular disease, as measured by an increased PVR, not only adversely affects right heart function, but also further impairs gas exchange, ventilatory efficiency, and in some ways, adds a “second disease” to the underlying respiratory condition. The result is that patients with PHD, with and without significant pulmonary vascular disease, often have different overall clinical presentations that cannot be distinguished on the basis of a single metric. Therefore, it seems best to describe the overall differences in the clinical, echocardiographic, hemodynamic, and exercise physiology manifestations of the two groups. We recognize that dichotomization of PHD is subject to oversimplification, given an intermediate phenotype is missed; however, by providing the boundaries of the two patient phenotypes, we trust the clinician will be able to recognize the patient with PHD whom may lie somewhere in between.
We have deliberately avoided separating the PHD phenotypes on the basis of PA pressure severity, given the presence of an increased PA pressure does not necessarily denote the presence of pulmonary vascular disease nor what comprises the pressure. It is common to consider the PA pressure in CRD as being “proportionate” or “disproportionate” to the underlying respiratory condition. For example, most patients with CRD have mild PH (i.e., mean PA pressure < 35 mmHg) and this is typically considered proportionate to their underlying condition. So called disproportionate PH typically refers to patients with more severe PH (i.e., mean PA pressure > 35 mmHg). This terminology has practical value, especially at the extremes of PH and also when used in real-time to describe relative changes in PA pressure (i.e., by echo-Doppler examination) relative to changes in respiratory function (i.e., pulmonary function tests or radiographic findings) in the individual patient. However, describing PHD or PH in terms of their proportionality to an underlying respiratory condition is limited by the inherent subjectivity of what one considers proportionate or not, and still does not provide sufficient pathophysiologic information about the PH itself, nor its effects on the right heart. Moreover, there is such substantial overlap in the degree of PH using the “proportionate” schema, that this approach is not especially useful when trying to differentiate one patient from another. Another pitfall of using PA pressure as the focal point of PHD is that the pressure is most commonly derived via echo-Doppler examination, which may differ significantly from invasively derived PA pressure.[35,36]
Type I PHD
Patients with Type I PHD typically have a clinical presentation that most closely resembles that of their primary underlying respiratory condition (Table 1). Therefore, increasing dyspnea will coincide with parallel deterioration of respiratory function and, in turn, will lead to transient, and typically modest, degrees of right heart congestion. Edema may or may not be present. Improvement in the underlying respiratory condition will typically coincide with improved edema and RV congestion. Exertional angina, syncope, or presyncope are distinctly uncommon, with a history of cough, wheezing, and respiratory infection often dominating the clinical picture. Aside from modest right heart congestion, the physical examination will typically reveal normal or increased arterial pulse volume and turgor. The left parasternal border is typically inactive, and the examination is more typically dominated by the respiratory findings than cardiovascular findings. The electrocardiogram may or may not manifest RVH, and the axis is either normal or vertical, typically without frank right axis deviation or RV strain. Oftentimes, the vertical axis is caused by lung hyperinflation and diaphragmatic flattening. Pulmonary function testing typically reveals relatively balanced reductions in the forced vital capacity (FVC) and diffusing capacity for carbon monoxide (DLCO), with the relative proportion of these measures remaining stable even in the context of worsening respiratory function.
Table 1.
Clinical history
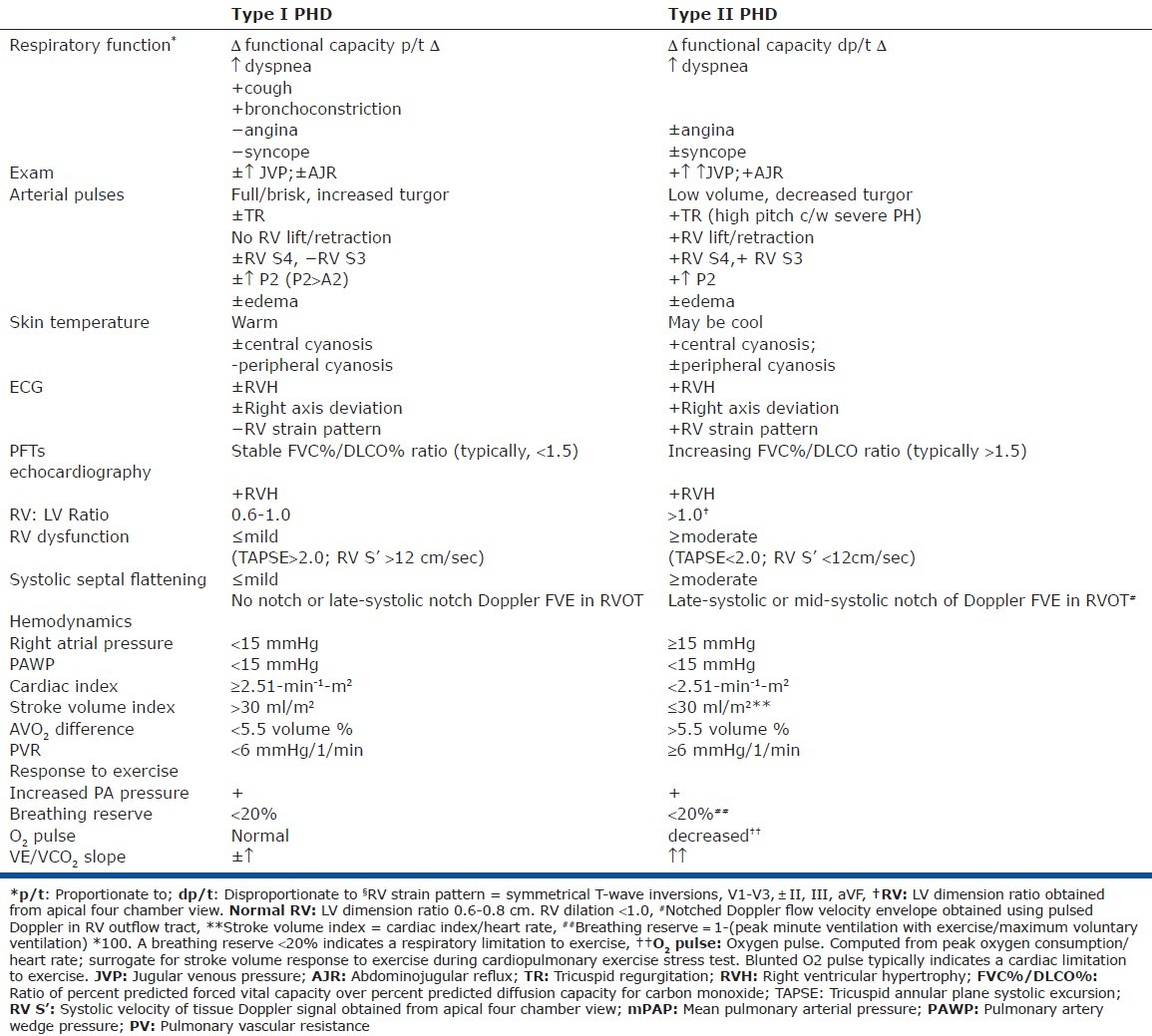
The echocardiogram of patients with Type I PHD will typically reveal variable degrees of increased RV wall thickness, although this is often difficult to quantify. The RV dimensions will typically remain within normal range or increase to a mild degree, with the ratio of RV to LV diastolic dimensions typically not exceeding 1.0. Importantly, right ventricular systolic function typically remains within normal limits or at most, mildly reduced. Right ventricular systolic function may be quantified using relatively simple, reproducible methods such as tricuspid annular plane systolic excursion (TAPSE) or by tissue Doppler imaging at the base of the RV. Both methods assess RV shortening along its longitudinal axis and do not require endocardial definition or volumetric estimates.[37,38] The degree of TR present is variable, depending on the degree of volume excess and annular dilatation at the time the study is performed.
Most patients with CRD have either normal PA pressure or mild pulmonary hypertension. More significant PH in the Type I PHD patient typically arises from a high cardiac output state and sometimes, concomitant left heart congestion. By Doppler estimate, the PA systolic pressure will usually not exceed 55 mmHg. In a large cohort of patients with severe COPD, the average RA pressure was 9 mmHg, with a cardiac index of 3.0 L/min/m2. The mean PA pressure was approximately 27 mmHg (systolic PA pressure 45-50 mmHg) with an average PVR of < 3 mmHg/l/min.[39] Similar hemodynamics were noted in the majority of patients with idiopathic pulmonary fibrosis.[40] Likewise, most patients with sleep apnea have either normal or mildly increased PA pressure, with a normal or increased cardiac index and a PVR that rarely exceeds 3 mmHg/L/min.[15] In one study of morbidly obese subjects at the time of gastric bypass surgery, the average right atrial pressure and pulmonary artery wedge pressure were approximately 18 and 22 mmHg, respectively, while the average PVR was < 4 mmHg/l/min with an average RVEF > 40%.[41] Importantly, the predictors of PH during waking hours in patients with sleep apnea are not predicted by nocturnal events, with daytime hypoxemia, hypercapnia, greater body mass index, and decreased lung function (i.e., low FVC) being important associates. Also, it cannot be overemphasized that many patients with OSA and obesity hypoveniltation syndrome have increased left atrial pressure, and in several studies, left heart congestion has been shown to be the single most important predictor of pulmonary hypertension.[20,21] In our experience, the vast majority of obese subjects with significant sleep-disordered breathing and moderate or greater degrees of PH have systemic hypertension and an increased left atrial pressure at the time of right heart catheterization, with a normal or increased cardiac output and a PVR that is within the normal range. This is often overlooked, and suggests that many cases of “right heart failure” in this setting are, in fact, due to left heart congestion.
Exercise induced PH is common in patients with COPD, resulting from a rise in the cardiac output and lack of fall of the PVR.[42] Dynamic hyperinflation may also contribute, by way of vascular compression. Importantly, patients with severe COPD and mild-to-moderate PH exhibit the same aerobic exercise capacity as COPD patients without PH; in both groups, exercise capacity was related to an exhausted ventilatory reserve and not to PA pressure, nor differences in the oxygen pulse, a surrogate for cardiac stroke volume.[43] Likewise, exercise-induced PH is common to sleep apnea, and was explained by a rise in the pulmonary artery wedge pressure with exertion, while the PVR remained within the normal range in all subjects.[20] Thus, the presence of exercise-induced PH should not necessarily be taken to indicate that pulmonary vascular disease is present, nor that the PH or RV function are in fact the source of the functional limitations. This is especially true when considering submaximal exercise.
Type II PHD
The presence of pulmonary vascular disease can greatly modify the overall clinical presentation of a patient with CRD and PHD. Often, the clinical course seems to become detached from changes in the underlying respiratory condition, as if a patient has taken on another condition. As such, dyspnea may progress in a manner that is disparate to objective studies of respiratory function, such as pulmonary function testing or imaging studies. The history may include reporting of exertional angina, syncope, or near syncope, similar to the presentation of patients with "pure" pulmonary vascular disease states such as PAH or chronic thromboembolic pulmonary hypertension.
As Paul Wood pointed out some four decades ago, “when the pulmonary vascular resistance is high in cor pulmonale the physical signs are different…”[44] These differences primarily reflect the presence of RV systolic dysfunction and decreased systemic perfusion. The jugular venous pulse is typically markedly elevated, while the arterial pulses are low in volume with less tension, indicative of a depressed stroke volume and pulse pressure. Cyanosis is often more prominent. The ECG will more commonly display RVH, right axis deviation, and RV strain, similar to that seen in PAH. Pulmonary function testing will often suggest pulmonary vascular disease, with an increasing FVC/DLCO ratio (Table 1). The falling DLCO likely belies the nearly universal progression of hypoxemia in these patients, which is unmasked further during ambulation. A stable, mild oxygen requirement makes Type II PHD far less likely.
With more advanced PH, and thus the Type II PHD phenotype, the cardiopulmonary exercise stress test (CPET) data more closely resembles that of a PAH patient (Table 1). Patients will demonstrate a ventilatory limitation to exercise (similar to Type I PHD); however, they will also exhibit a significant circulatory limitation to exercise (i.e., lower oxygen pulse, lower peak systolic blood pressure to exercise) and a markedly increased VE/VCO2 relationship as compared to the Type I PHD patient.[45–47]
The echocardiographic findings in Type II PHD are often indistinguishable from PAH, with marked RV dilation, right to left septal flattening in systole, and depressed RV systolic function. The flow velocity envelope (FVE) obtained with pulsed Doppler interrogation in the RV outflow tract often demonstrates a “notched” configuration, representing early arrival of reflected arterial waves from the distal pulmonary vasculature, with the notch representing temporary flow interruption due to increased RV impedence.[48] We recently showed that a notched FVE in the RV outflow tract was highly sensitive and specific for underlying pulmonary vascular disease in patients with pulmonary hypertension of varying etiologies.[49] In patients with CRD and pulmonary hypertension, subjects without a notched FVE had an average mean PA pressure of 37 mmHg and PVR of 4.5 mmHg/l/min versus patients with a notched FVE, whom demonstrated a mean PA pressure of 48 mmHg, a PVR of 9 mmHg/l/min, and significantly greater echocardiographic evidence of RV systolic dysfunction.[50]
Approximately 1-5% of patients with COPD demonstrate hemodynamics that are comparable to patients with PAH, with a mean PA pressure > 50 mmHg in the context of a markedly elevated PVR and decreased cardiac index.[39,51] Interestingly, the subjects with COPD and pulmonary vascular disease are significantly more hypoxic than those with mild-moderate PH with a normal PVR, and have a pCO2 less than 40 mmHg; these arterial blood gas findings are more consistent with the findings of PAH than COPD. Moreover, these patients demonstrated lesser degrees of pulmonary obstruction than subjects without pulmonary vascular disease. Pulmonary sarcoid seems to have a particular predilection for pulmonary vascular involvement, and represents the diagnosis that we most commonly see in our Type II PHD patients. Other parenchymal lung disorders that present a higher risk of significant pulmonary vascular involvement include Langerhans cell histiocytosis and interstitial lung disease associated with scleroderma.[52–54] Combined pulmonary fibrosis with emphysema can also be associated with a severe PH phenotype. A subgroup of idiopathic pulmonary fibrosis patients can also manifest a more severe PHD phenotype, with the degree of PH, pulmonary vascular disease, and right heart dysfunction more closely resembling PAH than a typical Group III PH patient.[55] Taken together, clinical observations as well as the published literature indicate that subgroups of many different forms of chronic respiratory disease can manifest a more severe PHD phenotype. This phenotypic distinction cannot be made by PA pressure estimation alone. Therefore, clinicians need be aware of the varying clinical, CPET, echocardiographic, and hemodynamic parameters that set a Type I PHD patient apart from a Type II patient.
Treatment
In patients with Type I and Type II PHD, if the initial diagnosis is made in the presence of an acute respiratory exacerbation, the focus is directed toward aggressive treatment of the underlying trigger—i.e., infection, bronchospasm. Patients should be re-evaluated in a steady state, as their initial assessment often overestimates the degree of PHD that will remain following resolution of the acute exacerbation. Hypoxemia and hypercapnia should be aggressively treated. For patients with suspected sleep-disordered breathing, prompt diagnosis and initiation of continuous positive airway pressure should be pursued. Diuretics should be used to treat volume excess, especially when associated with right heart congestion. Long term oxygen therapy is the cornerstone of treatment of PHD in patients with COPD and, along with smoking cessation, remains the only intervention associated with improved survival in COPD.[56,57] Physicians treating patients with COPD over the past three decades will attest to the observed decline in PHD seen in the post oxygen therapy era. Patients should also be treated for ambulatory and nocturnal hypoxemia whenever possible.
With the rapid expansion of therapies available for the treatment of patients with PAH, it is tempting to try and extend our PH specific therapies to patients with PH and related PHD. However, the efficacy and safety of PH specific therapy in patients with WHO Group III pulmonary hypertension and PHD remain unproven. Thus, we will discuss the use of PH specific therapies within a specific clinical and pathophysiologic context that may provide a framework for more tailored investigation and rational application of these therapies in PHD.
Conceptually, for PH specific therapy to be safe and efficacious in the setting of PHD, it seems that a minimum of four conditions must be met. First, patients must have underlying pulmonary vascular disease and thus increased RV afterload (i.e., increased PVR). Second, patients must have evidence of RV systolic dysfunction on the basis of the increased PVR. This combination of an increased PVR and depressed RV function indicates relative uncoupling of the RV and pulmonary vasculature, and represents the basic physiologic paradigm that lends to both the circulatory limitation to exertion as well as the propensity toward hemodynamic improvements in response to pulmonary vasodilating therapy.[58,59] Patients with Type I PHD have mild pulmonary vascular disease and mild RV dysfunction, and thus, a relatively small separation or uncoupling between RV function and afterload. This does not lend toward a significant circulatory limitation to exertion or a hemodynamic benefit from PH specific therapies. In contrast, patients described as Type II PHD meet both criteria, with RV-PA uncoupling that more closely resembles that seen in patients with PAH.
The third condition that must be met is that pulmonary vasodilation should not worsen gas exchange to the point that the drop in oxygen saturation occurs in disproportion to an improvement in cardiac output. If the relative degree of hypoxia exceeds the relative rise in cardiac output, then the net effect may be reduced peripheral oxygen delivery. In contrast, if there is a mild reduction in oxygen saturation in response to PH specific therapy, yet a marked rise in cardiac output owed to improved right heart function, net oxygen delivery to tissue may still be significantly elevated despite a modest increase in hypoxia. It seems logical that an essential element to avoiding the scenario where PH specific therapy would lead to hypoxia in disproportion to improved cardiac function is to select the appropriate PH phenotype. In patients with PAH, pulmonary vasodilating therapy often improves oxygenation by improving V/Q mismatch (via a drop in PVR), improving the transport of more saturated mixed venous blood to the alveolar-capillary interface (via the increase in cardiac output), and occasionally, by alleviating a right to left shunt through a patent foraman ovale as the right atrial pressure falls. In a Type II PHD patient, the circulatory physiology resembles PAH. As such, even if there is some degree of intrapulmonary shunt that occurs in response to pulmonary vasodilatation, an improved cardiac output (and thus, delivery of more oxygenated mixed venous blood to the alveolar-capillary interface) and falling right atrial pressure (and alleviation of a right to left shunt) can offset the effect of the shunt, leading to stable or even improved oxygenation.
In most patients with PHD, the degree of pulmonary vascular disease is mild and relates to hypoxic pulmonary vasoconstriction, remodeling, and loss of lung surface area. The use of pulmonary vasodilators in patients with relatively mild PH in association to CRD tends to make patients more hypoxic, as was seen in a cohort of patients with COPD treated for 12 weeks with bosentan, where the alveolar-arterial oxygen gradient increased and functional capacity worsened.[60] In contrast, patients with Type II PHD exhibit PVR elevations that are well beyond that seen from HPV alone. Moreover, due to a reduced cardiac output, these subjects often have reduced mixed venous oxygen saturation. Therefore, a drop in the PVR may leave HPV relatively intact, and also improve mixed venous oxygen saturation, thus preventing a net fall in arterial oxygen saturation, similar to what occurs in PAH. The consequent reduction in right atrial pressure may also improve oxygenation via alleviation of right to left shunting through a patent foramen ovale. Therapy selection may also matter, as inhaled therapies as well as oral sildenafil are less apt to cause increased hypoxemia through vasodilation than calcium channel blockers or acute administration of intravenous epoprostenol.[61] The inhaled therapies deliver drug to ventilated areas and thus tend to preserve V/Q matching, while sildenafil is relatively more pulmonary selective as compared to calcium channel blockers and parenteral prostacyclins. Of note, no studies have investigated the effects of chronic, gradual titration of parenteral prostacyclin therapy in a severe pulmonary heart disease phenotype. It may be that slow, steady titration of parenteral prostacyclin therapies in this context do not lead to increases in intrapulmonary shunting as was seen with acute intravenous administration. This may be especially true when the PH phenotype is robust, such that the improved PVR and right heart function improve gas exchange, thus offsetting the chance that a modest increase in intrapulmonary shunt would lead to a significant increase in hypoxia.
The fourth condition that must be met is that pulmonary vasodilation, improved right heart function, and improved hemodynamics should translate into functional improvements and decreased symptoms for the patient. The translation of improved circulatory reserve into overall clinical and functional improvements represents a complex physiologic algorithm, where the net improvement in functional capacity should occur due to a change in the balance of circulatory and ventilatory dependent functional limitations following treatment. Indeed, it is challenging to precisely predict the extent to which improved cardiac function will improve overall functional capacity in the context of significant pulmonary disease. However, it is logical that there must be a requisite baseline level of right heart dysfunction and increased pulmonary arterial afterload present, such that there is a coexisting cardiovascular limitation to exercise as well.
Without a sufficient disruption of cardiac reserve on the basis of abnormal RV-PA interaction, the chances of demonstrating a treatment effect with PH therapies would be nil. Considering the relatively minor disruption in the RV-PA relationship that occurs at baseline in the Type I PHD patient, it stands to reason that patients with Type I PHD are not poised to significantly benefit from a hemodynamic perspective from pulmonary vasodilator therapy. As such, whether there is no change or a modest improvement in circulatory function following PH specific therapy, the relative change in circulatory reserve is not sufficient to offset the predominant and uncorrected ventilatory limitation. As a result, the functional capacity will remain essentially unchanged. If gas exchange worsens at the same time, overall functional capacity may even decline.
In contrast, patients with Type II PHD are poised to improve hemodynamically with PH therapies, typically without worsening gas exchange. In these patients, liberalizing cardiac reserve may serve to alleviate the superimposed cardiac limitation to their functional impairment, leaving only the ventilatory limitation behind. The net functional improvement then depends on the extent of the circulatory limitation imposed by the PH at baseline, its response to PH therapy, and the relative balance of this limitation to the background ventilatory limitation. Interestingly, a recent post hoc analysis of a placebo-controlled randomized clinical trial in IPF showed that the subgroup of patients with RV dysfunction and RV hypertrophy improved their six minute walk distance in response to sildenafil, while subjects without RV dysfunction and hypertrophy did not respond to therapy.[62] Figure 2 illustrates the relative balance of respiratory disease and corresponding ventilatory reserve as well as the degree of circulatory impairment and reserve in subjects with Type I and Type II PHD at baseline and in response to PH specific therapy. Predicted changes each of these parameters in addition to hypoxia are compared among the PHD I and II phenotypes and how PH specific therapy would potentially affect overall functional status.
Figure 2.
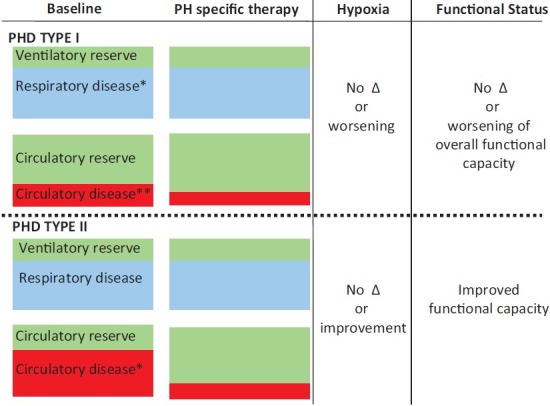
Illustrates the qualitative degree of respiratory disease (blue) and ventilatory reserve (green) as well as circulatory disease (red) and reserve (green) of subjects with Type I (top half of figure) and Type II pulmonary heart disease (bottom half of figure). Note that in the Type I phenotype, there is significant respiratory disease at baseline; the degree of respiratory disease >degree of circulatory disease. Therefore, the circulatory reserve is >degree of ventilatory reserve. In response to PH specific therapy, the balance of respiratory disease and ventilatory reserve remain unchanged. The degree of circulatory disease decreases modestly, with a small increase in circulatory reserve. Given no change in the respiratory disease in response to therapy and only a small improvement in circulatory function, there is no net change in functional capacity. If oxygenation worsens significantly, the functional status may even decline. Note that in the Type II phenotype, there is a similar degree of baseline respiratory disease and ventilatory reserve as seen in the Type I subject. However, there is a much greater degree of circulatory disease, such that the degree of circulatory disease is comparable to the degree of respiratory disease. As such, the degree of circulatory reserve is much lower than in the type I PHD example. In response to PH specific therapy, the balance of respiratory disease and ventilatory reserve remain unchanged. However, the degree of circulatory disease decreases substantially while there is a significant improvement in circulatory reserve. The degree of hypoxia may not change, or may even improve. This most often leads to a net improvement in functional status. *Respiratory disease-may refer to obstructive or restrictive respiratory disorders. **Circulatory disease-refers to the severity of right ventricular afterload (pulmonary vascular resistance) and the degree of right ventricular dysfunction.
With a mild or moderate baseline ventilatory limitation, the functional improvements in response to treatment of type II PHD are more predictable and may be very impressive. In patients with truly end-stage lung disease, treatment of the PHD often provides a temporary, but critically important “bridge” that allows a patient to survive and remain relatively physiologically intact awaiting lung transplantation.
CASE ILLUSTRATION
Figure 3 shows representative echo-Doppler images, hemodynamics and PFTs from a patient with Type I PHD (panel A) and type II PHD (panel B). Both patients had significant COPD and PH, and both were treated with oral PH-specific therapy for six months. Patient A had near normal baseline RV systolic function, which further improved in response to PH therapy (TAPSE increased from 2.0 cm to 2.4 cm). The BNP level also fell. In spite of these improvements, patient A had a fall in 6 MWD from 320 to 300 m and a worsening in NYHA Functional Class from 3 to 4. Patient B had a comparable degree of PH (mean PA pressure), yet significantly worse baseline RV function and a much higher PVR as compared to Patient A. The TAPSE improved significantly in response to PH therapy and the BNP fell. However, in contrast to Patient A, in Patient B, the improved RV function paralleled a relatively marked increase in the 6MWD from 238 m to 320 m, and a fall in NYHA Functional Class from 4 to 3. Thus, the RV function improvements correlated with improved clinical status in the Type II PHD patient only. This likely relates to the relatively greater burden of pulmonary vascular disease, RV dysfunction, and RV-PA uncoupling in this patient, which should predict a greater baseline circulatory limitation to exercise, and thus a more robust clinical response to improved right heart-pulmonary vascular interaction.
Figure 3.
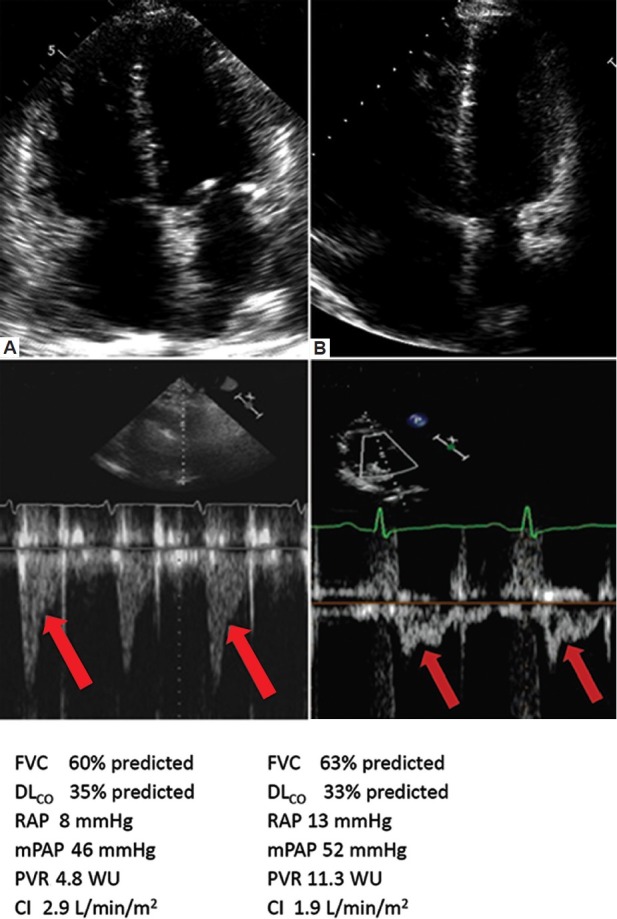
Representative apical four chamber view and Doppler flow velocity envelope (FVE) in right ventricular outflow tract (RVOT). Data were taken from two separate patients: (A) one with PHD I phenotype and (B) one with PHD II phenotype. Below each panel are representative pulmonary function tests and invasive hemodynamics for each patient. Note the greater degree of RVH, and lesser degree of RV dilatation and dysfunction in the patient in panel A vs. panel B. Similarly, the subject in panel A has a late-systolic notch pattern RV outflow tract Doppler pattern, which is consistent with a modest and comparatively lesser degree of RV afterload as compared to the mid-systolic notch pattern seen in the Doppler pattern of the subject in panel B. The subjects had a similar degree of respiratory disease. FVC = forced vital capacity; DLCO = carbon monoxide diffusing capacity; mPAP = mean pulmonary artery pressure; PVR = pulmonary vascular resistance; WU = Wood units; CI = cardiac index. Both patients in Figure 1 had COPD and both were treated with oral PH-specific therapy for 6 months. Patient A (PHD I phenotype) had a fall in 6 MW from 320 m to 300 m and a worsening in NYHA functional class from 3 to 4. RV function improved as evidenced by an increase in TAPSE from 2.0 cm to 2.4 cm and a 40% fall in the BNP level. Patient B (PHD II phenotype) had an increase in 6 MW from 238 m to 320 m with a fall in NYHA functional class from 4 to 3. RV function also improved, as evidenced by an increase in TAPSE from 1.4 cm to 1.9 cm and a 40% fall in the BNP level. Note that in both patients A and B, BNP fell and RV function improved, however only in patient B with the PHD II phenotype did the RV function improvement correlate with overall improvements in clinical status. The differences in clinical response likely relate to the fact the subject with the type II PHD phenotype had a greater degree of RV-PA uncoupling, and thus, a greater degree of functional impairment on the basis of a circulatory limitation. As such, direct PH treatment in this context translates into overall improved functional capacity, even though the background ventilatory limitation remains.
With this in mind, it is not likely coincidence that evidence from small case series and case reports of the effectiveness of PH therapies in the context of CRD are almost exclusively seen in patients with severe PVD, RV dysfunction, and a phenotype closely resembling that of our proposed Type II PHD patient.[55,63–66]
SUMMARY
Pulmonary heart disease is a heterogeneous condition with its clinical, echocardiographic and hemodynamic manifestations varying in proportion to the ways in which abnormal lung function can affect the heart. Most patients with PHD have relatively mild pulmonary vascular disease and cardiac dysfunction, mostly the direct or indirect results of abnormal pulmonary gas exchange. In these patients, the PHD signifies the underlying respiratory condition, but the PHD does not dominate the clinical picture. In these patients, it is the lungs, and not the heart that remains the primary therapeutic target.
In contrast, a smaller subset of patients with CRD can develop severe PVD and right heart dysfunction, similar to patients with PAH. These patients typically clinically deteriorate despite optimization of treatment of their underlying CRD, suggesting they have taken on a second disease. Recognizing PHD as distinct phenotypes may assist in the recognition of these very ill patients, and may also provide a clinical and pathophysiological framework that may prove useful in patient selection for future studies investigating the role of PH specific therapies in PHD. The failure to recognize the different phenotypes of PHD serves to oversimplify the condition, which increases the potential for misapplication of therapy, and also, the potential for a missed opportunity to adequately refine clinical trial design to ascertain whether certain subgroups of patients with PHD may indeed benefit from PH specific therapy.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Fishman AP. State of the art: Chronic cor pulmonale. Am Rev Respir Dis. 1976;114:775–94. doi: 10.1164/arrd.1976.114.4.775. [DOI] [PubMed] [Google Scholar]
- 2.Girgis RE, Mathai SC. Pulmonary hypertension associated with chronic respiratory disease. Clin Chest Med. 2007;28:219–32. doi: 10.1016/j.ccm.2006.11.006. x. [DOI] [PubMed] [Google Scholar]
- 3.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 4.Hughes JM, Glazier JB, Maloney JE, West JB. Effect of lung volume on the distribution of pulmonary blood flow in man. Respir Physiol. 1968;4:58–72. doi: 10.1016/0034-5687(68)90007-8. [DOI] [PubMed] [Google Scholar]
- 5.von Euler US, Liljestrand G. Observations on the pulmonary arterial blood pressure in the cat. Acta Physiol Scand. 1946;12:301–20. [Google Scholar]
- 6.Mark Evans A, Ward JP. Hypoxic pulmonary vasoconstriction-invited article. Adv Exp Med Biol. 2009;648:351–60. doi: 10.1007/978-90-481-2259-2_40. [DOI] [PubMed] [Google Scholar]
- 7.Naito T, Miyahara Y, Ikeda S. Ventilatory and pulmonary vascular responses to acute hypoxia are nonuniform in healthy man. Respiration. 1995;62:185–9. doi: 10.1159/000196445. [DOI] [PubMed] [Google Scholar]
- 8.Weitzenblum E, Schrijen F, Mohan-Kumar T, Colas des Francs V, Lockhart A. Variability of the pulmonary vascular response to acute hypoxia in chronic bronchitis. Chest. 1988;94:772–8. doi: 10.1378/chest.94.4.772. [DOI] [PubMed] [Google Scholar]
- 9.Fishman AP, McClement J, Himmelstein A, Cournand A. Effects of acute anoxia on the circulation and respiration in patients with chronic pulmonary disease studied during the steady state. J Clin Invest. 1952;31:770–81. doi: 10.1172/JCI102662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bartsch P, Gibbs JS. Effect of altitude on the heart and the lungs. Circulation. 2007;116:2191–202. doi: 10.1161/CIRCULATIONAHA.106.650796. [DOI] [PubMed] [Google Scholar]
- 11.Weitzenblum E, Chaouat A. Cor pulmonale. Chron Respir Dis. 2009;6:177–85. doi: 10.1177/1479972309104664. [DOI] [PubMed] [Google Scholar]
- 12.Penaloza D, Sime F, Bachero N, Gamboa R. Pulmonary hypertension in healthy men born and living at high altitude. Med Thorac. 1962;19:449–60. [Google Scholar]
- 13.Zielinski J, Tobiasz M, Hawrylkiewicz I, Sliwinski P, Palasiewicz G. Effects of long-term oxygen therapy on pulmonary hemodynamics in COPD patients: A 6-year prospective study. Chest. 1998;113:65–70. doi: 10.1378/chest.113.1.65. [DOI] [PubMed] [Google Scholar]
- 14.Lloyd TC., Jr Influence of blood pH on hypoxic pulmonary vasoconstriction. J Appl Physiol. 1966;21:358–64. doi: 10.1152/jappl.1966.21.2.358. [DOI] [PubMed] [Google Scholar]
- 15.Chaouat A, Weitzenblum E, Krieger J, Oswald M, Kessler R. Pulmonary hemodynamics in the obstructive sleep apnea syndrome.Results in 220 consecutive patients. Chest. 1996;109:380–6. doi: 10.1378/chest.109.2.380. [DOI] [PubMed] [Google Scholar]
- 16.Harris P, Segel N, Green I, Housley E. The influence of the airways resistance and alveolar pressure on the pulmonary vascular resistance in chronic bronhcitis. Cardiovasc Res. 1968;2:84–92. doi: 10.1093/cvr/2.1.84. [DOI] [PubMed] [Google Scholar]
- 17.Jardin F, Farcot JC, Boisante L, Prost JF, Gueret P, Bourdarias JP. Mechanism of paradoxic pulse in bronchial asthma. Circulation. 1982;66:887–94. doi: 10.1161/01.cir.66.4.887. [DOI] [PubMed] [Google Scholar]
- 18.Jellinek H, Krenn H, Oczenski W, Veit F, Schwarz S, Fitzgerald RD. Influence of positive airway pressure on the pressure gradient for venous return in humans. J Appl Physiol. 2000;88:926–32. doi: 10.1152/jappl.2000.88.3.926. [DOI] [PubMed] [Google Scholar]
- 19.Nichols WM, O’Rourke MF, editors. McDonald's Blood Flow in Arteries. 3rd ed. Philadelphia, PA: Lea and Febiger; 1990. The nature of flow of a fluid; pp. 12–53. [Google Scholar]
- 20.Hetzel M, Kochs M, Marx N, Woehrle H, Mobarak I, Hombach V, et al. Pulmonary hemodynamics in obstructive sleep apnea: Frequency and causes of pulmonary hypertension. Lung. 2003;181:157–66. doi: 10.1007/s00408-003-1017-y. [DOI] [PubMed] [Google Scholar]
- 21.O’Hearn DJ, Gold AR, Gold MS, Diggs P, Scharf SM. Lower extremity edema and pulmonary hypertension in morbidly obese patients with obstructive sleep apnea. Sleep Breath. 2009;13:25–34. doi: 10.1007/s11325-008-0200-z. [DOI] [PubMed] [Google Scholar]
- 22.Davidson D, Stalcup SA, Mellins RB. Systemic hemodynamics affecting cardiac output during hypocapnic and hypercapnic hypoxia. J Appl Physiol. 1986;60:1230–6. doi: 10.1152/jappl.1986.60.4.1230. [DOI] [PubMed] [Google Scholar]
- 23.Marshall JM. Peripheral chemoreceptors and cardiovascular regulation. Physiol Rev. 1994;74:543–94. doi: 10.1152/physrev.1994.74.3.543. [DOI] [PubMed] [Google Scholar]
- 24.Stewart AG, Waterhouse JC, Billings CG, Baylis PH, Howard P. Hormonal, renal, and autonomic nerve factors involved in the excretion of sodium and water during dynamic salt and water loading in hypoxaemic chronic obstructive pulmonary disease. Thorax. 1995;50:838–45. doi: 10.1136/thx.50.8.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campbell EJ, Short DS. The cause of oedema in ‘corpulmonale’. Lancet. 1960;1:1184–6. doi: 10.1016/s0140-6736(60)91062-x. [DOI] [PubMed] [Google Scholar]
- 26.Anand IS, Chandrashekhar Y, Ferrari R, Sarma R, Guleria R, Jindal SK, et al. Pathogenesis of congestive state in chronic obstructive pulmonary disease.Studies of body water and sodium, renal function, hemodynamics, and plasma hormones during edema and after recovery. Circulation. 1992;86:12–21. doi: 10.1161/01.cir.86.1.12. [DOI] [PubMed] [Google Scholar]
- 27.Baudouin SV. Oedema and cor pulmonale revisited. Thorax. 1997;52:401–2. doi: 10.1136/thx.52.5.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mannix ET, Dowdeswell I, Carlone S, Palange P, Aronoff GR, Farber MO. The effect of oxygen on sodium excretion in hypoxemic patients with chronic obstructive lung disease. Chest. 1990;97:840–4. doi: 10.1378/chest.97.4.840. [DOI] [PubMed] [Google Scholar]
- 29.de Leeuw PW, Dees A. Fluid homeostasis in chronic obstructive lung disease. Eur Respir J Suppl. 2003;46:33s–40s. doi: 10.1183/09031936.03.00000603a. [DOI] [PubMed] [Google Scholar]
- 30.Golbin JM, Somers VK, Caples SM. Obstructive sleep apnea, cardiovascular disease, and pulmonary hypertension. Proc Am Thorac Soc. 2008;5:200–6. doi: 10.1513/pats.200708-143MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kerbaul F, Brimioulle S, Rondelet R, Dewachter C, Hubloue I, Naeije R. How prostacyclin improves cardiac output in right heart failure in conjunction with pulmonary hypertension. Am J Respir Crit Care Med. 2007;175:846–50. doi: 10.1164/rccm.200611-1615OC. [DOI] [PubMed] [Google Scholar]
- 32.Kuehne T, Yilmaz S, Steendijk P, Moore P, Groenink M, Saaed M, et al. Magnetic resonance imaging analysis of right ventricular pressure-volume loops: In vi vo validation and clinical application in patients with pulmonary hypertension. Circulation. 2004;110:2010–6. doi: 10.1161/01.CIR.0000143138.02493.DD. [DOI] [PubMed] [Google Scholar]
- 33.Butler J. The heart is not always in good hands. Chest. 1990;97:453–60. doi: 10.1378/chest.97.2.453. [DOI] [PubMed] [Google Scholar]
- 34.Settle HP, Jr, Engel PJ, Fowler NO, Allen JM, Vassallo CL, Hackworth JN, et al. Echocardiographic study of the paradoxical arterial pulse in chronic obstructive lung disease. Circulation. 1980;62:1297–1307. doi: 10.1161/01.cir.62.6.1297. [DOI] [PubMed] [Google Scholar]
- 35.Arcasoy SM, Christie JD, Ferrari VA, Sutton MS, Zisman DA, Blumenthal NP, et al. Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. Am J Respir Crit Care Med. 2003;167:735–40. doi: 10.1164/rccm.200210-1130OC. [DOI] [PubMed] [Google Scholar]
- 36.Fisher MR, Forfia PR, Chamera E, Housten-Harris T, Champion HC, Girgis RE, et al. Accuracy of Doppler echocardiography in the hemodynamic assessment of pulmonary hypertension. Am J Respir Crit Care Med. 2009;179:615–21. doi: 10.1164/rccm.200811-1691OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forfia PR, Fisher MR, Mathai SC, Housten-Harris T, Hemnes AR, Borlaug BA, et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med. 2006;174:1034–41. doi: 10.1164/rccm.200604-547OC. [DOI] [PubMed] [Google Scholar]
- 38.Saxena N, Rajagopalan N, Edelman K, Lopez-Candales A. Tricuspid annular systolic velocity: A useful measurement in determining right ventricular systolic function regardless of pulmonary artery pressures. Echocardiography. 2006;23:750–5. doi: 10.1111/j.1540-8175.2006.00305.x. [DOI] [PubMed] [Google Scholar]
- 39.Thabut G, Dauriat G, Stern JB, Logeart D, Levy A, Marrash-Chahla R, et al. Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest. 2005;127:1531–6. doi: 10.1378/chest.127.5.1531. [DOI] [PubMed] [Google Scholar]
- 40.Patel NM, Lederer DJ, Borczuk AC, Kawut SM. Pulmonary hypertension in idiopathic pulmonary fibrosis. Chest. 2007;132:998–1006. doi: 10.1378/chest.06-3087. [DOI] [PubMed] [Google Scholar]
- 41.Nakatsuka M. Pulmonary vascular resistance and right ventricular function in morbid obesity in relation to gastric bypass surgery. J Clin Anesth. 1996;8:205–9. doi: 10.1016/0952-8180(95)00231-6. [DOI] [PubMed] [Google Scholar]
- 42.Chaouat A, Naeije R, Weitzenblum E. Pulmonary hypertension in COPD. Eur Respir J. 2008;32:1371–85. doi: 10.1183/09031936.00015608. [DOI] [PubMed] [Google Scholar]
- 43.Pynnaert C, Lamotte M, Naeije R. Aerobic exercise capacity in COPD patients with and without pulmonary hypertension. Respir Med. 2010;104:121–6. doi: 10.1016/j.rmed.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 44.Cor Pulmonale. Diseases of the Heart and Circulation. In: Wood Paul Hamilton., editor. 3rd ed. London: Eyre and Spottiswoode; 1968. [Google Scholar]
- 45.Boutou AK, Pitsiou GG, Trigonis I, Papakosta D, Kontou PK, Chavouzis N, et al. Exercise capacity in idiopathic pulmonary fibrosis: The effect of pulmonary hypertension. Respirology. 2011;16:451–8. doi: 10.1111/j.1440-1843.2010.01909.x. [DOI] [PubMed] [Google Scholar]
- 46.Glaser S, Noga O, Koch B, Opitz CF, Schmidt B, Temmesfeld B, et al. Impact of pulmonary hypertension on gas exchange and exercise capacity in patients with pulmonary fibrosis. Respir Med. 2009;103:317–24. doi: 10.1016/j.rmed.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 47.Vonbank K, Funk GC, Marzluf B, Burian B, Ziesche R, Stiebellehner L, et al. Abnormal pulmonary arterial pressure limits exercise capacity in patients with COPD. Wien Klin Wochenschr. 2008;120:749–55. doi: 10.1007/s00508-008-1103-5. [DOI] [PubMed] [Google Scholar]
- 48.Naeije R, Huez S. Reflections on wave reflections in chronic thromboembolic pulmonary hypertension. Eur Heart J. 2007;28:785–7. doi: 10.1093/eurheartj/ehm040. [DOI] [PubMed] [Google Scholar]
- 49.Arkles JS, Opotowsky AR, Ojeda J, Rogers F, Liu T, Prassana V, et al. Shape of the right ventricular Doppler envelope predicts hemodynamics and right heart function in pulmonary hypertension. Am J Respir Crit Care Med. 2011;183:268–76. doi: 10.1164/rccm.201004-0601OC. [DOI] [PubMed] [Google Scholar]
- 50.Prasanna V, Raina A, Arkles J, Opotowsky A, Kawut S, Akaya Smith K, et al. Echo-Doppler scoring system identifies ‘proportionate’ and ‘disproportionate’ pulmonary hypertension in patients with chronic left heart disease and respiratory disease. Am J Respir Crit Care Med. 2011;183:A2294. [Google Scholar]
- 51.Chaouat A, Bugnet AS, Kadaoui N, Schott R, Enache I, Ducolone A, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172:189–94. doi: 10.1164/rccm.200401-006OC. [DOI] [PubMed] [Google Scholar]
- 52.Fartoukh M, Humbert M, Capron F, Maitre S, Parent F, Le Gall C, et al. Severe pulmonary hypertension in histiocytosis X. Am J Respir Crit Care Med. 2000;161:216–23. doi: 10.1164/ajrccm.161.1.9807024. [DOI] [PubMed] [Google Scholar]
- 53.Shorr AF, Helman DL, Davies DB, Nathan SD. Pulmonary hypertension in advanced sarcoidosis: Epidemiology and clinical characteristics. Eur Respir J. 2005;25:783–8. doi: 10.1183/09031936.05.00083404. [DOI] [PubMed] [Google Scholar]
- 54.Trad S, Amoura Z, Beigelman C, Haroche J, Costedoat N, Boutin le TH, et al. Pulmonary arterial hypertension is a major mortality factor in diffuse systemic sclerosis, independent of interstitial lung disease. Arthritis Rheum. 2006;54:184–91. doi: 10.1002/art.21538. [DOI] [PubMed] [Google Scholar]
- 55.Saggar R, Shapiro SS, Ross DJ, Fishbein MC, Zisman DA, Lynch JP, 3rd, et al. Treprostinil to reverse pulmonary hypertension associated with idiopathic pulmonary fibrosis as a bridge to single-lung transplantation. J Heart Lung Transplant. 2009;28:964–7. doi: 10.1016/j.healun.2009.05.017. [DOI] [PubMed] [Google Scholar]
- 56.Nocturnal Oxygen Therapy Trial Group. Continuous or nocturnal oxygen therapy in hypoxemic chronic obstructive lung disease: A clinical trial. Ann Intern Med. 1980;93:391–8. doi: 10.7326/0003-4819-93-3-391. [DOI] [PubMed] [Google Scholar]
- 57.Medical Research Council Working Party. Long term domiciliary oxygen therapy in chronic hypoxic cor pulmonale complicating chronic bronchitis and emphysema. Report of the Medical Research Council Working Party. Lancet. 1981;1:681–6. [PubMed] [Google Scholar]
- 58.Kerbaul F, Brimioulle S, Rondelet B, Dewachter C, Hubloue I, Naeije R. How prostacyclin improves cardiac output in right heart failure in conjunction with pulmonary hypertension. Am J Respir Crit Care Med. 2007;175:846–50. doi: 10.1164/rccm.200611-1615OC. [DOI] [PubMed] [Google Scholar]
- 59.Sun XG, Hansen JE, Oudiz RJ, Wasserman K. Exercise pathophysiology in patients with primary pulmonary hypertension. Circulation. 2001;104:429–35. doi: 10.1161/hc2901.093198. [DOI] [PubMed] [Google Scholar]
- 60.Stolz D, Rasch H, Linka A, Di Valentino M, Meyer A, Brutsche M, et al. A randomised, controlled trial of bosentan in severe COPD. Eur Respir J. 2008;32:619–28. doi: 10.1183/09031936.00011308. [DOI] [PubMed] [Google Scholar]
- 61.Olschewski H, Ghofrani HA, Walmrath D, Schermuly R, Temmesfeld- Wollbruck B, Grimminger F, et al. Inhaled prostacyclin and iloprost in severe pulmonary hypertension secondary to pulmonary fibrosis. Pneumologie. 2000;54:133–42. doi: 10.1055/s-2000-9076. [DOI] [PubMed] [Google Scholar]
- 62.Han MK, Bach D, Hagan P, Schmidt SL, Flaherty KR, Toews GB, et al. Presence of right ventricular dysfunction modifies treatment response to sildenafil in the step-ipf trial [abstract] Am J Respir Crit Care Med. 2011;183:A5301. [Google Scholar]
- 63.Ghofrani HA, Wiedemann R, Rose F, Schermuly RT, Olschewski H, Weissmann N, et al. Sildenafil for treatment of lung fibrosis and pulmonary hypertension: A randomised controlled trial. Lancet. 2002;360:895–900. doi: 10.1016/S0140-6736(02)11024-5. [DOI] [PubMed] [Google Scholar]
- 64.Dernaika TA, Beavin M, Kinasewitz GT. Iloprost improves gas exchange and exercise tolerance in patients with pulmonary hypertension and chronic obstructive pulmonary disease. Respiration. 2010;79:377–82. doi: 10.1159/000242498. [DOI] [PubMed] [Google Scholar]
- 65.Mercurio V, Carlomagno G, Fazio S. Response to pulmonary vasodilator treatment in a former smoker with combined interstitial lung disease complicated by pulmonary hypertension: Case report and review of the literature. Heart Lung. 2012;41:512–7. doi: 10.1016/j.hrtlng.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 66.Barnett CF, Bonura EJ, Nathan SD, Ahmad S, Shlobin OA, Osei K, et al. Treatment of sarcoidosis-associated pulmonary hypertension. A two-center experience. Chest. 2009;135:1455–61. doi: 10.1378/chest.08-1881. [DOI] [PMC free article] [PubMed] [Google Scholar]