

Abstract
Pulmonary arterial hypertension (PAH) is a progressive disease characterized by increased pulmonary arterial resistance and vessel remodeling. Patients living with human immunodeficiency virus-1 (HIV-1) have an increased susceptibility to develop severe pulmonary hypertension (PH) irrespective of their CD4+ lymphocyte counts. While the underlying cause of HIV-PAH remains unknown, the interaction of HIV-1 proteins with the vascular endothelium may play a critical role in HIV-PAH development. Hypoxia promotes PH in experimental models and in humans, but the impact of HIV-1 proteins on hypoxia-induced pulmonary vascular dysfunction and PAH has not been examined. Therefore, we hypothesize that the presence of HIV-1 proteins and hypoxia synergistically augment the development of pulmonary vascular dysfunction and PH. We examined the effect of HIV-1 proteins on pulmonary vascular resistance by measuring pressure-volume relationships in isolated lungs from wild-type (WT) and HIV-1 Transgenic (Tg) rats. WT and HIV-1 Tg rats were exposed to 10% O2 for four weeks to induce experimental pulmonary hypertension to assess whether HIV-1 protein expression would impact the development of hypoxia-induced PH. Our results demonstrate that HIV-1 protein expression significantly increased pulmonary vascular resistance (PVR). HIV-1 Tg mice demonstrated exaggerated pulmonary vascular responses to hypoxia as evidenced by greater increases in right ventricular systolic pressures, right ventricular hypertrophy and vessel muscularization when compared to wild-type controls. This enhanced PH was associated with enhanced expression of HIF-1α and PCNA. In addition, in vitro studies reveal that medium from HIV-infected monocyte derived macrophages (MDM) potentiates hypoxia-induced pulmonary artery endothelial proliferation. These results indicate that the presence of HIV-1 proteins likely impact pulmonary vascular resistance and exacerbate hypoxia-induced PH.
Keywords: human immunodeficiency virus, chronic hypoxia, pulmonary hypertension
An estimated 1.2 million United States residents are infected with human immunodeficiency virus-1 (HIV-1) and nearly 600,000 people with acquired immune deficiency syndrome (AIDS) have died since the epidemic began.[1] Patients with HIV have increased susceptibility to develop infectious pulmonary complications such as bacterial pneumonia and tuberculosis. The advent of highly active antiretroviral therapy (HAART) and subsequent restored immune function has greatly reduced the incidence of infectious disorders and improved survival. However, noninfectious complications of HIV-1 such as pulmonary hypertension (PH) are now being recognized with increasing frequency.
Pulmonary arterial hypertension (PAH) is defined as a sustained elevation of pulmonary arterial pressure greater than 25 mmHg and a pulmonary capillary wedge pressure or left ventricular end diastolic pressure less than 15 mmHg.[2,3] Pathogenic vascular alterations in PH are characterized by abnormal muscularization of small pulmonary arteries and progressive intimal hyperplasia. Patients with severe PH may develop obstructive plexiform lesions in the distal pulmonary circulation.[4] These occlusive lesions are associated with decreased lumen cross-sectional area and progressive increases in pulmonary vascular resistance, which leads to the development of right ventricular hypertrophy (RVH) and PH. Although the underlying cause of PH remains unknown, endothelial activation and proliferation are implicated as major contributors to PH pathogenesis and progression.[5,6] Significant endothelial alterations have been identified in the pulmonary arteries of PH patients.[7] Abnormal endothelial cell growth patterns have also been documented in the vascular wall and in cultured pulmonary artery endothelial cells isolated from patients with idiopathic pulmonary arterial hypertension (IPAH).[8]
HIV-associated pulmonary arterial hypertension (HIV-PAH) was first identified in 1987[9] and has been increasingly diagnosed thereafter.[10,11] According to the most updated clinical classification of PH, HIV is designated as a Group I disease associated with the development of PAH.[12] In 2001, the incidence of PH in HIV-1 patients was estimated to be 1:200, whereas PH in the general population occurs in approximately 1-2 in one million patients.[13] More recent studies estimate that up to 1% of HIV-1 patients will develop PH.[10] This increase is still likely to underestimate the true incidence of HIV-PAH because the complication is not routinely evaluated in patients and is often misdiagnosed.[11] HIV-PAH also progresses more rapidly[14] and is associated with a poorer prognosis than PH in the general population.[15] Furthermore, epidemiological studies demonstrate that the survival of patients with HIV-PAH is significantly less than other patients classified with Group I PAH, including IPAH-, collagen vascular disease-, and congenital heart disease-associated PAH.[16]
The pathogenic mechanisms that predispose HIV patients to develop PAH are unclear. However, studies show that HIV-PAH occurs in the absence of any apparent lung disease and PAH disease severity does not correlate with CD4+ lymphocyte count.[13,14] In addition, HIV has not been found in endothelial cells of patients who develop PAH[17,18] nor has HIV DNA, RNA, or p24 antigen been detected in the pulmonary vessels of HIV-PAH patients.[19–21] These data suggest the pathogenesis of HIV-PAH is unrelated to infection or immune dysfunction and may be partially attributable to the indirect actions of HIV-1 proteins on the vasculature.[18] Supporting evidence demonstrate that the HIV proteins Tat, Nef and gp120 are able to enter the endothelium[22,23] and alter cell function. HIV-1 proteins may also act as a “priming” mechanism to increase susceptibility of vascular endothelial cells to a “second hit” which promotes PH development.[24–27]
Previous studies in an HIV-1 Tg model indicate that the expression of HIV-1 proteins results in the development of PH.[28,29] The present study extends these results by examining the response of the HIV-1 Tg to chronic hypoxia in a strain that is more resistant to PH. Therefore, these studies address how HIV-1 proteins may impact the response to conditions that promote PH. Our results indicate that HIV-1 protein expression alters pulmonary vascular resistance (PVR), promotes cellular proliferation, and exacerbates the development of hypoxia-induced pulmonary hypertension.
MATERIALS AND METHODS
Animals
Male Fischer 344 Wild-type (WT) and HIV-1 Transgenic (Tg) rats were obtained from Harlan (Indianapolis, Ind.) and bred in the animal facility at the Atlanta VA under a 12:12 light-dark cycle. All studies were completed in compliance with protocols approved by the Atlanta VA Animal Care and Use Committee. The HIV-1 Tg rats for this study were generated from established lines of an HIV provirus. This HIV-1 Tg rat model was developed at UMD using the NL4-3 gag/pol HIV-1 transgene[30] and has proviral DNA with deleted gag and pol but intact env and tat, nef, rev, vif, vpr, and vpu accessory genes.[31,32] HIV-1 transgenic rats have dense cataracts at birth but otherwise appear healthy. However, by six months of age, they begin to display evidence of systemic disease including poor weight gain and muscle atrophy that progresses over time.[30] HIV-1 transgene expression has been detected in the intestines and at low levels in kidney, lymph nodes, lung and spleen.[30,33] Hemizygous rats, ages seven to nine months, were used for this study.
Chronic hypoxia model
A chronic hypoxia model that has been previously used by our lab to induce experimental PH, RVH, and pulmonary vascular remodeling was employed in the current study.[34] Rats were housed in normoxic (21% O2) or hypoxic (10% O2) conditions. Hypoxic conditions were created by infusing nitrogen gas into an enclosed chamber until the desired oxygen tension was reached (Biospherix). Rats placed in the hypoxia chamber remained there for four weeks to induce experimental pulmonary hypertension. Food and water were provided ad libitum.
Isolated perfused lung
Rats were anesthetized with isoflourane and mechanically ventilated after tracheal cannulation at a rate of ~60 strokes per minute with a tidal volume of 2.5 mL/breath. The heart and lungs were exposed by thoracotomy, and heparin was administered. The pulmonary artery was cannulated with a 14 G cannula connected to a pressure transducer (AD Instruments, Colorado Springs, Colo., USA). The pulmonary artery was then perfused with 37°C Hanks’ Balanced Salt Solution (Sigma Chemical, St Louis, Mo.) at a rate of 7 ml/min for five minutes while the right atrium was incised to allow removal of the perfusate. Pressure/volume relationships were generated using a calibrated peristaltic pump at flow rates of 7, 16, 26, and 35 ml/min. Data were collected using the Power Lab digital acquisition and analyzed using Chart software (AD Instruments, Colorado Springs, Colo., USA).
Hematocrit measurements
Whole blood was collected and placed in heparinized, microcapillary tubes (Fisher). Blood was subjected to centrifugation using an Adams MHCT II. Hematocrit levels were determined using an International Micro-Capillary Reader. Values are expressed as percentage of red blood cells in the blood sample.
Right ventricular systolic pressure
Right ventricular systolic pressure (RVSP) was assessed using a 0.8 F microtip pressure transducer (Millar Instruments) as previously described.[34] In brief, rats were anesthetized with isoflourane and the 0.8 F microtip pressure transducer was inserted into the right jugular vein and advanced to the right ventricle. Right ventricular pressure was monitored for a period of 10 minutes. Data were analyzed using the Powerlab system (AD Instruments, Denver, Colo., USA).
Right ventricular hypertrophy measurements
Right ventricular hypertrophy was assessed by determining the Fulton index of right ventricle/left ventricle + septum (RV/LV + S) gross weight ratios as previously described.[34]
Histology
The pulmonary circulation was perfused with phosphate buffered saline (PBS) to remove red blood cells followed by perfusion with calcium-free PSS. Lungs were inflated with 4% paraformaldehyde via the trachea. Perfused lungs were immersed in 4% paraformaldehyde, embedded in paraffin, and sectioned for analysis. Medial thickness of pulmonary arteries was quantified by measuring wall thickness, as delineated by the internal and external elastic lamina, and expressing it as a percentage of the vessel diameter. In addition, the percentage of smooth muscle within the media was also determined by staining with antibodies to smooth muscle α-actin (Sigma, St Louis, Mo., USA). Immunohistochemistry samples were quantitated using Scion software (Scion Corporation, Frederick, Md., USA). Photomicrographs were obtained using a Leica DM4000B microscope.
Western blot analysis
Rat lung homogenates were subjected to Western Blot analysis as reported.[35] Briefly, 40 micrograms of protein were loaded per lane and subjected to SDS-PAGE. Protein samples were then transferred to nitrocellulose membranes using the Fast Semi-Dry Blotter according to manufacturer's instruction (Thermo Scientific). After blocking in 5% nonfat dried milk (NFDM), membranes were placed either (1) in a goat primary antibody solution (1:400 dilution) raised against HIF-1alpha (Novus Biologicals), (2) overnight at 4°C then fluorescent anti-goat, or (3) in a 1:500 dilution of rabbit primary antibody solution raised against PCNA (Abcam). Membranes were incubated overnight at 4°C, washed with TBST, and placed in solutions containing fluorescent anti-goat or anti-rabbit secondary antibodies. Immunoreactive bands were detected using the Licor system and the proteins of interest were quantified by densitometry and normalized to beta-actin levels within the same sample.
Cell proliferation assays
Human pulmonary arterial endothelial cell (HPAEC) proliferation was assessed using MTT Assay (ATCC). Briefly, proliferating cells reduce the tetrazolium MTT resulting in intracellular formazan. Detergent reagent was added to cell to solubilize formazan. Supernatants were then collected and quantified using a spectrophotometer at 562 nm. HPAEC were seeded at 10,000 cells/well in 24-well plates. Selected cells were cultured in an incubator under normoxic conditions (21% O2, 5% CO2), and others were exposed to hypoxic conditions (1% O2, 5% CO2) in a hypoxia chamber (Biospherix, Lacona, N.Y., USA) for 72 hours. Following exposure to normoxic or hypoxic conditions, cells were subjected to MTT assay according to the manufacturer's protocol.
Monocyte-derived macrophages
Medium from infected and control monocyte-derived macrophages were obtained from Dr. William Tyor of the Atlanta VA Medical Center. Primary human monocyte-derived macrophages (MDM) were cultured at 37°C with 5% CO2 in DMEM containing 10% human serum, L-glutamine, penicillin-streptomycin, and macrophage colony stimulating factor (M-CSF) for seven days. MDM (5 × 106) were infected with HIV-1ADA (clade B) at a multiplicity of infection (MOI) of 0.1 for one hour. Following infection, MDM were resuspended in medium devoid of M-CSF and cultured for 14 days with media changes every three days.[36] HIV-1 p24 levels were measured in media (1:10,000 dilutions) by ELISA (Advanced BioScience Laboratories, Kensington, Md., USA).
Exosome isolation
Exosomes were isolated from HIV-MDM medium using the ultra-centrifugation method as previously described.[37] Briefly, medium was subjected to repeated centrifugations to remove dead cells and debris at 200 and 3,000 rpm at 4°C, respectively. Medium was then spun down at 100,000 rpm for 70 minutes at 10°C using a Beckman XL-90 ultracentrifuge and Type 50.4 TI rotor. Supernatants and exosome-containing preparations were carefully collected and stored at 4°C.
Statistical analysis
A student's t-test analysis was used for comparison of two groups. One-way ANOVA with Tukey's post-test was used for the comparison of multiple groups. Statistical significance was defined as P < 0.05.
RESULTS
Effects of HIV-1 protein expression on pulmonary vascular resistance
Pressure-flow measurements were obtained to examine pulmonary vascular reactivity in WT and HIV-1 Tg pulmonary arteries. In response to increases in pulmonary flow, lungs from HIV-1 Tg rats showed significantly greater elevations in pressure when compared to wild-type controls (Fig. 1). These data indicate that vessels from HIV-1 Tg animals are less able to regulate pressure responses due to alterations in blood flow (n = 4-5; P < 0.0001). Furthermore, this data indirectly demonstrates that HIV-1 proteins can affect pulmonary vascular resistance.
Figure 1.

HIV-1 Tg expression (dotted line) increases pulmonary vascular resistance when compared to WT rats (solid line) (n = 4-5). Rats were anesthetized with isoflourane, mechanically ventilated and the pulmonary artery was cannulated with a 14G cannula connected to a pressure transducer. Pressure/volume relationships were generated using a calibrated peristaltic pump at flow rates of 7, 16, 26 and 35 ml/min. *denotes P < 0.0001 when compared to pulmonary arteries of wild-type controls.
Effect of HIV-1 proteins on hypoxia-induced HIF-1alpha expression
Hematocrit levels were measured to determine whether hypoxia exposure affected red blood cell counts in WT and HIV-1 Tg rats similarly. Increases in serum red blood cell counts occur in response to decreased oxygen tension. Following the 4 weeks of normoxia or hypoxia exposure, rats were sacrificed and blood was collected via cardiac puncture. Blood from hypoxic wild-type and HIV Tg mice exhibited a significant increase in the percentage of red blood cells when compared to normoxic controls (45.1% following normoxia vs. 58.2% following hypoxia; P < 0.0001; n = 12-16). Hematocrit levels between normoxic wild-type and HIV-1 Tg rats were not significantly different and there was no difference between hematocrit levels of hypoxic wild-type and HIV-1 Tg animals (data not shown). We also assessed whether HIV-1 protein expression affects hypoxia-induced HIF-1alpha protein expression. While no differences in lung HIF-1alpha expression were found between WT or HIV-1 Tg animals exposed to normoxic conditions, the increase in HIF-1alpha expression in HIV-1 Tg rats exposed to hypoxia was significantly greater when compared to normoxic animals and hypoxic wild-type controls (Fig. 2).
Figure 2.
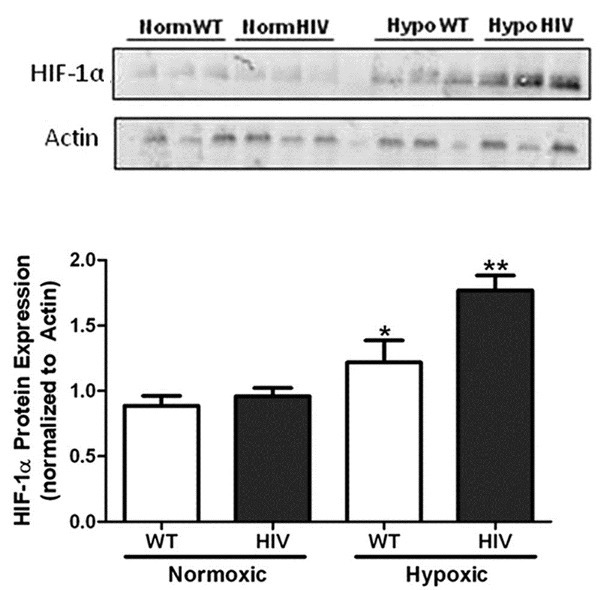
Hypoxia exposure induces greater HIF-1alpha expression in HIV-1 Tg animals (n = 3). Wild-type and HIV-1 Tg rats were housed in either normoxic or hypoxic conditions for four weeks. Lung homogenates were subjected to SDS-PAGE, transferred to nitrocellulose membranes and exposed to anti-HIF-1alpha antibodies overnight at 4°C, rinsed and incubated in anti-rabbit fluorescent antibody solution. Hypoxia exposure stimulates HIF- 1alpha protein expression in rat lung homogenates when analyzed by Western blot analysis (A). Lung homogenates from HIV-1 Tg animals exhibit a 2-fold increase in HIF-1alpha protein expression following hypoxia exposure (B). *denotes P < 0.01 when compared to normoxic groups. **denotes P < 0.05 when compared to hypoxic wild-types.
Effects of HIV-1 protein expression on hypoxia-induced right ventricular systolic pressures and right heart hypertrophy
Right ventricular systolic pressures (RVSP) and right ventricular hypertrophy (RVH) are indices used to confirm the presence of PH. To determine whether HIV-1 protein expression exacerbates hypoxia-induced PH, we assessed RVSP and RVH following four weeks of hypoxia exposure in WT and HIV-1 Tg rats. HIV-1 Tg rats between 7-12 months of age demonstrate no change in RVSP and RVH following normoxia when compared to wild-type animals (data not shown). HIV-1 Tg rats (P < 0.0001) exhibit greater elevations in RVSP in response to hypoxia when compared to hypoxic wild-type and normoxic control rats Fig. 3A. Hypoxic HIV-1 Tg rats also exhibit marked increases in RVH when compared to hypoxic wild-type and normoxic control rats Fig. 3B. These results demonstrate that the expression of the HIV-1 transgene causes exaggerated pulmonary vascular responses to hypoxia and structural alterations to the right ventricle.
Figure 3.
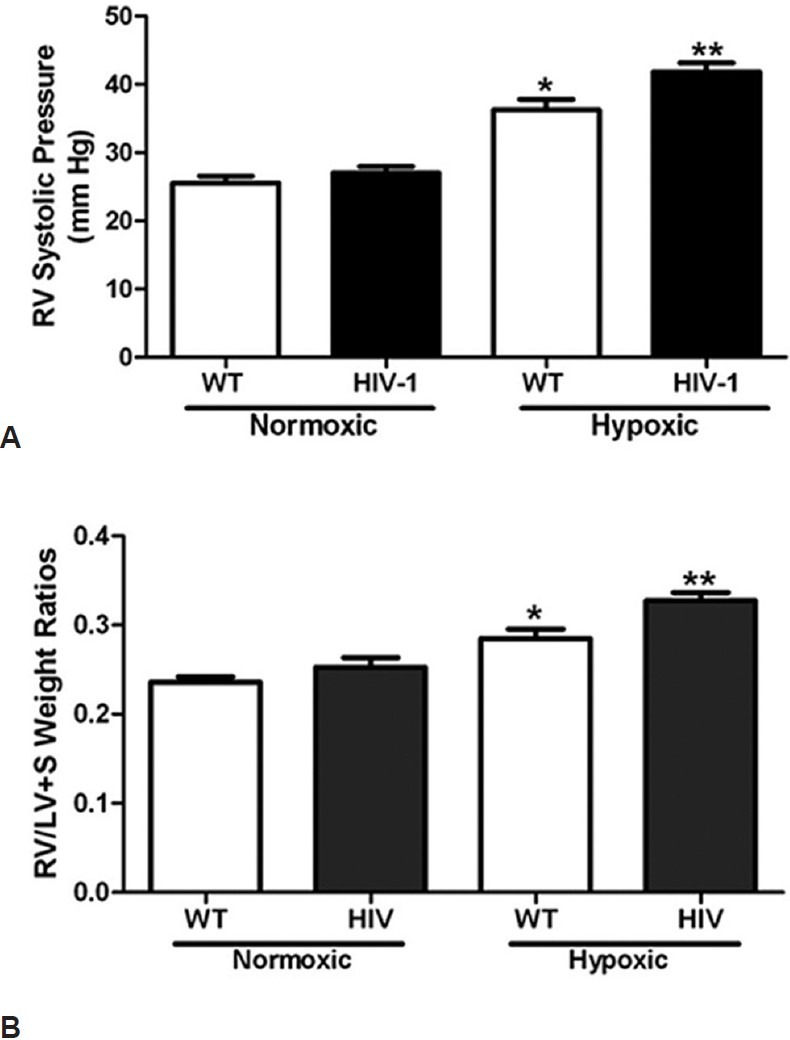
HIV-1 transgene expression exacerbates PH development. Hypoxic HIV-1 rats have significantly greater RVSP (A) (n = 6-11) and right ventricular hypertrophy (B) (n = 7-9) than normoxic controls and hypoxic wild-types. Wild-type and HIV-1 Tg rats were housed in either normoxic or hypoxic conditions for four weeks. Rats were anesthetized with isoflourane and right ventricular pressure was monitored using a microtip pressure transducer. * denotes P < 0.0001 when compared to normoxic groups. ** denotes a P < 0.01 when compared to hypoxic wild-types.
Effect of HIV-1 on PH-induced vessel wall thickness
PH is associated with enhanced vascular wall cell proliferation and vascular remodeling. These alterations progressively narrow the vascular lumen and increase the pulmonary vascular resistance. To determine if HIV-1 transgene expression causes vascular remodeling as evidenced by increased vessel wall thickness, the smooth muscle actin-alpha content was measured in rat pulmonary arteries by immunostaining. In representative images showing smooth muscle actin-alpha Fig. 4A, pulmonary arteries from hypoxic HIV-1 Tg rats exhibit a greater staining intensity when compared to normoxic HIV-1 or WT pulmonary arteries. Fig. 4B shows a graphical representation of smooth muscle actin-alpha staining in pulmonary vessels. Pulmonary arteries from hypoxia-exposed HIV Tg rats demonstrated significant increases in vessel thickness when compared to all other groups. There was no difference in pulmonary artery vessel thickness between normoxic wild-type and HIV-1 Tg rats.
Figure 4.
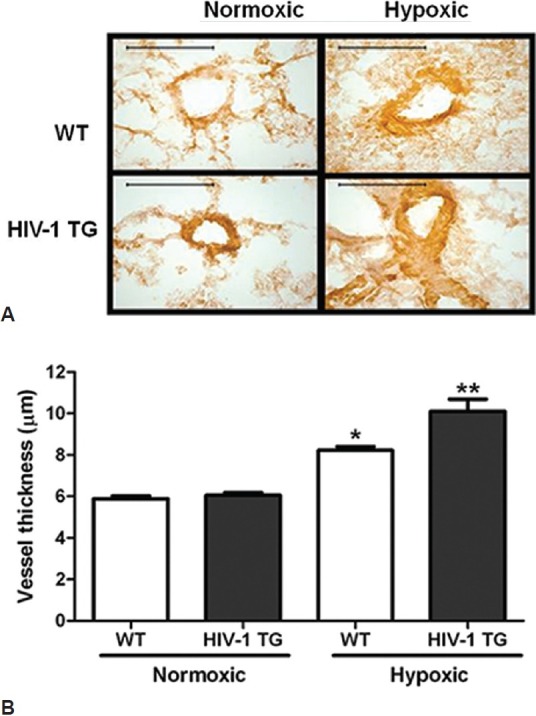
HIV-1 Tg expression increases hypoxia-induced vessel muscularization (A) (n = 3). Rats were exposed to normoxia or hypoxia for four weeks. Rat lungs were isolated, pressure perfused, inflated and immersed with 4% paraformaldehyde and embedded in paraffin. The percentage of smooth muscle within the media was determined by staining with antibodies to smooth muscle α-actin (α-SMA).Scale bar in each image = 50 μm. Hypoxia exposure increases HIV-1 Tg rat vessel thickness (B). **denotes P < 0.0001 when compared to normoxic groups. **denotes P < 0.01 when compared to hypoxic controls.
Effect of HIV-1 proteins on cell proliferation
The excessive proliferation of pulmonary endothelial and smooth muscle cells is believed to contribute to the abnormal vascular phenotype characteristic of PH. To examine whether HIV-1 proteins exacerbate hypoxia-induced cellular proliferation in vivo, the proliferating cell nuclear antigen (PCNA) content in lung homogenates from HIV-1 Tg rats was assessed by Western blot. Representative Western blot images show that lungs from hypoxic HIV-1 Tg rats express significantly greater PCNA than normoxic WT or HIV-1 Tg animals (Fig. 5A). To assess whether HIV-1 proteins exacerbate hypoxia-induced proliferation in vivo, we exposed HPAEC to 1% oxygen for 72 hours in the presence or absence of medium of HIV-infected monocyte derived macrophage (HIV-MDM) diluted to approximate p24 levels observed in infected patients or 50 pg/ml.[38] Our data indicated that hypoxia-exposed HPAEC cultured in HIV-MDM medium exhibit significant increases in cell proliferation when compared to all other treatment groups (Fig. 5B). Results also indicated that exosomes mediate the HIV-MDM-induced potentiating in cellular proliferation following hypoxia exposure. Whereas HIV-MDM medium devoid of exosomes (MDM-) produces no additive effect on hypoxia-induced proliferation, HIV-MDM medium containing exosomes (MDM+) induce further increases HPAEC proliferation following hypoxia exposure (Fig. 5C).
Figure 5.
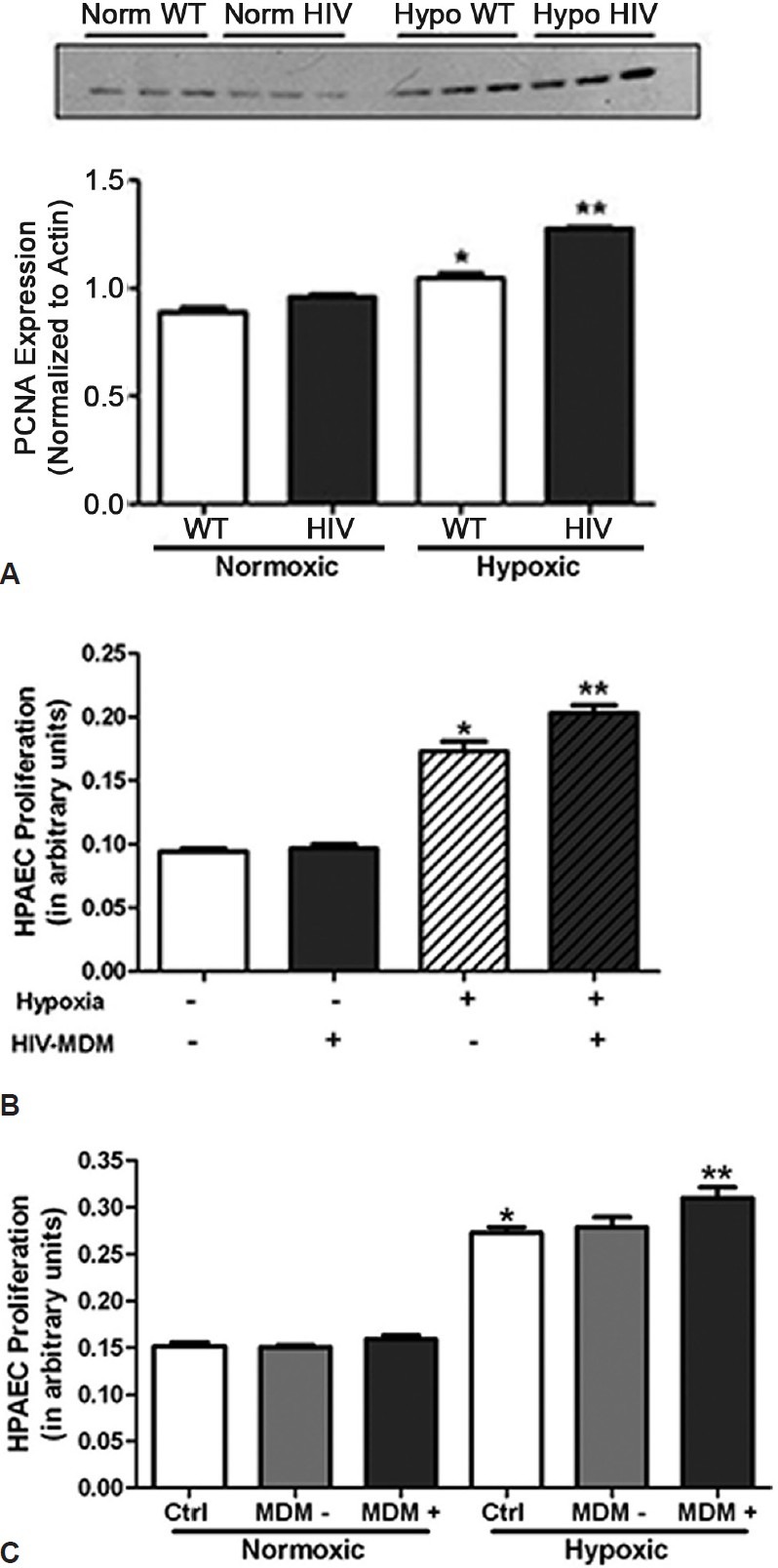
HIV-1 protein exposure exacerbates pulmonary cellular proliferation. Lung homogenates from HIV-1 Tg rats express significantly greater PCNA than normoxic WT or HIV-1 Tg rats (A) (n = 4). Following four weeks of normoxic or hypoxic conditions, rats were sacrificed and lungs removed for protein expression analysis. Lung homogenates were subjected to SDS-PAGE, transferred to nitrocellulose membranes and stained with anti-PCNA antibodies. *denotes P < 0.05 when compared to normoxic controls. ** denotes P < when compared to all other groups. Incubation of HPAEC treated with media from HIV-infected Monocyte derived Macrophages exacerbates hypoxia-induced proliferation of human pulmonary artery endothelial cells (B) (n = 4). Exosome-containing fractions of HIV-MDM (MDM+) potentiate hypoxia-induced HPAEC proliferation when compared to HIV-MDM devoid of exosomes (MDM-) (C) (n = 4). HPAEC were exposed to 1% oxygen for 72 hours in the presence or absence of HIV-MDM media. Following exposure to normoxic or hypoxic conditions, cells were subjected to MTT assay to assess cell proliferation. *denotes P < 0.0001 when compared to treated and untreated normoxic groups. **denotes P < 0.01 when compared to untreated hypoxic groups.
DISCUSSION
In this study, we demonstrate that HIV-1 protein expression promotes PH development following prolonged hypoxia exposure. Using an HIV-1 transgenic rat model, we show that HIV-1 protein expression alters pulmonary vascular resistance and exacerbates hypoxia-induced increases in right ventricular systolic pressures, right ventricular hypertrophy, and vessel muscularization. In addition, in vivo and in vivo studies demonstrate that HIV-1 protein exposure exacerbates pulmonary cell proliferation following prolonged hypoxia exposure.
Our experimental design employed the NL4-3 ∆ gag/pol HIV-1 transgenic (Tg) rat model. The HIV-1 transgenic model allows the study of the physiological effects of HIV-1 proteins in vivo in a noninfectious and relevant manner. Due to the deletion of gag and pol regions of the viral genome, this animal model expresses a non-replicative HIV-1 provirus under the viral promoter. The transgene encodes for the viral genes env, tat, nef, rev, vif, vpr and vpu.[30] These levels of viral gene products are consistent with those observed in the blood and lymphoid tissue of HIV-1 patients. Moreover, HIV-1 gp 120 has been detected in bronchoalveolar lavage fluid (BALF) and serum of HIV-1 Tg rats at approximate levels of 10 ng/ml and 28 ng/ml, respectively.[39] The transgenic rat is also a useful model for studying HIV-1-associated neurological and cardiac pathologies because it displays clinical manifestations that resemble those seen in HIV-1 patients.[30] Thus, the transgenic HIV-1 model is a useful and appropriate tool for examining the physiological consequences of HIV-1 proteins on cells of the vasculature.
In these studies, HIV-1 Tg rats were exposed to normobaric hypoxia to examine the contribution of HIV-1 proteins to PH development. These studies are clinically relevant as chronic hypoxia occurs as a result of pulmonary parenchymal disease, sleep disordered breathing and severe chronic obstructive pulmonary disease (COPD).[40] Additionally, pulmonary hypertension is usually of mild-to-moderate severity in COPD patients but is nonetheless associated with increased risk of exacerbations and decreased survival.[41] Consistent with clinical studies and statistics, our results showed that the presence of HIV-1 exacerbates PH development following hypoxia exposure. HIV-1 Tg rats demonstrated increased systolic pressures and right heart hypertrophy when compared to wild-type controls. In addition, pulmonary arteries of hypoxic HIV-1 Tg rats exhibited a 65% increase in vessel muscularization and cellular proliferation when compared to normoxic controls.
Interestingly, in our model, the expression of HIV-1 proteins failed to induce PH under normoxic conditions. Yet, hypoxia exposure markedly exacerbates PH development in wild-type and HIV-1 Tg animals. These results conflict with data recently published demonstrating that HIV-1 Tg animals spontaneously exhibit PH.[28,29] Several potential factors may provide an explanation for these conflicting results. Studies published by Lund et al. were performed in Albuquerque, New Mexico, USA, which is located at an elevation of 5,312 feet above sea level. This altitude could result in hypoxic challenge and is associated with altitude-related lung diseases such as high altitude pulmonary edema and chronic mountain sickness.[42] Additionally, other research indicated that HIV-1 Tg animals display increases in pulmonary vessel wall thickness and HIF-1α expression independent of hypoxia exposure.[29] However, strain differences may account for the varied results.[43–46] The animals used in the Mermis et al.[29] studies were completed in HIV Tg rats in the Sprague Dawley strain, whereas our studies were completed in rats on the Fischer 344 background. Research demonstrated that this strain is more resistant to hypoxia-induced PH. For example, rats on the Wistar-Kyoto background exhibit an exacerbated PH following prolonged hypoxia exposure compared to those on the Fischer 344 background.[43] Moreover, isolated perfused lung preparations from Fischer 344 rats demonstrate a reduced pulmonary vascular response to alveolar hypoxia when compared to lungs from Sprague Dawley rats.[47] These studies suggest that Fischer 344 rats may be more resistant to vascular injury and likely explain the differences in outcome.
The underlying mechanism of HIV-related PH is unknown. However, research suggests that endothelial cell dysfunction and excessive proliferation may contribute to HIV-PH pathogenesis. Studies show that the endothelium is continually exposed to actively secreted viral proteins due to its position between the blood and the vascular wall.[48] Clinical studies revealed that antiretroviral-naïve HIV-1 patients display markers of endothelial activation. Plasma levels of von Willebrand factor, plasminogen activator inhibitor-1 antigen, and tissue-type plasminogen activator are significantly elevated in HIV-1 positive patients.[49,50] In addition, comparison of flow-mediated dilation (FMD) of the brachial artery in 75 HIV-1 positive and 223 control subjects revealed significantly impaired endothelial function in the HIV-1-infected population.[51] A smaller study also showed that both treated and HAART-naïve HIV-1 infected children between 3.5 to 19.5 years old have significantly reduced FMD compared to non-infected age- and sex-matched controls.[52] These studies suggest that HIV-1 significantly affects endothelial cell function in the absence of cardiovascular risk factors and irrespective of age or duration of infection.
In addition to the cardiovascular effects, HIV-1 also significantly impairs lung function.[53] Despite the availability of combination antiretroviral therapies, respiratory complications and chronic lung disease remain common among HIV-infected individuals,[54–56] and rates of hospitalization and deaths from pulmonary obstructive diseases are growing.[57,58] Lung cancer[59] and chronic obstructive pulmonary disease (COPD)[60,61] diagnoses are significantly higher in the HIV-1 population. Recent studies also report that HIV-1 patients are almost three times more likely to develop asthma than the general population, with no correlation between the risk of asthma and viral load or CD4 lymphocyte count.[62] These studies highlight the targeted effects of HIV-1 on the lung and the contribution of HIV-1 to pulmonary disease.
Although we utilized a noninfectious, replication-incompetent animal model, we report that HIV-1, particularly HIV-1 proteins, contributes to PH pathogenesis. These data also indicate that HIV-1 protein exposure potentiates hypoxia-induced endothelial proliferation. Although previous studies report that hypoxia exposure fails to promote endothelial cell proliferation,[63] our findings demonstrating that 72 hours of hypoxia promotes endothelial cell proliferation and are consistent with published data.[64,65] Furthermore, our studies suggest that exosomes contribute to the HIV-MDM-induced potentiation of pulmonary artery endothelial proliferation following hypoxia exposure. We postulate that the increase in cellular proliferation is linked to the dysregulation of HIF-1α, as hypoxia significantly increases HIF-1alpha expression in HIV-1 Tg animals. This finding is interesting as HIF-1alpha is suggested to regulate the metabolic shift to glycolysis in pulmonary hypertensive endothelial cells associated with abnormal proliferation and apoptosis-resistance.[66] Hypoxia-induced increases in HIF-1α also promote reductions in Nrf2 expression and activity in human lung endothelial cells.[67] Moreover, inhibition of HIF-1α by the cardiac glycoside digoxin attenuates hypoxia-induced increases in RV pressures, RV hypertrophy, and vascular remodeling.[68] These data suggest that hypoxia-induced HIF may contribute to PH pathogenesis by stimulating endothelial cell proliferation, reducing antioxidant availability, and altering pulmonary vascular function.
At this time, it is unclear how HIV-1 proteins mediate the effects seen in this study. However, the HIV-1 proteins (tat, gp 120, and/or Nef) have been shown to affect endothelial cell function and are suggested to mediate PH pathogenesis and progression in HIV-1 patients. The HIV-1 protein Tat is a transcriptional transactivator with the ability to upregulate viral gene expression by increasing the rates of transcription initiation and elongation.[69] As a secreted protein from HIV-1 infected cells, Tat has been shown to enter non-infected cells from the circulation and alter normal cellular function.[48] With this capacity, Tat is known to induce endothelial activation,[70,71] inflammation,[72] cell growth,[73] and injury. Porcine coronary arteries exhibit significant decreases in eNOS expression and alterations in endothelium-dependent relaxation when incubated in HIV-1 Tat.[74] Tat transgenic mice also exhibit marked increases in oxidant levels and oxidative stress genes when compared to wild-type mice.[24] Additionally, HIV-1 Tat stimulates the production of proinflammatory cytokines, such as IL-6, which has been found in the lungs of patients with severe PAH.[75]
Another potential mediator is the HIV-1 accessory protein Nef. Nef is found in HIV-infected patients at a level of 10 ng/mL[76] and may mediate PH development by altering pulmonary morphology. For example, Marecki reported that simian HIV carrying a functional Nef protein promotes vascular remodeling[77,78] and induces PAH in macaques. The same study also showed an enhanced accumulation of Nef in endothelial cells in plexiform lesions in lungs of patients with HIV-PAH. In addition, Nef administration is shown to alter endothelium-dependent relaxation, decrease HPAEC eNOS mRNA levels, and increase superoxide production in porcine pulmonary arteries.[79]
The HIV-1 surface protein gp120 is also implicated in the development of pulmonary vascular disorders.gp120 is actively secreted into the bloodstream and is readily detectable in sera of HIV-1 infected patients at concentrations ranging from 0.24 ng/mL to 92 ng/mL.[80] In 2005, Kanmogne showed that gp 120 significantly increases the secretion of endothelin-1 by human lung endothelial cells.[81] Similarly to Tat, gp 120 induces monocyte adhesion and increased ICAM-1 gene expression, but not VCAM-1 or E-selectin in human endothelial cells.[82] Furthermore, gp120-induced generation of reactive oxygen species (ROS) has been implicated in endothelial cell toxicity.[83]
Exosomes released by HIV-infected cells may also contribute to vascular injury as evidenced by the HIV-MDM-induced potentiation of hypoxia-induced proliferation. Exosomes are released by most cell types and act as intercellular signaling molecules influencing the physiology of neighboring cells.[84] Although the mechanism of action remains unclear, exosomes are thought to alter cellular function by binding to cell-surface receptors of nearby cells or by transferring both mRNA and microRNAs to recipient cells.[85] Further studies are needed to determine the role of exosomes in HIV-associated pathologies.
Oxidative stress may also mediate the development of pulmonary hypertension in our HIV-1 model. Our group has previously shown that aortas from HIV-1 Tg demonstrated increases in superoxide and 3-nitrotyrosine due to a diminished antioxidant capacity particularly GSH and Cu/Zn superoxide dismutase activity.[35] Similarly, lung lavage fluid from HIV-1 Tg rats showed decreases in GSH and increased ratios of glutathione disulfide (GSSG) to GSH.[86] In addition, hydrogen peroxide (H2O2) was increased in lung tissue from HIV-1 Tg rats.[86] These results are consistent with clinical data which shows that lung homogenates from severe PH patients demonstrate decreased total SOD activity, reduced MnSOD protein levels and increased nitrotyrosine expression.[87]
Although HIV-1 transgene expression induces significant oxidative stress, HIV-1 Tg animals do not show signs of chronic inflammation. Previous studies showed no difference in HIV-1 Tg cytokine levels when compared to wild-type controls. Lassiter et al.[86] showed similar concentrations of interleukin-2, TNF-α, and interleukin-4 in lung lavage fluids of HIV-1 Tg rats and wild-type controls. These results are consistent with previous studies performed by our group which showed no difference in GM-CSF, IL-12, IL-6, IL-10, IL-4, IL-2, IL-1β or TNF-α in HIV-1 Tg mouse serum when compared to wild-type controls.[88]
In summary, our results establish that expression of HIV-1 proteins impairs pulmonary vascular function and exacerbates hypoxia-induced PH development. Collectively, these studies underscore the potential contribution of HIV-1 proteins to HIV-PAH. Furthermore, using an HIV-1 transgenic rat model, we demonstrated that HIV-1 protein expression alters pulmonary vascular resistance and exacerbates hypoxia-induced changes in right ventricular pressure, right ventricular hypertrophy, and pulmonary vascular remodeling. In addition, in vitro and in vivo studies demonstrate that HIV-1 protein exposure exacerbates cell proliferation. These results indicate that similar to the HIV-1 patient, HIV-1 Tg rats are more susceptible to the development of pulmonary hypertension. Therefore, these studies highlight a contributing role of HIV-1 proteins in HIV-PAH pathogenesis.
ACKNOWLEDGMENTS
The authors sincerely thank Dr. Nnenna Adimora Finn for her assistance with the HIV-MDM exosome isolation studies. This research was supported by HL070892 to RLS. National Science Foundation Award #0450303 Sub award # I-66-606-63 to Emory University. Pharmacological Sciences Training Grant TM GM 008602.National Institute of Allergy and Infectious Diseases 1-F31-AI084460.
Footnotes
Source of Support: This research was supported by HL070892 to RLS, the National Science Foundation Award #0450303, Subaward # I.66-606-63 to Emory University, the Pharmacological Sciences Training Grant TM GM 008602 and the National Institute of Allergy and Infectious Diseases 1.F31.AI084460
Conflict of Interest: None declared.
REFERENCES
- 1.Centers for Disease Control and Prevention, HIV Surveillance Report: Diagnoses of HIV infection and AIDS in the United States and Dependent Areas. 2010;22 [Google Scholar]
- 2.McGoon M, Gutterman D, Steen V, Barst R, McCrory DC, Fortin TA, et al. Screening, early detection and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126:14S–34S. doi: 10.1378/chest.126.1_suppl.14S. [DOI] [PubMed] [Google Scholar]
- 3.Galie N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2009;34:1219–63. doi: 10.1183/09031936.00139009. [DOI] [PubMed] [Google Scholar]
- 4.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2008;118:2372–9. doi: 10.1172/JCI33452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang J, Wolk JH, Gewitz MH, Mathew R. Progressive endothelial cell damage in an inflammatory model of pulmonary hypertension. Exp Lung Res. 2010;36:57–66. doi: 10.3109/01902140903104793. [DOI] [PubMed] [Google Scholar]
- 6.Sakao S, Tatsumi K, Voelkel NF. Endothelial cells and pulmonary arterial hypertension: Apoptosis, proliferation, interaction and transdifferentiation. Respir Res. 2009;10:95. doi: 10.1186/1465-9921-10-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rabinovitch M, Bothwell T, Hayakawa BN, Williams WG, Trusler GA, Rowe RD, et al. Pulmonary artery endothelial abnormalities in patients with congenital heart defects and pulmonary hypertension.A correlation of light with scanning electron microscopy and transmission electron microscopy. Lab Invest. 1986;55:632–53. [PubMed] [Google Scholar]
- 8.Masri FA, Xu W, Comhair SA, Asosingh K, Koo M, Vasanji A, et al. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293:L548–54. doi: 10.1152/ajplung.00428.2006. [DOI] [PubMed] [Google Scholar]
- 9.Kim KK, Factor SM. Membranoproliferative glomerulonephritis and plexogenic pulmonary arteriopathy in a homosexual man with acquired immunodeficiency syndrome. Hum Pathol. 1987;18:1293–6. doi: 10.1016/s0046-8177(87)80417-3. [DOI] [PubMed] [Google Scholar]
- 10.Nunes H, Humbert M, Sitbon O, Morse JH, Deng Z, Knowles JA, et al. Prognostic factors for survival in human immunodeficiency virus-associated pulmonary hypertension. Am J Respir Crit Care Med. 2003;167:1433–9. doi: 10.1164/rccm.200204-330OC. [DOI] [PubMed] [Google Scholar]
- 11.Petrosillo N, Chinello P, Cicalini S. Pulmonary hypertension in individuals with HIV infection. AIDS. 2006;20:2128–9. doi: 10.1097/01.aids.0000247569.03504.8b. [DOI] [PubMed] [Google Scholar]
- 12.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 13.Speich R, Jenni R, Opravil M, Pfab M, Russi EW. Primary pulmonary hypertension in HIV infection. Chest. 1991;100:1268–71. doi: 10.1378/chest.100.5.1268. [DOI] [PubMed] [Google Scholar]
- 14.Seoane L, Shellito J, Welsh D, de Boisblanc BP. Pulmonary Hypertension Associated With HIV Infection. South Med J. 2001;94:635–9. [PubMed] [Google Scholar]
- 15.Opravil M, Pechere M, Speich R, Joller-Jemelka HI, Jenni R, Russi EW, et al. HIV-associated primary pulmonary hypertension. A case control study. Swiss HIV Cohort Study. Am J Respir Crit Care Med. 1997;155:990–5. doi: 10.1164/ajrccm.155.3.9117037. [DOI] [PubMed] [Google Scholar]
- 16.McLaughlin VV, Presberg KW, Doyle RL, Abman SH, McCrory DC, Fortin T, et al. Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126:78S–92S. doi: 10.1378/chest.126.1_suppl.78S. [DOI] [PubMed] [Google Scholar]
- 17.Humbert M, Monti G, Fartoukh M, Magnan A, Brenot F, Rain B, et al. Platelet-derived growth factor expression in primary pulmonary hypertension: Comparison of HIV seropositive and HIV seronegative patients. Eur Respir J. 1998;11:554–9. [PubMed] [Google Scholar]
- 18.Mette SA, Palevsky HI, Pietra GG, Williams TM, Bruder E, Prestipino AJ, et al. Primary pulmonary hypertension in association with human immunodeficiency virus infection.A possible viral etiology for some forms of hypertensive pulmonary arteriopathy. Am Rev Respir Dis. 1992;145:1196–200. doi: 10.1164/ajrccm/145.5.1196. [DOI] [PubMed] [Google Scholar]
- 19.Kanmogne GD, Kennedy RC, Grammas P. Analysis of Human Lung Endothelial Cells for Susceptibility to HIV Type 1 Infection, Coreceptor Expression and Cytotoxicity of gp120 Protein. AIDS Res Hum Retroviruses. 2001;17:45–53. doi: 10.1089/088922201750056771. [DOI] [PubMed] [Google Scholar]
- 20.Klings ES, Farber HW. The pathogenesis of HIV-associated pulmonary hypertension. Adv Cardiol. 2003;40:71–82. doi: 10.1159/000073176. [DOI] [PubMed] [Google Scholar]
- 21.Pellicelli AM, D’Ambrosio C, Vizza CD, Borgia MC, Tanzi P, Pino P, et al. HIV-related pulmonary hypertension.From pathogenesis to clinical aspects. Acta Cardiol. 2004;59:323–30. doi: 10.2143/AC.59.3.2005190. [DOI] [PubMed] [Google Scholar]
- 22.Liu NQ, Lossinsky AS, Popik W, Li X, Gujuluva C, Kriederman B, et al. Human Immunodeficiency Virus Type 1 Enters Brain Microvascular Endothelia by Macropinocytosis Dependent on Lipid Rafts and the Mitogen-Activated Protein Kinase Signaling Pathway. J Virol. 2002;76:6689–700. doi: 10.1128/JVI.76.13.6689-6700.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gujuluva C, Burns AR, Pushkarsky T, Popik W, Berger O, Bukrinsky M, et al. HIV-1 penetrates coronary artery endothelial cells by transcytosis. Mol Med. 2001;7:169–76. [PMC free article] [PubMed] [Google Scholar]
- 24.Cota-Gomez A, Flores AC, Ling XF, Varella-Garcia M, Flores SC. HIV-1 Tat increases oxidant burden in the lungs of transgenic mice. Free Radic Biol Med. 2011;51:1697–707. doi: 10.1016/j.freeradbiomed.2011.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Almodovar S, Hsue PY, Morelli J, Huang L, Flores SC. Pathogenesis of HIV-associated pulmonary hypertension: Potential role of HIV-1 Nef. Proc Am Thorac Soc. 2011;8:308–12. doi: 10.1513/pats.201006-046WR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voelkel NF, Cool CD, Flores S. From viral infection to pulmonary arterial hypertension: A role for viral proteins? AIDS. 2008;22(Suppl 3):S49–53. doi: 10.1097/01.aids.0000327516.55041.01. [DOI] [PubMed] [Google Scholar]
- 27.Zietz C, Hotz B, Sturzl M, Rauch E, Penning R, Lohrs U. Aortic endothelium in HIV-1 infection: Chronic injury, activation and increased leukocyte adherence. Am J Pathol. 1996;149:1887–98. [PMC free article] [PubMed] [Google Scholar]
- 28.Lund AK, Lucero J, Herbert L, Liu Y, Naik JS. Human immunodeficiency virus transgenic rats exhibit pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2011;301:L315–26. doi: 10.1152/ajplung.00045.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mermis J, Gu H, Xue B, Li F, Tawfik O, Buch S, et al. Hypoxia-inducible factor-1 alpha/platelet derived growth factor axis in HIV-associated pulmonary vascular remodeling. Respir Res. 2011;12:103. doi: 10.1186/1465-9921-12-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reid W, Sadowska M, Denaro F, Rao S, Foulke J, Jr, Hayes N, et al. An HIV-1 transgenic rat that develops HIV-related pathology and immunologic dysfunction. Proc Natl Acad Sci USA. 2001;98:9271–6. doi: 10.1073/pnas.161290298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dickie P, Felser J, Eckhaus M, Bryant J, Silver J, Marinos N, et al. HIV-associated nephropathy in transgenic mice expressing HIV-1 genes. Virology. 1991;185:109–19. doi: 10.1016/0042-6822(91)90759-5. [DOI] [PubMed] [Google Scholar]
- 32.Kopp JB, Klotman ME, Adler SH, Bruggeman LA, Dickie P, Marinos NJ, et al. Progressive glomerulosclerosis and enhanced renal accumulation of basement membrane components in mice transgenic for human immunodeficiency virus type 1 genes. Proc Natl Acad Sci USA. 1992;89:1577–81. doi: 10.1073/pnas.89.5.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bruggeman LA, Thomson MM, Nelson PJ, Kopp JB, Rappaport J, Klotman PE, et al. Patterns of HIV-1 mRNA expression in transgenic mice are tissue-dependent. Virology. 1994;202:940–8. doi: 10.1006/viro.1994.1416. [DOI] [PubMed] [Google Scholar]
- 34.Nisbet RE, Bland JM, Kleinhenz DJ, Mitchell PO, Walp ER, Sutliff RL, et al. Rosiglitazone attenuates chronic hypoxia-induced pulmonary hypertension in a mouse model. Am J Respir Cell Mol Biol. 2010;42:482–90. doi: 10.1165/rcmb.2008-0132OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kline ER, Kleinhenz DJ, Liang B, Dikalov S, Guidot DM, Hart CM, et al. Vascular oxidative stress and nitric oxide depletion in HIV-1 transgenic rats are reversed by glutathione restoration. Am J Physiol Heart Circ Physiol. 2008;294:H2792–804. doi: 10.1152/ajpheart.91447.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rao VR, Sas AR, Eugenin EA, Siddappa NB, Bimonte-Nelson H, Berman JW, et al. HIV-1 clade-specific differences in the induction of neuropathogenesis. J Neurosci. 2008;28:10010–6. doi: 10.1523/JNEUROSCI.2955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Théry C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2001 doi: 10.1002/0471143030.cb0322s30. [DOI] [PubMed] [Google Scholar]
- 38.Reddy MM, Sorrell SJ, Lange M, Grieco MH. Tumor necrosis factor and HIV P24 antigen levels in serum of HIV-infected populations. J Acquir Immune Defic Syndr. 1988;1:436–40. [PubMed] [Google Scholar]
- 39.Joshi PC, Raynor R, Fan X, Guidot DM. HIV-1-transgene expression in rats decreases alveolar macrophage zinc levels and phagocytosis. Am J Respir Cell Mol Biol. 2008;39:218–26. doi: 10.1165/rcmb.2007-0344OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1013–32. doi: 10.1152/ajplung.00217.2009. [DOI] [PubMed] [Google Scholar]
- 41.Chaouat A, Naeije R, Weitzenblum E. Pulmonary hypertension in COPD. Eur Respir J. 2008;32:1371–85. doi: 10.1183/09031936.00015608. [DOI] [PubMed] [Google Scholar]
- 42.Schoene RB. Lung disease at high altitude. Adv Exp Med Biol. 1999;474:47–56. doi: 10.1007/978-1-4615-4711-2_3. [DOI] [PubMed] [Google Scholar]
- 43.Aguirre JI, Morrell NW, Long L, Clift P, Upton PD, Polak JM, et al. Vascular remodeling and ET-1 expression in rat strains with different responses to chronic hypoxia. Am J Physiol Lung Cell Mol Physiol. 2000;278:L981–7. doi: 10.1152/ajplung.2000.278.5.L981. [DOI] [PubMed] [Google Scholar]
- 44.Pan L, Wilson DW, Segall HJ. Strain differences in the response of Fischer 344 and Spague-Dawley rats to monocrotaline induced pulmonary vascular disease. Toxicology. 1993;79:21–35. doi: 10.1016/0300-483x(93)90203-5. [DOI] [PubMed] [Google Scholar]
- 45.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thebaud B, Haromy A, et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation. 2006;113:2630–41. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 46.Sato K, Webb S, Tucker A, Rabinovitch M, O’Brien RF, McMurtry IF, et al. Factors influencing the idiopathic development of pulmonary hypertension in the fawn hooded rat. Am Rev Respir Dis. 1992;145:793–7. doi: 10.1164/ajrccm/145.4_Pt_1.793. [DOI] [PubMed] [Google Scholar]
- 47.He LS, Chang SW, Voelkel NF. Pulmonary vascular reactivity in Fischer rats. J Appl Physiol. 1991;70:1861–6. doi: 10.1152/jappl.1991.70.4.1861. [DOI] [PubMed] [Google Scholar]
- 48.Chang HC, Samaniego F, Nair BC, Buonaguro L, Ensoli B. HIV-1 Tat protein exits from cells via a leaderless secretory pathway and binds to extracellular matrix-associated heparan sulfate proteoglycans through its basic region. AIDS. 1997;11:1421–31. doi: 10.1097/00002030-199712000-00006. [DOI] [PubMed] [Google Scholar]
- 49.Schved JF, Gris JC, Arnaud A, Martinez P, Sanchez N, Wautier JL, et al. von Willebrand factor antigen, tissue-type plasminogen activator antigen and risk of death in human immunodeficiency virus 1-related clinical disease: Independent prognostic relevance of tissue-type plasminogen activator. J Lab Clin Med. 1992;120:411–9. [PubMed] [Google Scholar]
- 50.Lafeuillade A, Alessi MC, Poizot-Martin I, Boyer-Neumann C, Zandotti C, Quilichini R, et al. Endothelial cell dysfunction in HIV infection. J Acquir Immune Defic Syndr. 1992;5:127–31. [PubMed] [Google Scholar]
- 51.Solages A, Vita JA, Thornton DJ, Murray J, Heeren T, Craven DE, et al. Endothelial Function in HIV-Infected Persons. Clin Infect Dis. 2006;42:1325–32. doi: 10.1086/503261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bonnet D, Aggoun Y, Szezepanski I, Bellal N, Blanche S. Arterial stiffness and endothelial dysfunction in HIV-infected children. AIDS. 2004;18:1037–41. doi: 10.1097/00002030-200404300-00012. [DOI] [PubMed] [Google Scholar]
- 53.Morris A, Gingo MR, George MP, Lucht L, Kessinger C, Singh V, et al. Cardiopulmonary function in individuals with HIV infection in the antiretroviral therapy era. AIDS. 2012;26:731–40. doi: 10.1097/QAD.0b013e32835099ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.George MP, Kannass M, Huang L, Sciurba FC, Morris A. Respiratory symptoms and airway obstruction in HIV-infected subjects in the HAART era. PLoS One. 2009;4:e6328. doi: 10.1371/journal.pone.0006328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gingo MR, George MP, Kessinger CJ, Lucht L, Rissler B, Weinman R, et al. Pulmonary function abnormalities in HIV-infected patients during the current antiretroviral therapy era. Am J Respir Crit Care Med. 2010;182:790–6. doi: 10.1164/rccm.200912-1858OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Diaz PT, Wewers MD, Pacht E, Drake J, Nagaraja HN, Clanton TL. Respiratory symptoms among HIV-seropositive individuals. Chest. 2003;123:1977–82. doi: 10.1378/chest.123.6.1977. [DOI] [PubMed] [Google Scholar]
- 57.Louie JK, Hsu LC, Osmond DH, Katz MH, Schwarcz SK. Trends in causes of death among persons with acquired immunodeficiency syndrome in the era of highly active antiretroviral therapy, San Francisco, 1994-1998. J Infect Dis. 2002;186:1023–7. doi: 10.1086/343862. [DOI] [PubMed] [Google Scholar]
- 58.Grubb JR, Moorman AC, Baker RK, Masur H. The changing spectrum of pulmonary disease in patients with HIV infection on antiretroviral therapy. AIDS. 2006;20:1095–107. doi: 10.1097/01.aids.0000226949.64600.f9. [DOI] [PubMed] [Google Scholar]
- 59.Sigel K, Wisnivesky J, Gordon K, Dubrow R, Justice A, Brown ST, et al. HIV as an independent risk factor for incident lung cancer. AIDS. 2012;26:1017–25. doi: 10.1097/QAD.0b013e328352d1ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crothers K, Butt AA, Gibert CL, Rodriguez-Barradas MC, Crystal S, et al. Increased COPD among HIV-positive compared to HIV-negative veterans. Chest. 2006;130:1326–33. doi: 10.1378/chest.130.5.1326. [DOI] [PubMed] [Google Scholar]
- 61.Magalhaes MG, Greenberg B, Hansen H, Glick M. Comorbidities in older patients with HIV: A retrospective study. J Am Dent Assoc. 2007;138:1468–75. doi: 10.14219/jada.archive.2007.0083. [DOI] [PubMed] [Google Scholar]
- 62.Gingo MR, Wenzel SE, Steele C, Kessinger CJ, Lucht L, Lawther T, et al. Asthma diagnosis and airway bronchodilator response in HIV-infected patients. J Allergy Clin Immunol. 2012;129:708–714.e708. doi: 10.1016/j.jaci.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu L, Hales CA. Hypoxia does neither stimulate pulmonary artery endothelial cell proliferation in mice and rats with pulmonary hypertension and vascular remodeling nor in human pulmonary artery endothelial cells. J Vasc Res. 2011;48:465–75. doi: 10.1159/000327005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kang BY, Kleinhenz JM, Murphy TC, Hart CM. The PPAR-gamma ligand rosiglitazone attenuates hypoxia-induced endothelin signaling in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol. 2011;301:L881–91. doi: 10.1152/ajplung.00195.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schaefer CA, Kuhlmann CR, Weiterer S, Fehsecke A, Abdallah Y, Schaefer C, et al. Statins inhibit hypoxia-induced endothelial proliferation by preventing calcium-induced ROS formation. Atherosclerosis. 2006;185:290–6. doi: 10.1016/j.atherosclerosis.2005.06.035. [DOI] [PubMed] [Google Scholar]
- 66.Fijalkowska I, Xu W, Comhair SA, Janocha AJ, Mavrakis LA, Krishnamachary B, et al. Hypoxia Inducible-Factor1α Regulates the Metabolic Shift of Pulmonary Hypertensive Endothelial Cells. Am J Pathol. 2010;176:1130–8. doi: 10.2353/ajpath.2010.090832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Loboda A, Stachurska A, Florczyk U, Rudnicka D, Jazwa A, Wegrzyn J, et al. HIF-1 induction attenuates Nrf2-dependent IL-8 expression in human endothelial cells. Antioxid Redox Signal. 2009;11:1501–17. doi: 10.1089/ars.2008.2211. [DOI] [PubMed] [Google Scholar]
- 68.Abud EM, Maylor J, Undem C, Punjabi A, Zaiman AL, Myers AC, et al. Digoxin inhibits development of hypoxic pulmonary hypertension in mice. Proc Natl Acad Sci U S A. 2012;109:1239–44. doi: 10.1073/pnas.1120385109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rice AP, Mathews MB. Transcriptional but not translational regulation of HIV-1 by the tat gene product. Nature. 1988;332:551–3. doi: 10.1038/332551a0. [DOI] [PubMed] [Google Scholar]
- 70.Urbinati C, Bugatti A, Giacca M, Schlaepfer D, Presta M, Rusnati M. Alpha(v) beta3-integrin-dependent activation of focal adhesion kinase mediates NF-kappaB activation and motogenic activity by HIV-1 Tat in endothelial cells. J Cell Sci. 2005;118:3949–58. doi: 10.1242/jcs.02518. [DOI] [PubMed] [Google Scholar]
- 71.Urbinati C, Mitola S, Tanghetti E, Kumar C, Waltenberger J, Ribatti D, et al. Intergirin alphavbeta3 as a target for blocking HIV-1 Tat-induced endothelial cell activation in vitro and angiogenesis in vivo. Arterioscler Thromb Vasc Biol. 2005;25:2315–20. doi: 10.1161/01.ATV.0000186182.14908.7b. [DOI] [PubMed] [Google Scholar]
- 72.Toborek M, Lee YW, Pu H, Malecki A, Flora G, Garrido R, et al. HIV-Tat protein induces oxidative and inflammatory pathways in brain endothelium. J Neurochem. 2003;84:169–79. doi: 10.1046/j.1471-4159.2003.01543.x. [DOI] [PubMed] [Google Scholar]
- 73.Barillari G, Sgadari C, Fiorelli V, Samaniego F, Colombini S, Manzari V, et al. The Tat protein of human immunodeficiency virus type-1 promotes vascular cell growth and locomotion by engaging the alpha5beta1 and alphavbeta3 integrins and by mobilizing sequestered basic fibroblast growth factor. Blood. 1999;94:663–72. [PubMed] [Google Scholar]
- 74.Paladugu R, Fu W, Conklin BS, Lin PH, Lumsden AB, Yao Q, et al. Hiv tat protein causes endothelial dysfunction in porcine coronary arteries. J Vasc Surg. 2003;38:549–55. doi: 10.1016/s0741-5214(03)00770-5. [DOI] [PubMed] [Google Scholar]
- 75.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grandeot-Keros L, et al. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;151:1628–31. doi: 10.1164/ajrccm.151.5.7735624. [DOI] [PubMed] [Google Scholar]
- 76.Fujii Y, Otake K, Tashiro M, Adachi A. Soluble Nef antigen of HIV-1 is cytotoxic for human CD4+ T cells. FEBS Lett. 1996;393:93–6. doi: 10.1016/0014-5793(96)00859-9. [DOI] [PubMed] [Google Scholar]
- 77.Marecki J, Cool C, Voelkel N, Luciw P, Flores S. Evidence for Vascular Remodeling in the Lungs of Macaques Infected With Simian Immunodeficiency Virus/HIV NEF Recombinant Virus*. Chest. 2005;128:621S–2S. doi: 10.1378/chest.128.6_suppl.621S-a. [DOI] [PubMed] [Google Scholar]
- 78.Marecki JC, Cool CD, Parr JE, Beckey VE, Luciw PA, Tarantal AF, et al. HIV-1 Nef is associated with complex pulmonary vascular lesions in SHIV-nef-infected macaques. Am J Respir Crit Care Med. 2006;174:437–45. doi: 10.1164/rccm.200601-005OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Duffy P, Wang X, Lin PH, Yao Q, Chen C. HIV Nef protein causes endothelial dysfunction in porcine pulmonary arteries and human pulmonary artery endothelial cells. J Surg Res. 2009;156:257–64. doi: 10.1016/j.jss.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oh SK, Cruikshank WW, Raina J, Blanchard GC, Adler WH, Walker J, et al. Identification of HIV-1 envelope glycoprotein in the serum of AIDS and ARC patients. J Acquir Immune Defic Syndr. 1992;5:251–6. [PubMed] [Google Scholar]
- 81.Kanmogne GD, Primeaux C, Grammas P. Induction of apoptosis and endothelin-1 secretion in primary human lung endothelial cells by HIV-1 gp120 proteins. Biochem Biophys Res Commun. 2005;333:1107–15. doi: 10.1016/j.bbrc.2005.05.198. [DOI] [PubMed] [Google Scholar]
- 82.Ren Z, Yao Q, Chen C. HIV-1 envelope glcoprotein 120 increases intracellular adhesion molecule-1 expression by human endothelial cells. Lab Invest. 2002;82:245–55. doi: 10.1038/labinvest.3780418. [DOI] [PubMed] [Google Scholar]
- 83.Price TO, Ercal N, Nakaoke R, Banks WA. HIV-1 viral proteins gp120 and Tat induce oxidative stress in brain endothelial cells. Brain Res. 2005;1045:57–63. doi: 10.1016/j.brainres.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 84.Ludwig AK, Giebel B. Exosomes: Small vesicles participating in intercellular communication. Int J Biochem Cell Biol. 2012;44:11–5. doi: 10.1016/j.biocel.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 85.Mathivanan S, Ji H, Simpson RJ. Exosomes: Extracellular organelles important in intercellular communication. J Proteomics. 2010;73:1907–20. doi: 10.1016/j.jprot.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 86.Lassiter C, Fan X, Joshi PC, Jacob BA, Sutliff R, Jones D, et al. HIV-1 transgene expression in rats causes oxidant stress and alveolar epithelial barrier dysfunction. AIDS Res Ther. 2009;6:1. doi: 10.1186/1742-6405-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC, et al. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med. 2004;169:764–9. doi: 10.1164/rccm.200301-147OC. [DOI] [PubMed] [Google Scholar]
- 88.Jacob BA, Porter KM, Elms SC, Cheng PY, Jones DP, Sutliff RL. HIV-1-induced pulmonary oxidative and nitrosative stress: Exacerbated response to endotoxin administration in HIV-1 transgenic mouse model. Am J Physiol Lung Cell Mol Physiol. 2006;291:L811–9. doi: 10.1152/ajplung.00468.2005. [DOI] [PubMed] [Google Scholar]