

Abstract
Metabolites of arachidonic acid play an important role in mediating inflammation, cell proliferation, and oxidative stress that contribute to many pulmonary diseases. We hypothesized that the substantial differences between rats and mice in their responses to experimental pulmonary hypertensive stimuli would be associated with parallel differences in their basal eicosanoid profile. Rat and mouse lung extracts were subjected to liquid chromatography tandem mass spectrometry that was optimized for simultaneous separation and rapid quantification of the major hydroxyeicosatetraenoic acids (HETEs) and prostaglandins (PGs). Basal levels (pg/μg protein) of arachidonic acid metabolites differed significantly between rat and mouse lungs. Median values of the following major eicosanoids were significantly higher in mouse than in rat lungs: 5-HETE, 8-HETE, 12-HETE, 15-HETE, PGE2, and PGI2, as well as isoprostane-E2 and -F2α. In addition, the PGI2/TXB2 ratio was increased in mouse relative to rat lungs. On the basis of the important roles that these compounds play in determining pulmonary vascular tone, the differences in select eicosanoid profiles, especially the PGI2/TXB2 ratio, between rat and mouse lungs may underlie the interspecies differences in susceptibility to the development of pulmonary hypertension.
Keywords: eicosanoids, LC/MS/MS, lipoxygenase, liquid chromatography, prostacyclin, prostaglandin, pulmonary hypertension, tandem mass spectrometry, thromboxane
The lung vasculature synthesizes various eicosanoids derived from arachidonic acid. The conversion of arachidonic acid into many metabolites is achieved in a highly regulated manner, and many arachidonic acid derivatives have been linked to various disease states. Hydroxyeicosatetraenoic acids (HETEs) are produced enzymatically from arachidonic acid and the principal mammalian enzymes for arachidonic acid metabolism are lipoxygenases (LOX), cytochrome P450 isoenzymes and cyclooxygenase.[1] Three major LOXs (5-, 12-, and 15-LOX) have been identified in various tissues[1] and several reports suggest that they participate in vascular remodeling in pulmonary hypertension.[2–4]
Similar to HETEs, prostaglandins (PGs) are products that are enzymatically derived from arachidonic acid and act as autocrine and paracrine effectors to regulate pulmonary vascular tone. The major PG generated by the endothelium of pulmonary arteries is prostacyclin (PGI2), a potent vasodilator which inhibits platelet aggregation and has antiproliferative and anti-inflammatory properties.[5] Conversely, thromboxane A2(TXA2), produced from arachidonic acid via the action of thromboxane synthase, opposes the actions of PGI2 by promoting vasoconstriction, platelet aggregation, and smooth muscle cell proliferation.[6]
Decreased local production and availability of PGI2 have been implicated in the pathogenesis of pulmonary hypertension. Using a rat model, Rabinovitch et al.[7] showed that induction of PGI2 release prevents hypoxic pulmonary hypertension and vascular remodeling. In addition, translational research showed that patients with pulmonary arterial hypertension (PAH) have an abnormally low ratio of urinary PGI2 to TXA2 metabolites, and this imbalance is thought to participate in the pulmonary vasoconstriction and local thrombosis that characterizes this condition.[8–9]
The importance of lipid mediators in modulating inflammation, cell proliferation, and pulmonary vascular remodeling is highlighted by the therapeutic use of various modulators of these pathways, with resultant amelioration of disease states. Replacement therapy with PGI2[10] or its analogues[11] is being used in the treatment of pulmonary hypertension. Therefore, there has been an ongoing interest in understanding the contribution of various lipid metabolites to the pathophysiology of pulmonary vascular disorders.
Experimental pulmonary hypertension studies in rodents have revealed remarkable interspecies differences. When exposed to hypoxia or monocrotaline, rats develop more severe pulmonary hypertension than mice.[12] Pulmonary vascular remodeling, inflammation, and pressure elevation are all more pronounced in rats, but the mechanisms explaining the interspecies differences have not been elucidated. Because of the evidence suggesting a role for bioactive lipids in the pathogenesis of PAH, we hypothesized that rat and mouse lungs would have different eicosanoid profiles associated with their differing pulmonary hypertensive responses.
MATERIALS AND METHODS
Materials
The following compounds were purchased from Cayman Chemical Co. (Ann Arbor, Mich., USA): 5-hydroxy-6E,8Z,11Z,14Z-eicosatetraenoic acid (5-HETE); 8-hydroxy-5Z,9E,11Z,14Z- eicosatetraenoic acid (8-HETE); 12-hydroxy-5Z,8Z,10E,14Z-eicosatetraenoic acid(12-HETE); 15-hydroxy- 5Z,8Z,11Z,13E-eicosatetraenoic acid(15-HETE); 9-oxo-11R,15S-dihydroxy-5Z,13E- prostadienoic acid (PGE2); 9-oxo-11S,15S-dihydroxy- 5Z,13E- prostadienoic acid(11β-PGE2); 9,15-dioxo-11R-hydroxy- 5Z,13E-prostadienoic acid (15-keto-PGE2); 9,15-dioxo-11R-hydroxy- 5Z-prostenoic acid(13,14-dihydro-15-keto-PGE2); 9S,15S-dihydroxy-11-oxo-5Z,13E-prostadienoic acid (PGD2); 11,15-dioxo-9S-hydroxy-5Z-prostenoic acid (13,14-dihydro-15-keto-PGD2); 9S,11R,15S- trihydroxy-5Z,13E-prostadienoic acid (PGF2α); 9S,11S,15S-trihydroxy-5Z,13E-prostadienoic acid(11β-PGF2α); 6-oxo-9S,11R,15S- trihydroxy-13E- prostenoic acid(6-keto-PGF1α); 9S,11,15S- trihydroxy-thromboxa-5Z,13E-dien-1-oic acid (TXB2); 9-oxo-11R,15S- dihydroxy-5Z,13E-prostadienoic acid-cyclo[8S,12R] (8-iso-PGE2), 9S, 11S, 15S- trihydroxy-(8β)- prosta-5Z, 13E- dien-1- oic acid(8-iso-PGF2α); 9S,11R-dihydroxy-15-oxo- 5Z-prostaenoic acid-cyclo [8S,12R] (8-iso-13,14-dihydro-15-keto-PGF2α).
Internal standards mixture (Cayman Chemical Co., Ann Arbor, Mich., USA): 5S-hydroxy-6E,8Z,11Z,14Z- eicosatetraenoic acid (5,6,8,9,11,12,14,15-d8) [5(S)-HETE-d8]; 12S- hydroxy- 5Z,8Z, 10E, 14Z- eicosatetraenoic acid-d8 [12(S)- HETE-d8]; 15S-hydroxy-5Z,8Z,11Z,13E- eicosatetraenoic acid-d8 [15(S)-HETE-d8]; 11R,15S-dihydroxy-9-oxo- 5Z,13E-prostadienoic acid(3,3,4,4-d4)(PGE2-d4); 9S,11,15S-trihydroxy-thromboxa-5Z,13E-dien-1-oic acid-d4 (TXB2-d4). Gradient grade methanol, water, hexane, and ammonium acetate for high-performance liquid chromatography (HPLC) ≥99.9% were purchased from Sigma Aldrich (St. Louis, MO).
Animals
Animals were purchased from Taconic, Germantown, N.Y., USA. Male mice (C 57 BL/6, 30-35 g) and rats (Sprague Dawley, 250-300 g) were maintained on standard laboratory chow diet and water ad libitum. After animals were euthanized with a pentobarbital injection (120 mg/kg ip), the thorax was opened immediately, the lungs were removed, weighed, frozen in liquid nitrogen, and stored at –80°C. The ages of the animals used for this study were between two and four months. The protocol was approved by the IACUC at Tufts Medical Center and was conducted in accordance with the National Institute of Health Guidelines for the Care and Use of Laboratory Animals.
Lung sample extraction and preparation
Lipid extraction of lung tissue was carried out as previously described, with modifications.[13,14] Briefly, lung tissue was homogenized for 30 seconds in 2 mL in 66% methanol. After 100 μL were removed for protein quantification, the samples were frozen in -80°C for 45 minutes for protein precipitation. The homogenates were then centrifuged at 4,500 rpm for 30 minutes at 4°C. The supernatant was diluted ≥ 10-fold with ice-cold HPLC grade water. The diluted supernatant was acidified on ice to a pH of 4.0 with 1N HCl. Using an Agilent 25 μL glass syringe, a 20μL aliquot of internal standard master mix (5-, 12-, 15-, HETE-d8, PGE2 -d4, TXB2 -d4) was added to each sample (total of 2 ng). The acidified supernatant containing the internal standard was loaded onto an Agilent C18 6 mL, 500 mg cartridge that was conditioned with 14 mL methanol, 14 mL water by low feeding speed in sequence. Following the addition of the sample, the cartridge was washed with 14 mL of HPLC grade water to remove any unabsorbed material and with 14 mL HPLC grade ice-cold hexane to remove any interfering lipids. The analytes were collected in glass Pyrex tubes by elution with 7 mL HPLC grade methanol. The solvent was evaporated under a gentle stream of nitrogen. The residue was reconstituted with 100 μL mobile phase, vortexed briefly and transferred to an autosampler vial insert for LC/MS/MS analysis. All extraction procedures were performed under minimum light conditions to avoid photodegradation of arachidonic acid metabolites . Protein concentrations were determined using the Bradford assay.[15]
HPLC simultaneous separation of arachidonic acid metabolites
A Phenomenex Luna 5 micron C18 (Phenomenex, Torrance, CA; 150 × 2.00 mm × 5 μm) was used for HPLC. The Agilent 1200 Series HPLC system consisted of a high-performance binary pump, a 108 well plate autosampler at 4°C, and column compartment set at 25°C.The solvent system consisted of 100% methanol and 10 mM ammonium acetate, pH 8.5. Prior to each run, the analytical column was pre-equilibrated with methanol for 60 seconds, and the injection syringe was washed for eight seconds in the flush port. A total of 25 μL of the sample was drawn up at a speed of 200 μL/min with a 100 μl syringe and was ejected onto the column at a rate of 400 μL/min. The flow rate of solvent through the column was 400 μL/min. The analytical column was primed with 100% ammonium acetate for 0.11 minutes and immediately switched to 50% methanol and 50% ammonium acetate at five minutes. At five minutes, methanol was programmed to increase linearly from 50% to 100% over a period of five minutes, after which the flow remained at 100% methanol for 20 minutes to ensure elution of all analytes. At 30.10 minutes, the flow immediately switched to 100% ammonium acetate for the remainder of the 35 minutes run, to flush the methanol entirely from the column. In order to get a complete separation of HETEs and PGs in the same run and under the same conditions, the column was flushed with 100% ammonium acetate for the last 4.90 minutes of each run.
MS/MS targeted profiling of hydroxyeicosatetraenoic acids and prostaglandins
Targeted profiling of HETEs and PGs was performed using a 5500 QTRAP (ABSciex) hybrid triple quadrupole linear ion trap mass spectrometer equipped with a turbo ion-spray ionization source.[16] Multiple reaction monitoring (MRM) experiments were performed using negative ion- spray mode. Detection of eicosanoids was separated into two periods and the dwell time used for MRM experiments was 400 msec for HETEs and 120 msec for PGs and TXB2, respectively. The collision exit potential was –11 V for HETEs and –13 V for PGs and TXB2. The declustering potential was –250 V for HETEs and –150 V for PGs and TXB2. The source-dependent MS parameters such as temperature and ion-spray voltage were set at 350°C and –4500 V, respectively. Collision energy was determined for each specific compound. Using a valco valve diverter, analytes were sent to waste for the first eight minutes of the run and then sent to the MS/MS where they were collected for 22 minutes of the run and then diverted back to waste for the remainder of the run. For details, see [Table 1S] and [Table 2S] in supplemental data.
Table 1S.
MRM specifications for measurement of select hydroxyeicosatetraenoic acids
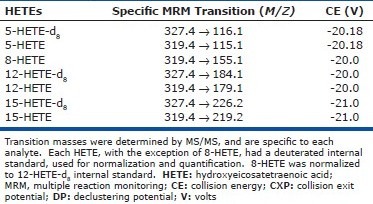
Table 2S.
MRM specifications for measurement of select prostaglandins and thromboxane
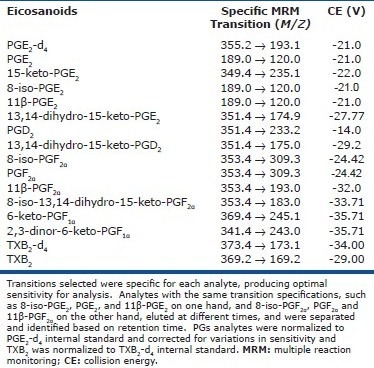
Statistical analysis
Values are reported in pg/μg protein, as median (25-75% confidence interval), or mean ± SD. Statistical analysis was performed using Sigma Stat 3.1 statistical software. Student's t-test or Wilcoxon-Mann-Whitney-U test was used to determine statistical significance when the normality passed or failed, respectively. P < 0.05 was considered statistically significant.
RESULTS
Lipid extraction from rodent lungs
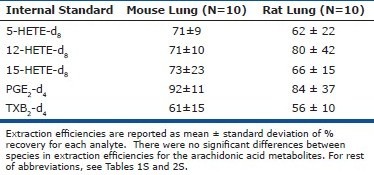
In order to ensure feasibility of comparison between species, lipid extraction was carried out under the same protocol for both rat and mouse lungs. Extraction efficiencies were 70.3 ± 17.0% for rat lung samples and 68.1 ± 26% (mean ± SD) for mouse lung samples. For detailed information on extraction efficiencies of various eicosanoids, see supplemental data [Table 3S].
Table 3S.
Extraction efficiency for select lipid metabolites in rodent lung tissue
Differences in HETEs between rats and mice
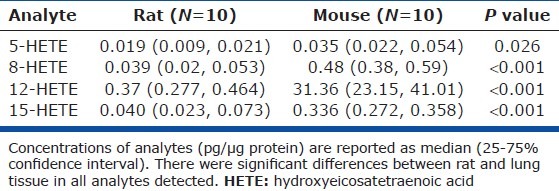
Analysis of lung samples from 10 rats and 10 mice showed significant interspecies differences. Basal levels of 5-, 8-, 12-, and 15-HETEs were detected in all samples from both rat and mouse lungs. MS/MS fragmentation characteristics are presented in [Table 1S]. While there was overlap in the values of various HETEs between species, mouse lungs had higher concentrations of all compounds measured Fig. 1, Table 1). Median values of the 5-, 8-, and 15-HETE were approximately two to 12 times higher in mouse than in rat lungs. The major HETE in both species was 12-HETE, whose levels were 80-fold higher in mouse samples.
Figure 1.

Differences in select hydroxyeicosatetraenoic acids (HETEs) between rat and mouse lungs. Concentrations of 5-HETE (A), 8-HETE (B), 12-HETE (C), and 15-HETE (D) are reported. * P < 0.05, ** P < 0.001
Table 1.
Concentrations of select hydroxyeicosatetraenoic acids in rodent lung tissue
Differences in prostaglandins, isoprostanes, and thromboxane between rat and mouse lungs
Based on characteristic MS/MS fragmentations [Table 2S] and retention times for different compounds, we identified the following compounds and their corresponding metabolites and isomers: PGE2 (with one isomer and two metabolites); PGD2 (with one metabolite); PGF2α(with one isomer); two PGI2 metabolites, one thromboxane metabolite (TXB2), and three compounds of the isoprostane pathway (Tables 2 and 3). Many PGs were nondetectable in the rat compared to mouse lungs [Table 2]. Among the compounds that were detectable in both species, PGI2 metabolites were significantly higher in mouse than in rat lungs and TXB2, the TXA2 metabolite had similar levels. Accordingly, there was a significantly higher PGI2/TXB2 ratio in mouse than in rat lungs (Fig. 2). Lastly, PGE2, a bronchodilator and vasodilator,[17] was also found in higher levels in mouse lungs.
Table 2.
Concentrations of select prostaglandins and thromboxane in rodent lung
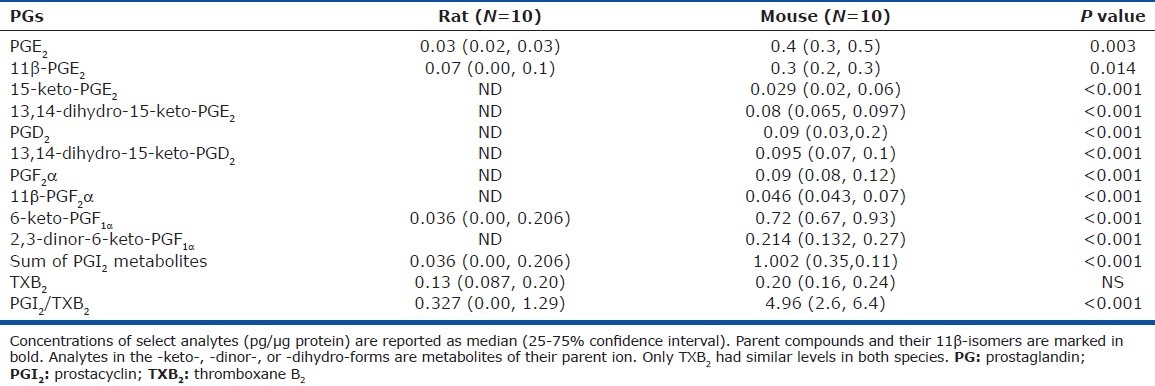
Table 3.
Concentrations of select isoprostanes in rodent lung
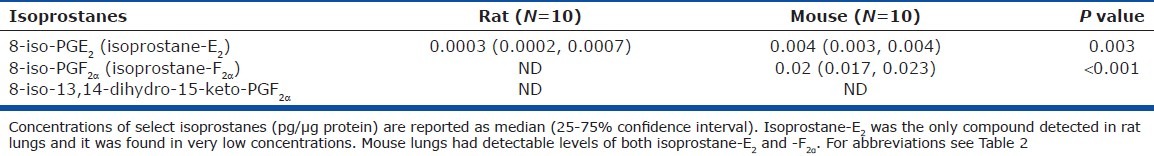
Figure 2.
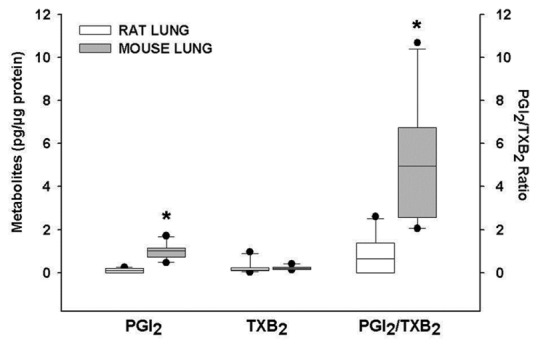
Concentrations of prostacyclin and thromboxane metabolites and their ratio in rat and mouse lungs. Mouse lungs had higher concentrations of PGI2 metabolites than rat lungs. Similarly, PGI2/TXB2 ratio was higher in mouse lungs. PGI2: Prostacyclin; TXB2: Thromboxane B2. *P < 0.001.
Interestingly, in rat lungs, isoprostane compounds, typically released under oxidative stress, were either not detectable or detected at low concentrations, whereas mouse lungs manifested significantly higher concentrations (Table 3).
DISCUSSION
We established that basal levels of select bioactive mediators derived from arachidonic acid are generated in the lungs of rats and mice and confirmed our hypothesis that there are interspecies pulmonary eicosanoid concentration differences that associate with interspecies differences in the severity of pulmonary hypertension.
5-, 8-, 12-, and 15-HETE were present in all samples and their levels were significantly higher in mouse lung tissue. HETEs can be synthesized by LOXs and cytochrome P450 isoenzymes. 5-HETE is an enzymatic product of 5-LOX, which can participate in the pathogenesis of experimental pulmonary hypertension. 5-LOX overexpression in rat lungs worsens the severity of pulmonary hypertension in the monocrotaline model and administration of 5-LOX inhibitors prevents the development of the disease.[18] In addition, 5-LOX inhibitors attenuate the growth of human pulmonary artery endothelial cells in culture, suggesting that this enzyme may modulate the response of the lung vasculature to experimental pulmonary hypertension.
12-HETE and 15-HETE are products of 12/15-LOX. 12-HETE is the major product of 12/15-LOX in rodents and is also generated by a dedicated 12-LOX. 12-HETE stimulates proliferation of pulmonary artery smooth muscle cells through a MAPK-dependent mechanism, possibly having a role in the remodeling process in experimental pulmonary hypertension. We previously reported an increase in 12-LOX gene expression and an upregulation of 12-LO protein in the lungs of hypoxic rats.[4] 15-HETE is a precursor for lipoxins, a family of proresolvin mediators[19] and has anti-inflammatory effects. 15-HETE effects on the vasculature, on the contrary, are conflicting. In some model systems, 15-HETE can mediate hypoxic pulmonary vasoconstriction[20,21] and displays antiapoptotic effects on pulmonary artery smooth muscle cells.[22] In other model systems, 15-HETE demonstrates vasodilator properties on both systemic and pulmonary beds.[23,24] Lastly, 8-HETE was also detected in both rat and mouse lungs, although its biologic properties on the lung vasculature are less extensively characterized.
Chronic hypoxia in mice is associated with only minimal vascular remodeling, certainly less than in rats.[12] The degree of remodeling parallels the degree of lung inflammation, with minimal inflammatory reaction in mice compared with rats. Similarly, monocrotaline causes severe pulmonary hypertension characterized by severe inflammation and remodeling in rats, but not in mice.[12] We found that two HETEs with vasoconstrictor and/or proliferative actions on the pulmonary vasculature, 5-and 12-HETE, are in higher concentrations in mouse than in rat lungs. While our results do not support the notion that basal HETE levels influence the response to pulmonary vascular stimuli, the significance of higher levels of 5-and 12-HETE as well as 15-HETE and 8-HETE in mouse lung under basal conditions is not known.
Human pulmonary hypertension is associated with an imbalance of urinary PGI2 and TXA2 metabolites favoring TXA2.[9] Moreover, expression of PGI2 synthase is decreased in the lungs of patients with pulmonary hypertension.[25] In addition, mice overexpressing PGI2 synthase are protected against the development of hypoxic pulmonary hypertension.[26] Therefore, it was of interest to determine lung concentrations of PGI2 and TXA2, as well as their ratio. TXA2 is unstable in vivo and is hydrolyzed within 30 seconds to TXB2. Similarly, PGI2 converts rapidly into several metabolites in vivo. We were able to measure TXB2 and two metabolites of PGI2, 6-keto-PGF1α and 2,3-dinor-6-keto-PGF1α. We determined that the total PGI2 metabolites and the ratio of the PGI2 metabolites and TXB2 were significantly higher in mouse lungs, and this increased ratio was mostly due to higher concentrations of PGI2 metabolites in mouse tissue. Of note, PGI2 is generated by the endothelium of pulmonary arteries, while TXA2 is mainly produced by activated platelets. The relative high concentration of PGI2 in the mouse suggests an increased cyclooxygenase-2 expression, or activity, or a decreased PGI2 metabolism in pulmonary endothelial cells. Since prior reports suggest an active role of PGI2 and TXA2 in the pathogenesis of pulmonary hypertension, it is possible that the mild degree of experimental pulmonary hypertension in mice is partly due to a high basal ratio of PGI2/TXB2.
Lastly, we determined basal levels of two products of the isoprostane pathway: 8-iso PGF2α and 8-iso PGE2. Isoprostane formation from arachidonyl moieties in phospholipids occurs in the setting of oxidative states and can disrupt biological membranes.[27] Under basal conditions, isoprostanes are rapidly cleaved, released, metabolized and excreted. 8-iso PGF2α is among the most abundant isoprostanes formed in vivo and is widely recognized as a biomarker of oxidative stress.[28] Our data suggest that mouse lungs had significantly higher basal levels of both 8-iso PGF2α and 8-iso PGE2 when compared with rat lungs, suggesting the existence of oxidative activity in mouse lungs under basal conditions. A third isoprostane, 8-iso-13,14-dihydro-15-keto-PGF2α, was not detected in any of the samples analyzed.
Limitations of our study include our inability to identify the cellular source of each eicosanoid in the lung, but reports from us and others suggest that the pulmonary vasculature is an active site of eicosanoid production. We did not determine which of the three pathways is more significant in the production of various HETEs in lung tissue: LOXs, cytochrome P450 isoenzymes, or cyclooxygenases. In addition, the column we selected did not permit chiral selective measurement of (R) and (S) enantiomers. More importantly, the present data cannot be directly extrapolated to susceptibility of these animals to the development of experimental PH. Further studies, such as determination of eicosanoid profiles with hypoxia-induced PH, are needed to clarify which of the eicosanoids described here modulate the vascular tone and cellular changes associated with PH.
In conclusion, we report for the first time, using a systematic screening approach through lipidomic analysis, a comparison of basal levels of select eicosanoids in rat and mouse lung. We show that arachidonic acid metabolism is active in rodent lung tissue and that there are significant interspecies differences in levels of select eicosanoids. These eicosanoids may participate in the maintenance and modulation of pulmonary vascular tone under basal conditions and during experimental vascular diseases. Lastly, we show that isoprostanes are present in mouse lung under basal conditions. Our determination of basal levels of select metabolites of arachidonic acid establishes the conditions with which to test whether differences in the levels of eicosanoids between species contribute to the differential responses to experimental pulmonary hypertension.
Footnotes
Source of Support: This work was funded by the following grants: National Institute of Health/NHLBI: K08 HL077341 (IRP); HL68669, GM095467 and HL086601 (BDL); and US Department of Agriculture 581950-7.707 (GGD).
Conflict of Interest: None declared.
REFERENCES
- 1.Funk CD. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science. 2001;294:1871–5. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 2.Casey LC, Fletcher JR, Zmudka MI, Ramwell PW. Prevention of endotoxin-induced pulmonary hypertension in primates by the use of a selective thromboxane synthetase inhibitor, OKY 1581. J Pharmacol Exp Ther. 1982;222:441–6. [PubMed] [Google Scholar]
- 3.Dusting GJ, Moncada S, Vane JR. Prostaglandins, their intermediates and precursors: Cardiovascular actions and regulatory roles in normal and abnormal circulatory systems. Prog Cardiovasc Dis. 1979;21:405–30. doi: 10.1016/0033-0620(79)90024-0. [DOI] [PubMed] [Google Scholar]
- 4.Preston IR, Hill NS, Warburton RR, Fanburg BL. Role of 12-lipoxygenase in hypoxia-induced rat pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol. 2006;290:L367–74. doi: 10.1152/ajplung.00114.2005. [DOI] [PubMed] [Google Scholar]
- 5.Vane JR, Botting RM. Pharmacodynamic profile of prostacyclin. Am J Cardiol. 1995;75:3A–10A. doi: 10.1016/s0002-9149(99)80377-4. [DOI] [PubMed] [Google Scholar]
- 6.Sachinidis A, Flesch M, Ko Y, Schror K, Bohm M, Dusing R, et al. Thromboxane A2 and vascular smooth muscle cell proliferation. Hypertension. 1995;26:771–80. doi: 10.1161/01.hyp.26.5.771. [DOI] [PubMed] [Google Scholar]
- 7.Rabinovitch M, Mullen M, Rosenberg HC, Maruyama K, O’Brodovich H, Olley PM. Angiotensin II prevents hypoxic pulmonary hypertension and vascular changes in rat. Am J Physiol. 1988;254(3 Pt 2):H500–8. doi: 10.1152/ajpheart.1988.254.3.H500. [DOI] [PubMed] [Google Scholar]
- 8.Christman BW. Lipid mediator dysregulation in primary pulmonary hypertension. Chest. 1998;114:205S–7S. doi: 10.1378/chest.114.3_supplement.205s. [DOI] [PubMed] [Google Scholar]
- 9.Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327:70–5. doi: 10.1056/NEJM199207093270202. [DOI] [PubMed] [Google Scholar]
- 10.Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334:296–302. doi: 10.1056/NEJM199602013340504. [DOI] [PubMed] [Google Scholar]
- 11.Simonneau G, Barst RJ, Galie N, Naeije R, Rich S, Bourge RC, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: A double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165:800–4. doi: 10.1164/ajrccm.165.6.2106079. [DOI] [PubMed] [Google Scholar]
- 12.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1013–32. doi: 10.1152/ajplung.00217.2009. [DOI] [PubMed] [Google Scholar]
- 13.Levy BD, Kohli P, Gotlinger K, Haworth O, Hong S, Kazani S, et al. Protectin D1 is generated in asthma and dampens airway inflammation and hyperresponsiveness. J Immunol. 2007;178:496–502. doi: 10.4049/jimmunol.178.1.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seki H, Fukunaga K, Arita M, Arai H, Nakanishi H, Taguchi R, et al. The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. J Immunol. 2010;184:836–43. doi: 10.4049/jimmunol.0901809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 16.Yang R, Chiang N, Oh SF, Serhan CN. Metabolomics-lipidomics of eicosanoids and docosanoids generated by phagocytes. Curr Protoc Immunol. 2011 doi: 10.1002/0471142735.im1426s95. Chapter 14:Unit 14.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ceserani R, Gandolfi C, Usardi MM, Bergamaschi M. PGE2:PGE-2: Prostaglandins. 1975;9:97–107. doi: 10.1016/s0090-6980(75)80121-3. [DOI] [PubMed] [Google Scholar]
- 18.Jones JE, Walker JL, Song Y, Weiss N, Cardoso WV, Tuder RM, et al. Effect of 5-lipoxygenase on the development of pulmonary hypertension in rats. Am J Physiol Heart Circ Physiol. 2004;286:H1775–84. doi: 10.1152/ajpheart.00281.2003. [DOI] [PubMed] [Google Scholar]
- 19.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–61. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li S, Ran Y, Zheng X, Pang X, Wang Z, Zhang R, et al. 15-HETE mediates sub-acute hypoxia-induced TRPC1 expression and enhanced capacitative calcium entry in rat distal pulmonary arterial myocytes. Prostaglandins Other Lipid Mediat. 2010;93:60–74. doi: 10.1016/j.prostaglandins.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 21.Zhu D, Medhora M, Campbell WB, Spitzbarth N, Baker JE, Jacobs ER. Chronic hypoxia activates lung 15-lipoxygenase, which catalyzes production of 15-HETE and enhances constriction in neonatal rabbit pulmonary arteries. Circ Res. 2003;92:992–1000. doi: 10.1161/01.RES.0000070881.65194.8F. [DOI] [PubMed] [Google Scholar]
- 22.Nie X, Song S, Zhang L, Qiu Z, Shi S, Liu Y, et al. 15-Hydroxyeicosatetraenoic acid (15-HETE) protects pulmonary artery smooth muscle cells from apoptosis via inducible nitric oxide synthase (iNOS) pathway. Prostaglandins Other Lipid Mediat. 2012;97:50–9. doi: 10.1016/j.prostaglandins.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 23.Aggarwal NT, Chawengsub Y, Gauthier KM, Viita H, Yla-Herttuala S, Campbell WB. Endothelial 15-lipoxygenase-1 overexpression increases acetylcholine-induced hypotension and vasorelaxation in rabbits. Hypertension. 2008;51:246–51. doi: 10.1161/HYPERTENSIONAHA.107.104125. [DOI] [PubMed] [Google Scholar]
- 24.Chawengsub Y, Aggarwal NT, Nithipatikom K, Gauthier KM, Anjaiah S, Hammock BD, et al. Identification of 15-hydroxy-11,12-epoxyeicosatrienoic acid as a vasoactive 15-lipoxygenase metabolite in rabbit aorta. Am J Physiol Heart Circ Physiol. 2008;294:H1348–56. doi: 10.1152/ajpheart.01326.2007. [DOI] [PubMed] [Google Scholar]
- 25.Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159:1925–32. doi: 10.1164/ajrccm.159.6.9804054. [DOI] [PubMed] [Google Scholar]
- 26.Geraci MW, Gao B, Shepherd DC, Moore MD, Westcott JY, Fagan KA, et al. Pulmonary prostacyclin synthase overexpression in transgenic mice protects against development of hypoxic pulmonary hypertension. J Clin Invest. 1999;103:1509–15. doi: 10.1172/JCI5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morrow JD, Awad JA, Boss HJ, Blair IA, Roberts LJ., 2nd Non-cyclooxygenase-derived prostanoids (F2-isoprostanes) are formed in situ on phospholipids. Proc Natl Acad Sci U S A. 1992;89:10721–5. doi: 10.1073/pnas.89.22.10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lawson JA, Rokach J, FitzGerald GA. Isoprostanes: Formation, analysis and use as indices of lipid peroxidation in vivo. J Biol Chem. 1999;274:24441–4. doi: 10.1074/jbc.274.35.24441. [DOI] [PubMed] [Google Scholar]