

Engineered extracellular matrices (eECMs) that broadly mimic the in vivo microenvironment while offering several tunable parameters provide physiological yet finely controlled matrix-cell signals for tissue engineering applications. The ideal eECM has nanofibrous architecture, tissue-like mechanics, and specific bioactive ligands, each of which should be adjustable over a physiological range for desired applications. To meet this need, a multivariately tunable family of recombinant elastin-like proteins (ELPs) is electrospun into an implantable, nanofibrous fabric from aqueous solution. Although electrospinning of recombinant proteins is not a new idea, previous efforts have relied on blending with synthetic polymers or crosslinking with non-specific fixatives to achieve stable fibrous matrices. These processes decrease the physiological verisimilitude and specificity of protein nanofibers, thus defeating the purpose of protein-based biomaterials. Here, by analyzing ELP's thermodynamic behavior, we devise a novel, sequence-specific, sequential process comprised of vapor-phase initiation and aqueous completion of crosslinks (Figure 1 a). Sequence-specific crosslinking (a) preserves arginine-glycine-aspartic acid (RGD) ligands, expressed along the protein's backbone, through nanofabrication and (b) provides stable nanotopology and native-like mechanics for cell-matrix interactions. Individual fibers are ribbon-like (thickness of 190 ± 60 nm) with tunable widths (1.2–1.8 μ m), and bulk implantable matrices have tissue-like compliance (tensile modulus of 62 ± 6 kPa). Applying coherent anti-Stokes Raman scattering (CARS) microscopy, which requires no labeling or chemical post-processing and thus preserves cell-matrix adhesions, we confirm the close interaction between rat marrow stromal cells (rMSCs) and the eECM.
Figure 1.

Stability of nanofibrous ELP. a) Comparison of protein loss in physiological conditions over eight days from untreated fabrics and two-stage crosslinked fabrics. b) CARS microscopy (2930 cm−1) 3D view of nanofibers after two weeks in physiological buffer. c) Schematic of two-stage crosslinking strategy. Crosslinking is initiated by exposing matrices to glutaraldehyde vapor. Complete crosslinks are formed after hydration in a concentrated sodium chloride buffer. d) LCST of ELP in varying buffer conditions. e) Comparison of overnight protein loss at physiological conditions for fabrics hydrated in varying w/v, ELP per 10× phosphate-buffered saline (PBS), during the aqueous crosslinking completion step.
Protein-engineered materials modularly incorporate extended peptide sequences to achieve functional and structural diversity.[1] Classical ELPs feature sequential repeats of valine-proline-glycine-X-glycine (VPGXG, where X is any amino acid) that enable facile purification, controlled crosslinking, and rubber-like elasticity.[2] ELP mechanically mimics native elastin,[3] which is an attractive target for nanofabrication in tissue-engineered constructs because of limited in vivo production of elastin fibers post-development. Likewise, many diseases involve mechanical dysregulation by degraded elastin fibers as both symptom and etiology.[4] Selection of lysine for the X-residue position enables crosslinking with amine-reactive small molecules, preserving elasticity while allowing stoichiometric control of crosslinking density and hence mechanical properties.[5] The position of lysine (Figure S1 of the Supporting Information (SI)) minimizes distortion of bioactive sequences, enabling simultaneous control of bioactivity and mechanics.[6] Specific bioactive domains designed into ELP hydrogels include fibronectin- and laminin-derived adhesion ligands, juxtacrine ligands,[7] and enzyme-specific cleavage sites.[8,9] While this compositional tunability results in materials whose mechanics, cell adhesion, and proteolytic degradation can be multifactorially optimized for specific tissue engineering applications, ELP hydrogels are typically amorphous and lack the nanofibrous architecture inherent in native extracellular matrix. Elastin-based protein nanofibers, which are decorated with bioactive, sequence-specific ligands in vivo, are an essential constituent of mammalian tissues and a goal for biomimetic eECMs.[10]
To impose biomimetic architecture on this eECM, we demonstrate electrospinning of pure ELPs into nanofibrous fabrics. Electrospinning is a versatile and scalable nanofabrication technique that has been applied to many medically relevant polymers.[11,12] Pure ELP has not been nanofabricated as a mimetic eECM because, without intervention, ELP fibers rapidly degrade under physiological conditions. Existing interventions to stabilize ELP fibers include blending with polycaprolactone before spinning,[13] genetically incorporating silk-like crosslinking domains,[14] and chemically incorporating methacrylate to enable radical-mediated photo-crosslinking.[15] Although effective, these approaches may negatively impact the biomimetic mechanical and bioactive properties of ELPs. Incorporation of other polymers or silk-like domains will likely alter the nanoscale mechanics of ELP fibers with unknown biological consequences. Furthermore, synthetic polymers can adsorb soluble proteins resulting in altered cell-substrate interactions.[16] Finally, radical chemistry, which is inherently non-specific, may interfere with both mechanical and bioactive properties. Extensive crosslinking within the VPGXG repeat may prevent proper folding into β-spirals, which are thought to be required for ELP rubber-like elasticity.[17] Similarly, crosslinking must be orthogonal to sequence-expressed ligands to preserve both bioactivity and reductive tunability. To achieve a tunable eECM with native-like nanoscale context, a crosslinking strategy that respects sequence specificity is needed. With a similar aim, pure solutions of recombinant tropoelastin in polyfluorinated solvents were electrospun and crosslinked with glutaraldehyde to form cytocompatible fabrics.[18,19] This previous work motivates exploration of glutaraldehyde, which preferentially reacts with lysine, as a fixation strategy for modular, protein-engineered ELPs.
Prior to electrospinning, ELPs (37.6 kDa) are expressed in Escherichia coli, purified, and suspended in deionized water (35% w/w). The resulting solutions are electrospun, producing a fibrous mesh. Without any crosslinking at all, fibers disassemble and loose their structural morphology almost immediately after exposure to a physiological buffer, phosphate-buffered saline (PBS, 0.137 mM sodium chloride (NaCl)) (Figure S2a of the SI). Although ELPs exhibit a lower-critical solution temperature (LCST) that is below physiological temperature (37 °C) in PBS,[8] the semi-soluble electrospun fibers disassemble and dissolve within one week at these conditions due to dynamic equilibrium and medium changes (Figure 1 a). These results are consistent with ELP's LCST behavior, which means that it remains fully soluble at temperatures below the LCST and that it partitions into polymer-rich and polymer-lean phases above the LCST. While this level of degradability may be appropriate for certain medical uses, the ability to maintain fabric stability over longer time periods is essential for tissue engineering.
To address this need, we have used thermodynamic analysis to develop a novel, two-stage crosslinking protocol that dramatically improves fiber stability: after crosslinking, fibers elute less than 2% of their total mass during one week of standard culture conditions (Figure 1 a). Non-linear CARS microscopy, which requires no labeling or post-processing of the ELP material, reveals the presence of distinct nanofiber morphology throughout the bulk after two weeks in physiological buffer (Figure 1 b). This effective and sequence-specific crosslinking is essential to the use of electrospun ELP as a tunable eECM.
In more detail, our two-step crosslinking process consists of a vapor-phase initiation followed by an aqueous-phase completion of crosslinks (Figure 1 c). During the first step, we treat fabrics with glutaraldehyde vapor, which has been used to stabilize electrospun fibers of other recombinant proteins.[20] Introduction of glutaraldehyde in the vapor-phase, as opposed to liquid-phase, prevents dissolution of ELP fibers or distortion of fiber morphology. In addition, glutaraldehyde vapor is restricted to the high vapor pressure monomeric species, which is a homobifunctional small molecule (100 Da). Importantly, monomeric glutaraldehyde preferentially reacts with the primary amine side chain of lysines.[21] This crosslinking specificity prevents conformational disruption of bioactive sequences and preserves the elastomeric secondary structure of VPGXG repeats. Lysine-specific crosslinks are also essential to formation of elastic fibers in vivo.[22] Conversely, aqueous glutaraldehyde is capable of forming high molecular weight oligomers, which react with less specificity for primary amines. Using glutaraldehyde vapor to initiate crosslinking avoids dissolution while targeting a specific sequence that tolerates modification without disrupting mechanical or bioactive sequences.
After this initial crosslinking step, hydrating the fibers in PBS results in partial disassembly, i.e., fibers appear to fray and to aggregate (Figure S2 of the SI). Based on this result, we hypothesized that during vapor-phase initiation of crosslinking, on average only one of glutaraldehyde's functional groups reacts with the protein, effectively conjugating a reactive aldehyde to the ELP. Monomeric glutaraldehyde is 7.5 Å in length, and during vapor-phase crosslinking, proteins are entrapped in dry fibers with little mobility. To form a complete crosslink, the other reactive group must find a primary amine on an adjacent protein. We reasoned that increasing the protein mobility by hydrating the nanofibers with a minimal volume of buffer would allow the reactive aldehyde to “find” another primary amine and hence complete the crosslinking reaction. As proof of this idea, a second crosslinking step was designed, during which nanofibers are hydrated in a small volume of concentrated NaCl buffer without any additional glutaraldehyde crosslinking molecules added to the aqueous phase. ELP nanofibers treated in this manner were observed to stay uniform and discrete even after replacement of the high salt buffer with a physiological medium (Figure 1 a,b; Figure S2 of the SI). This increase in nanofiber stability demonstrates that the glutaraldehyde crosslinks that were initiated in the vapor phase were able to be completed in the aqueous phase.
To determine the buffer requirements for this crosslinking completion step, we evaluated the ELP's thermodynamics-driven LCST behavior. The hydration buffer must be selected to minimize ELP dissolution while enabling chain mobility to promote crosslinking completion. Consistent with results on other ELP molecules, [23] the LCST is found to decrease in more concentrated NaCl solutions (from 0.1–0.7 M) and in more concentrated ELP solutions (from 0.005 to 0.025 mass ELP per volume buffer) (Figure 1 d). These data led us to compare two hydration volumes of a high-salt buffer (10× phosphate-buffered saline, PBS, with 1.4 M NaCl), corresponding to ELP concentrations of 0.005 and 0.083 w/v. As predicted by the LCST data, fiber stability is significantly improved upon use of the lesser hydration volume for the crosslinking completion step, which decreases the ELP solubility (Figure 1 e). Taken together, these results demonstrate that this novel, two-stage crosslinking protocol results in preservation of the nanofibrous, 3D architecture over a period relevant for in vitro studies of cell-matrix interactions.
Stable ELP nanofibers are characterized to determine if nanofiber morphology, mechanics, and cell adhesion can be tuned to fall within the range necessary for use as tissue engineering scaffolds. Fibers are spun from pure ELP solutions of two concentrations: 25% and 35% w/w, both of which produced ribbon-like nanofibers (Figure 2 a,b). Further quantitation using scanning electron microscopy (SEM, n > 100 fibers) shows that nanofiber width and thickness exhibit Gaussian distributions (Figure 2 c). Increasing the ELP fraction in solution increases the anisotropy of the fibers. While fiber thickness, defined as the cross-section's minor axis, is similar (180 ± 50 nm and 190 ± 60 nm), fiber width, i.e., the cross-section's major axis, increases for higher ELP weight fractions (1.2 ± 0.3 μ m and 1.8 ± 0.4 μm for 25% w/w and 35% w/w, respectively). Mechanistically, protein chain entanglements increase in density with polymer mass fraction. These chain entanglements increase fiber cross-section by sterically resisting electrostatic-driven narrowing of the fluidic electrospinning jet.[24] Fibers reach their final conformation once solvent fully evaporates from the jet, entrapping constituent polymers. Difference in the width and thickness of fibers is thought to be a consequence of drying inhomogeneity: the inchoate fiber's cross-section becomes elliptical as the dry, entrapped shell collapses into the still fluid, extending core.[25] Overall, we can control the nanoscale architecture of electrospun ELP by simply changing the polymer fraction in the pre-spinning solution.
Figure 2.
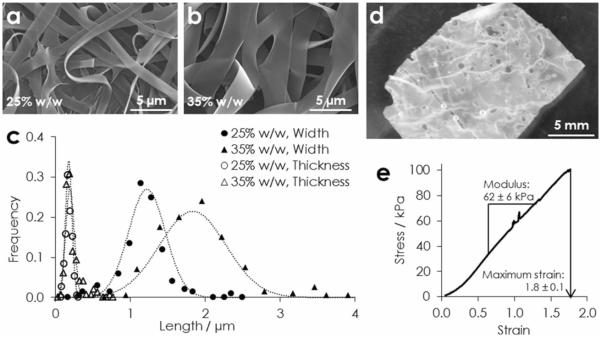
Physical characterization of electrospun ELP. a) SEM of fibers spun from aqueous solutions of a) 20%, and b) 35% w/w ELP. c) Histograms and Gaussian fits of nanofiber width and thickness (n > 100) from both solutions. d) An implantable bulk fabric in PBS, and e) a representative tensile stress-strain curve.
Appropriately for a tunable, bioactive material, ribbon-like fibers have greater surface area than cylindrical fibers of the same cross-sectional area. This fabrication increases the potential number of ligands accessible for cell-matrix interaction, enabling a greater effective range of matrix-displayed ligand density. Moreover, native elastin fibers are often ribbon-like, including those in the smooth muscle of the vascular tunica media (width 0.9 – 1.7 μ m)[26] and the dermis (1.0 – 3.5 μ m).[27] This similarity to native tissues makes nanofibrous ELP an architecturally relevant eECM for a variety of tissue engineering applications.
To be clinically relevant, eECMs must have the capacity to be fabricated as bulk, tissue-scale constructs. Deposition of ~4 mg ELP per cm2 produces a free-standing, implantable fabric (Figure 2 d). Tensile testing (n = 5) indicates an elastic modulus of 62 ± 6 kPa, as measured in the linear range of deformation from 0.7 to 1.3 strain. Fabrics are highly extensible and, after a toe region, deform linearly to rupture, with maximum strain of 1.8 ± 0.1 (Figure 2 e). These mechanical properties are relevant to soft tissues, such as smooth muscle, which has a tensile modulus of 100 ± 20 kPa at rest.[28] Tissue-like mechanical properties are critical for achieving functional outcomes, such as differentiation.[29] Although not explored here, mechanical properties of ELP constructs can be tailored by controlling crosslink density through glutaraldehyde stoichiometry.[21]
Modular protein-engineering of the ELP family enables, in addition to sequence-targeted crosslinking, reductionist tuning of bioactive ligand density. By blending the RGD-bearing ELP (ELP-RGD) with an otherwise identical ELP variant that includes a non-cell-adhesive scrambled ligand sequence (ELP-RDG),[30] the exact concentration of RGD ligands in the nanofibers is controlled without altering any of the other material properties, including swelling ratio, surface charge, fiber morphology, or crosslinking density. To confirm that the RGD ligand retains its bioactivity during electrospinning and crosslinking, the adhesion and metabolic activity of rMSCs is evaluated. After one day in serum-free medium, metabolic activity on the non-adhesive ELP-RDG fabric is significantly less than the ELP-RGD fabric and the glass control (n = 3, p < 0.02) (Figure 3 a). Comparing the glass control to ELP-RDG, these data suggest that ELP-RDG reduces non-specific cell adhesion and survival. As a β1 integrin-dependent cell type,[31] it is not surprising that rMSCs have a pro-survival response to the presence of the RGD ligand, which is derived from a domain in fibronectin that activates the β1-family integrins.[32] Specific control of signaling from these integrins is expected to be of clinical relevance because MSCs are an expandable, autologous source of tissue-plastic cells.
Figure 3.

Bioactivity of the RGD ligand. a) Metabolic activity of rMSCs after 24 h in serum-free medium on ELP-RGD (8.4 mM RGD) or ELP-RDG (0 mM RGD) fabrics or glass control (n = 3). Maximal projection images obtained by CARS microscopy of rMSC after 24 h in standard conditions on electrospun ELP-RGD: b) nanofibers, c) rMSCs, d) overlay. Close contact is suggested by imprints of nanofibers on the cell membrane, white arrow.
The capacity to investigate cell-fiber interactions unaltered by any labeling chemistries whatsoever in a nanofibrous context is an advantage of this eECM. Using CARS microscopy, a label-free imaging modality that is compatible with 3D studies (Figure 1 c) and live imaging,[33] we see close adhesion between cell membranes and fibers of the ELP matrix, with cells spanning multiple fibers (Figure 3 b–d). For these experiments, in which excitation wavenumbers are varied to optimize protein/lipid contrast, ELP-RGD was chosen as the constituent protein to increase the likelihood of cell-fiber interaction. By probing the carbon-hydrogen vibrations at 2845 cm−1 and 2930 cm−1, contrast is obtained for lipid- and protein-rich structures, respectively. Thus, the cell membrane, internal lipid stores, and the nanofibrous ELP can be resolved. Imprints of nanofibers in the cell membrane, i.e., relative decreases in the lipid signal that coincide with enhanced protein fiber signal, are evident (Figure 3 c), proving close interactions between rMSCs and nanofibers under native-like conditions without any artificial contrast enhancement that may affect the scaffold architecture or cell-scaffold adhesions. In addition to confirming cell-fiber interactions after electrospinning and the novel two-step crosslinking procedure, these results foreshadow the exciting possibility of real-time analysis of tissue dynamics within a nanofibrous eECM using CARS microscopy, which is free of potential artifacts caused by cell labeling.
Overall, our results demonstrate the successful sequence-targeted stabilization of mimetic eECMs nanofabricated from a highly tunable family of ELPs. Taking advantage of the thermodynamic behavior of glutaraldehyde and ELP, we have developed a novel, two-stage protocol that preserves native-like mechanics and matrix-displayed ligands while adding tunable and physiological nanotopology. Within the context of the broader field of protein electrospinning, this advance obviates the need for synthetic polymeric additives or non-specific fixatives, which countervail both the bio-compatibility and specificity of protein-based biomaterials. Although previous engineering of the ELP family enabled tuning of elastic modulus, biodegradation rate, and density of matrix-displayed ligands within amorphous hydro-gels, nanofabrication and crosslinking enables tuning of the matrix morphology to achieve a more mimetic, nanofibrous microenvironment. By simply changing the mass fraction of ELP in the spinning solution, we gain additional control over fiber dimension. Reductive control of multiple parameters is essential for optimizing materials for specific tissue-engineering applications. Ultimately, the ability to orthogonally stabilize nanofibrous protein matrices that are electrospun from tunable ELPs has the potential to deliver a new level of control over the behavior of cells and engineered tissues.
Experimental Section
Scanning electron microscopy, quantification of LCST, tensile testing, culture of rMSCs, and confocal fluorescent microscopy
The techniques are performed according to standard protocols and are described in the SI.
ELP synthesis and processing
All protein sequences were cloned, expressed, and purified as previously described, unless otherwise noted.[8] To de-salt before lyophilization, solutions were dialyzed three times (10,000 molecular weight cutoff, 4 h, 4 ° C, deionized water). For electrospinning (NaBond electrospinning unit), protein solutions were extruded (0.2 mL h−1) through a blunt needle (stainless steel, 32 gauge, room temperature). An electric field (15 kV, 15 cm) was applied from extruder to a plate collector. Fabrics were collected on grounded stainless steel plate (8 h) or on aminopropyltriethoxysilane-aminated glass slides (40 min). Nanofibrous eECMs were treated in a vacuum chamber containing two separate solutions (10 mL glutaraldehyde and 10 mL water saturated with potassium chloride, 24 h). For the second crosslinking step, fabrics were hydrated with varying buffers (1 h, 37 ° C). To quantify protein stability, the hydration buffer is diluted to 300 μL with PBS. At each time point, soluble ELP is measured by bicinchoninic acid assay (BCA assay, Sigma). For cell culture, fabrics are sterilized and reactive aldehydes are quenched by autoclaving with 2% w/v lysine.
CARS microscopy
The set-up for CARS microscopy is described in the SI and in detail elsewhere.[33] Samples were imaged in unlabeled, hydrated conditions. Both cells and ELP generate a CARS signal at 2845 cm−1, whereas ELP only displays a distinct resonance at 2930 cm−1. With imaging software (ImageJ) the cells were identified by subtracting the 2930 cm−1 from the 2845 cm−1 image.
Supplementary Material
Acknowledgements
We acknowledge Ivan Wong for technical training on and Marc Levenston for provision of tensile testing equipment. The authors acknowledge funding from NIH F31-HL114315-01 (P.L.B.), NSF DMR-0846363, NIH R21-AR062359, NIH DP2-OD-006477, and Stanford Cardiovascular Institute Younger Grant (S.C.H.), European Commission/VINNOVA Marie-Curie Fellowship (A.E.), and Indo-US Science and Technology Forum (K.P.C. and S.V.N.).
Footnotes
Supporting Information Supporting Information is available from the Wiley Online Library or from the author.
References
- [1].Sengupta D, Heilshorn SC. Tissue Eng. Part B. Rev. 2010;16:285. doi: 10.1089/ten.teb.2009.0591. [DOI] [PubMed] [Google Scholar]
- [2].Chilkoti A, Christensen T, MacKay JA. Curr. Opin. Chem. Biol. 2006;10:652. doi: 10.1016/j.cbpa.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lee J, Macosko CW, Urry DW. Macromolecules. 2001;34:5968. [Google Scholar]
- [4].Brooke BS, Bayes-Genis A, Li DY. Trends Cardiovasc. Med. 2003;13:176. doi: 10.1016/s1050-1738(03)00065-3. [DOI] [PubMed] [Google Scholar]
- [5].Di Zio K, Tirrell DA. Macromolecules. 2003;36:1553. [Google Scholar]
- [6].Heilshorn SC, Liu JC, Tirrell DA. Biomacromolecules. 2005;6:318. doi: 10.1021/bm049627q. [DOI] [PubMed] [Google Scholar]
- [7].Straley KS, Heilshorn SC. Front. Neuroeng. 2009;2:9. doi: 10.3389/neuro.16.009.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Straley KS, Heilshorn SC. Soft Matter. 2009;5:114. [Google Scholar]
- [9].Straley KS, Heilshorn SC. Adv Mater. 2009;21:4148. [Google Scholar]
- [10].Kielty CM. Expert Rev. Mol. Med. 2006;8:1. doi: 10.1017/S146239940600007X. [DOI] [PubMed] [Google Scholar]
- [11].Agarwal S, Wendorff JH, Greiner A. Polymer. 2008;49:5603. [Google Scholar]
- [12].Liu W, Thomopoulos S, Xia Y. Advanced Healthcare Materials. 2012;1:2. doi: 10.1002/adhm.201100021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lee S, Kim J, Chu HS, Kim G, Won J, Jang J. Acta Biomaterialia. 2011;7:3868. doi: 10.1016/j.actbio.2011.06.035. [DOI] [PubMed] [Google Scholar]
- [14].Qiu W, Huang Y, Teng W, Cohn CM, Cappello J, Wu X. Biomacromolecules. 2010;11:3219. doi: 10.1021/bm100469w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nagapudi K, Brinkman WT, Leisen JE, Huang L, McMillan RA, Apkarian RP, Conticello VP, Chaikof EL. Macromolecules. 2002;35:1730. [Google Scholar]
- [16].Horbett TA. In: Biomaterials: Interfacial Phenomena and Applications. Cooper Stuart L., Peppas Nicholas A., Hoffman Allan S., Ratner Buddy D., editors. Vol. 199. American Chemical Society; Washington, DC, USA: 1982. Ch. 17. [Google Scholar]
- [17].Venkatachalam CM, Urry DW. Macromolecules. 1981;14:1225. [Google Scholar]
- [18].Li M, Mondrinos MJ, Gandhi MR, Ko FK, Weiss AS, Lelkes PI. Biomaterials. 2005;26:5999. doi: 10.1016/j.biomaterials.2005.03.030. [DOI] [PubMed] [Google Scholar]
- [19].Rnjak-Kovacina J, Wise SG, Li Z, Maitz PKM, Young CJ, Wang Y, Weiss AS. Biomaterials. 2011;32:6729. doi: 10.1016/j.biomaterials.2011.05.065. [DOI] [PubMed] [Google Scholar]
- [20].Li M, Mondrinos MJ, Gandhi MR, Ko FK, Weiss AS, Lelkes PI. Biomaterials. 2005;26:5999. doi: 10.1016/j.biomaterials.2005.03.030. [DOI] [PubMed] [Google Scholar]
- [21].Welsh ER, Tirrell DA. Biomacromolecules. 2000;1:23. doi: 10.1021/bm0002914. [DOI] [PubMed] [Google Scholar]
- [22].Miller EJ, Martin GR, Piaz KA. Biochem. Biophys. Res. Commun. 1964;17:248. doi: 10.1016/0006-291x(64)90392-4. [DOI] [PubMed] [Google Scholar]
- [23].Reguera J, Urry DW, Parker TM, McPherson DT, Rodríguez-Cabello JC. Biomacromolecules. 2007;8:354. doi: 10.1021/bm060936l. [DOI] [PubMed] [Google Scholar]
- [24].McKee MG, Wilkes GL, Colby RH, Long TE. Macromolecules. 2004;37:1760. [Google Scholar]
- [25].Koombhongse S, Liu W, Reneker DH. Journal of Polymer Science Part B: Polymer Physics. 2001;39:2598. [Google Scholar]
- [26].Crissman RS, Guilford W. Am. J. Anat. 1984;171:401. doi: 10.1002/aja.1001710405. [DOI] [PubMed] [Google Scholar]
- [27].Tsuji T, Lavker RM, Kligman AM. J. Microsc. 1979;115:165. doi: 10.1111/j.1365-2818.1979.tb00166.x. [DOI] [PubMed] [Google Scholar]
- [28].Bergel DH. J. Physiol. 1961;156:445. doi: 10.1113/jphysiol.1961.sp006686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Engler AJ, Griffin MA, Sen S, Bonnemann CG, Sweeney HL, Discher DE. J. Cell Biol. 2004;166:877. doi: 10.1083/jcb.200405004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cook AD, Hrkach JS, Gao NN, Johnson IM, Pajvani UB, Cannizzaro SM, Langer R. J. Biomed. Mater. Res. 1997;35:513. doi: 10.1002/(sici)1097-4636(19970615)35:4<513::aid-jbm11>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- [31].Daley WP, Peters SB, Larsen M. J. Cell. Sci. 2008;121:255. doi: 10.1242/jcs.006064. [DOI] [PubMed] [Google Scholar]
- [32].Plow EF, Haas TA, Zhang L, Loftus J, Smith JW. J. Biol. Chem. 2000;275:21785. doi: 10.1074/jbc.R000003200. [DOI] [PubMed] [Google Scholar]
- [33].Hellerer T, Axang C, Brackmann C, Hillertz P, Pilon M, Enejder A. Proc. Natl. Acad. Sci. U. S. A. 2007;104:14658. doi: 10.1073/pnas.0703594104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.